
Scaffold Morphing and In Silico Design of Potential BACE-1 (β-Secretase) Inhibitors: A Hope for a Newer Dawn in Anti-Alzheimer Therapeutics
 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Scaffold Morphing through Bioisosteric Replacement
2.2. In Silico Pharmacokinetic Studies
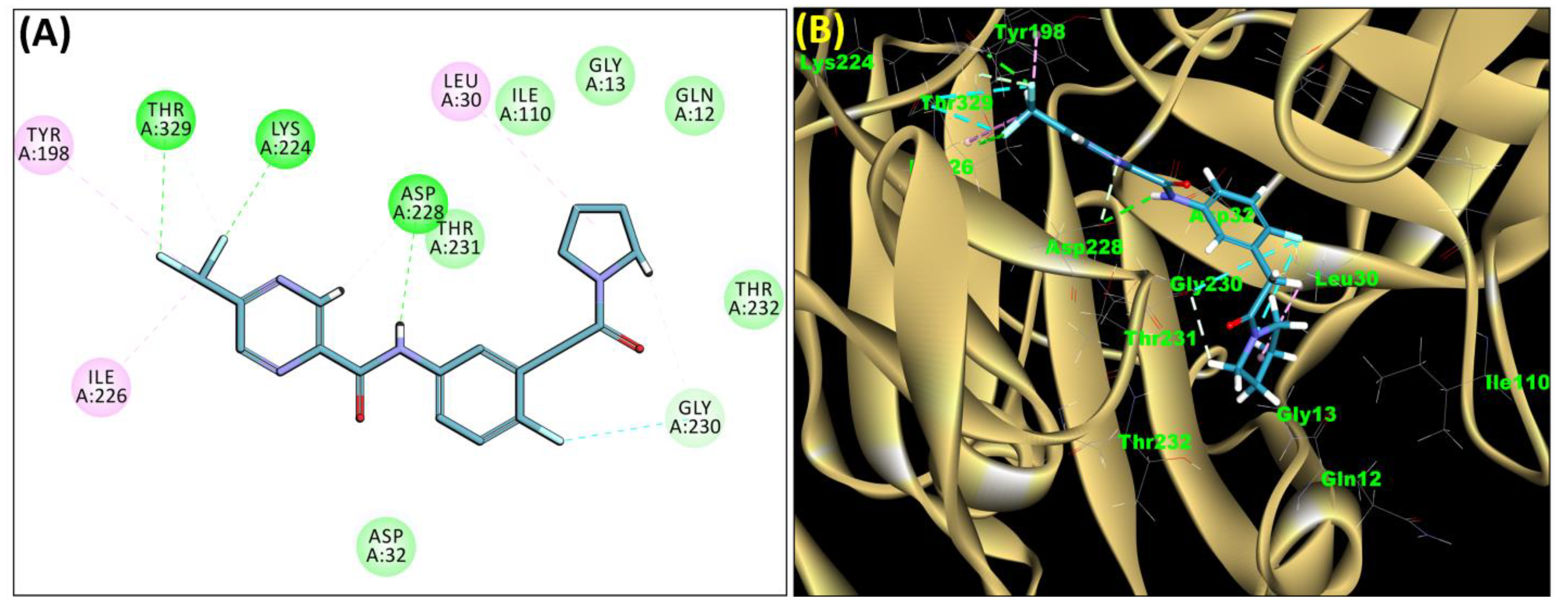
2.3. Molecular Docking
3. Molecular Dynamics Simulations
4. Material and Methods
4.1. Clinical Trials Screening
4.2. Scaffold Morphing
4.3. In Silico Pharmacokinetic Predictions
4.4. Molecular Docking Studies
4.5. Molecular Dynamic Simulations
5. Conclusions and Future Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wiley, J. Alzheimer’s disease facts and figures. J. Alzheimers Dement. 2021, 17, 327–406. [Google Scholar]
- Alzheimer’s Association. 2012 Alzheimer’s disease facts and figures. Alzheimers Dement. 2012, 8, 131–168. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, M.; Slavin, J.; Cao, Y.; Basbaum, A.I.; Neoptolemos, J.P. Preprotachykinin-A gene deletion protects mice against acute pancreatitis and associated lung injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 284, G830–G836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naskar, S.; Gour, N. Realization of Amyloid like Aggregation as a Common Cause for the Pathogenesis in Diseases. Life 2023, 13, 1523. [Google Scholar] [CrossRef]
- Plascencia-Villa, G.; Perry, G. Neuropathological changes provide insights into key mechanisms related to Alzheimer’s disease and related dementia. Am. J. Pathol. 2022, 192, 1340–1346. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, D.; Sehgal, A.; Singh, S.; Sharma, N.; Zengin, G.; Andronie-Cioara, F.L.; Toma, M.M.; Bungau, S.; Bumbu, A.G. Role of monoamine oxidase activity in Alzheimer’s disease: An insight into the therapeutic potential of inhibitors. Molecules 2021, 18, 3724. [Google Scholar] [CrossRef] [PubMed]
- Palop, J.J.; Mucke, L. Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2016, 17, 777–792. [Google Scholar] [CrossRef] [PubMed]
- Tampi, R.R. Paired associative stimulation (PAS) and Alzheimer’s disease (AD). Int. Psychogeriatr. 2023, 35, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Vassar, R.; Kovacs, D.M.; Yan, R.; Wong, P.C. The beta-secretase enzyme BACE in health and Alzheimer’s disease: Regulation, cell biology, function, and therapeutic potential. J. Neurosci. 2009, 29, 12787–12794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, R.; Vassar, R. Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol. 2014, 13, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Cole, S.L.; Vassar, R. The Alzheimer’s disease beta-secretase enzyme, BACE1. Mol. Neurodegener. 2007, 2, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iraji, A.; Khoshneviszadeh, M.; Firuzi, O.; Khoshneviszadeh, M.; Edraki, N. Novel small molecule therapeutic agents for Alzheimer disease: Focusing on BACE1 and multi-target directed ligands. Bioorg. Chem. 2020, 97, 103649. [Google Scholar] [CrossRef] [PubMed]
- Grover, P.; Rohilla, S.; Bhardwaj, M.; Mehta, L.; Malhotra, A. Piperidine Nucleus as a Promising Scaffold for Alzheimer’s Disease: Current Landscape and Future Perspective. Curr. Top. Med. Chem. 2023, 23, 1221–1259. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.L.; Vassar, R. The Basic Biology of BACE1: A Key Therapeutic Target for Alzheimer’s Disease. Curr. Genom. 2007, 8, 509–530. [Google Scholar] [CrossRef]
- Kocak, A.; Erol, I.; Yildiz, M.; Can, H. Computational insights into the protonation states of catalytic dyad in BACE1-acyl guanidine based inhibitor complex. J. Mol. Graph. Model. 2016, 70, 226–235. [Google Scholar] [CrossRef]
- Mermelstein, D.J.; McCammon, J.A.; Walker, R.C. pH-dependent conformational dynamics of beta-secretase 1: A molecular dynamics study. J. Mol. Recognit. 2019, 32, e2765. [Google Scholar] [CrossRef]
- R, M.M.; Shandil, R.K.; Panda, M.; Sadler, C.; Ambady, A.; Panduga, V.; Kumar, N.; Mahadevaswamy, J.; Sreenivasaiah, M.; Narayan, A.; et al. Scaffold Morphing To Identify Novel DprE1 Inhibitors with Antimycobacterial Activity. ACS Med. Chem. Lett. 2019, 10, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Ji, C. MolOpt: A Web Server for Drug Design using Bioisosteric Transformation. Curr. Comput. Aided Drug Des. 2020, 16, 460–466. [Google Scholar] [CrossRef]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Das, B.; Yan, R. A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment. CNS Drugs 2019, 33, 251–263. [Google Scholar] [CrossRef]
- Langdon, S.R.; Ertl, P.; Brown, N. Bioisosteric Replacement and Scaffold Hopping in Lead Generation and Optimization. Mol Inform. 2010, 29, 366–385. [Google Scholar] [CrossRef] [PubMed]
- Dick, A.; Cocklin, S. Bioisosteric Replacement as a Tool in Anti-HIV Drug Design. Pharmaceuticals 2020, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Silakari, O. Scaffold morphing of arbidol (umifenovir) in search of multi-targeting therapy halting the interaction of SARS-CoV-2 with ACE2 and other proteases involved in COVID-19. Virus Res. 2020, 289, 198146. [Google Scholar] [CrossRef]
- Singh, M.; Kaur, M.; Singh, N.; Silakari, O. Corrigendum to “Exploration of multi-target potential of chromen-4-one based compounds in Alzheimer’s disease: Design, synthesis and biological evaluations” [Bioorg. Med. Chem. 25 (2017) 6273–6285]. Bioorg. Med. Chem. 2018, 26, 4360–4361. [Google Scholar] [CrossRef]
- Shah, K.; Mujwar, S.; Krishna, G.; Gupta, J.K. Computational Design and Biological Depiction of Novel Naproxen Derivative. ASSAY Drug Dev. Technol. 2020, 18, 308–317. [Google Scholar] [CrossRef]
- Kaur, A.; Mujwar, S.; Adlakha, N. In-silico analysis of riboswitch of Nocardia farcinica for design of its inhibitors and pharmacophores. Int. J. Comput. Biol. Drug Des. 2016, 9, 261–276. [Google Scholar] [CrossRef]
- Pradhan, P.; Soni, N.K.; Chaudhary, L.; Mujwar, S.; Pardasani, K.R. In-silico prediction of riboswitches and design of their potent inhibitors for H1N1, H2N2 and H3N2 strains of influenza virus. Biosci. Biotechnol. Res. Asia 2015, 12, 2173–2186. [Google Scholar] [CrossRef] [Green Version]
- Client BDS. v19. 1.0. 18287. Accelrys Discovery Studio. 2019.
- Wu, G.; Robertson, D.H.; Brooks, C.L., 3rd; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Gupta, S.; Parihar, D.; Shah, M.; Yadav, S.; Managori, H.; Bhowmick, S.; Patil, P.C.; Alissa, S.A.; Wabaidur, S.M.; Islam, A. Computational screening of promising beta-secretase 1 inhibitors through multi-step molecular docking and molecular dynamics simulations-Pharmacoinformatics approach. J. Mol. Struct. 2020, 1205, 127660. [Google Scholar] [CrossRef]
- Oehlrich, D.; Peschiulli, A.; Tresadern, G.; Van Gool, M.; Vega, J.A.; De Lucas, A.I.; de Diego, S.A.A.; Prokopcova, H.; Austin, N.; Van Brandt, S.; et al. Evaluation of a Series of β-Secretase 1 Inhibitors Containing Novel Heteroaryl-Fused-Piperazine Amidine Warheads. ACS Med. Chem. Lett. 2019, 10, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Mujwar, S.; Harwansh, R.K. In silico bioprospecting of taraxerol as a main protease inhibitor of SARS-CoV-2 to develop therapy against COVID-19. Struct Chem. 2022, 33, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Mujwar, S.; Sun, L.; Fidan, O. In silico evaluation of food-derived carotenoids against SARS-CoV-2 drug targets: Crocin is a promising dietary supplement candidate for COVID-19. J. Food Biochem. 2022, 46, e14219. [Google Scholar] [CrossRef]
- Fidan, O.; Mujwar, S.; Kciuk, M. Discovery of adapalene and dihydrotachysterol as antiviral agents for the Omicron variant of SARS-CoV-2 through computational drug repurposing. Mol. Divers. 2022, 27, 463–475. [Google Scholar] [CrossRef]
- Kciuk, M.; Mujwar, S.; Szymanowska, A.; Marciniak, B.; Bukowski, K.; Mojzych, M.; Kontek, R. Preparation of Novel Pyrazolo [4, 3-e] tetrazolo [1, 5-b][1,2,4] triazine Sulfonamides and Their Experimental and Computational Biological Studies. Int. J. Mol. Sci. 2022, 23, 5892. [Google Scholar] [CrossRef]
- Mujwar, S. Computational repurposing of tamibarotene against triple mutant variant of SARS-CoV-2. Comput. Biol. Med. 2021, 136, 104748. [Google Scholar] [CrossRef] [PubMed]
- Mujwar, S. Computational bioprospecting of andrographolide derivatives as potent cyclooxygenase-2 inhibitors. Biomed. Biotechnol. Res. J. 2021, 5, 446. [Google Scholar] [CrossRef]
- Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2021. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2023.
- Mujwar, S.; Tripathi, A. Repurposing Benzbromarone as Antifolate to Develop Novel Antifungal Therapy for Candida Albicans. J. Mol. Model. 2021, 28, 193. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Ullah, H.; Taha, M.; Rahim, F.; Sarfraz, M.; Iqbal, R.; Iqbal, N.; Hussain, R.; Ali Shah, S.A.; Ayub, K.; et al. Synthesis, DFT Studies, Molecular Docking and Biological Activity Evaluation of Thiazole-Sulfonamide Derivatives as Potent Alzheimer’s Inhibitors. Molecules 2023, 28, 559. [Google Scholar] [CrossRef]
- Ullah, H.; Zada, H.; Khan, F.; Hayat, S.; Rahim, F.; Hussain, A.; Manzoor, A.; Wadood, A.; Ayub, K.; Rehman, A.U.; et al. Benzimidazole bearing thiourea analogues: Synthesis, β-glucuronidase inhibitory potential and their molecular docking study. J. Mol. Struct. 2022, 1270, 133941. [Google Scholar] [CrossRef]
- Khan, S.; Ullah, H.; Rahim, F.; Nawaz, M.; Hussain, R.; Rasheed, L. Synthesis, in vitro α-amylase, α-glucosidase activities and molecular docking study of new benzimidazole bearing thiazolidinone derivatives. J. Mol. Struct. 2022, 1269, 133812. [Google Scholar] [CrossRef]
- Ullah, H.; Liaqat, A.; Khan, Q.U.; Taha, M.; Khan, F.; Rahim, F.; Uddin, I.; Rehman, Z.U. Synthesis, in vitro thymidine phosphorylase activity and molecular docking study of thiadiazole bearing isatin analogs. Chem. Pap. 2021, 76, 213–224. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, G.; Sehgal, A.; Bhardwaj, S.; Singh, S.; Buhas, C.; Judea-Pusta, C.; Uivarosan, D.; Munteanu, M.A.; Bungau, S. Multifaceted role of matrix metalloproteinases in neurodegenerative diseases: Pathophysiological and therapeutic perspectives. Int. J. Mol. Sci. 2021, 22, 1413. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Compound ID | Structure | S. No. | Compound ID | Structure |
|---|---|---|---|---|---|
| 1. | SB6 |  | 26. | SB213 |  |
| 2. | SB12 |  | 27. | SB220 |  |
| 3. | SB19 |  | 28. | SB234 |  |
| 4. | SB20 |  | 29. | SB242 |  |
| 5. | SB21 |  | 30. | SB247 |  |
| 6. | SB22 |  | 31. | SB262 |  |
| 7. | SB35 |  | 32. | SB282 |  |
| 8. | SB37 |  | 33. | SB283 |  |
| 9. | SB38 |  | 34. | SB306 |  |
| 10. | SB41 |  | 35. | SB335 |  |
| 11. | SB42 |  | 36. | SB339 |  |
| 12. | SB63 |  | 37. | SB340 |  |
| 13. | SB92 |  | 38. | SB342 |  |
| 14. | SB139 |  | 39. | SB353 |  |
| 15. | SB153 |  | 40. | SB357 |  |
| 16. | SB171 |  | 41. | SB362 |  |
| 17. | SB195 |  | 42. | SB363 |  |
| 18. | SB196 |  | 43. | SB364 |  |
| 19. | SB203 |  | 44. | SB367 |  |
| 20. | SB204 |  | 45. | SB375 |  |
| 21. | SB205 |  | 46. | SB381 |  |
| 22. | SB206 |  | 47. | SB382 |  |
| 23. | SB208 |  | 48. | SB395 |  |
| 24. | SB209 |  | 49. | SB523 |  |
| 25. | SB210 |  | 50. | SB856 |  |
| S. No. | Compound ID | MW | HBA | HBD | TPSA (Å) | Consensus Log P | Ali Log S | Lipinski Violations | BBB Permeant | GI Absorption |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | [SB6] | 344.29 | 7 | 1 | 67.77 | 2.85 | −3.32 | 0 | Yes | High |
| 2 | [SB12] | 378.35 | 7 | 1 | 75.19 | 2.47 | −2.78 | 0 | Yes | High |
| 3 | [SB19] | 325.29 | 7 | 2 | 75.11 | 2.37 | −2.83 | 0 | Yes | High |
| 4 | [SB20] | 311.26 | 7 | 2 | 75.11 | 2.01 | −2.46 | 0 | Yes | High |
| 5 | [SB21] | 406.4 | 7 | 1 | 75.19 | 3 | −3.45 | 0 | Yes | High |
| 6 | [SB22] | 358.32 | 7 | 1 | 67.77 | 3.09 | −3.66 | 0 | Yes | High |
| 7 | [SB35] | 351.32 | 7 | 1 | 71.95 | 2.99 | −3.29 | 0 | Yes | High |
| 8 | [SB37] | 337.3 | 7 | 1 | 71.95 | 2.71 | −2.93 | 0 | Yes | High |
| 9 | [SB38] | 347.29 | 7 | 1 | 72.7 | 2.35 | −2.75 | 0 | Yes | High |
| 10 | [SB41] | 360.29 | 7 | 1 | 76.88 | 2.44 | −2.96 | 0 | Yes | High |
| 11 | [SB42] | 398.34 | 7 | 1 | 75.19 | 2.82 | −3.52 | 0 | Yes | High |
| 12 | [SB63] | 350.34 | 7 | 2 | 66.91 | 2.57 | −2.84 | 0 | Yes | High |
| 13 | [SB92] | 375.35 | 7 | 1 | 72.7 | 2.73 | −3.22 | 0 | Yes | High |
| 14 | [SB139] | 348.32 | 7 | 2 | 66.91 | 2.47 | −2.46 | 0 | Yes | High |
| 15 | [SB153] | 351.32 | 7 | 1 | 64.11 | 3 | −3.27 | 0 | Yes | High |
| 16 | [SB171] | 349.31 | 7 | 1 | 64.11 | 2.81 | −2.69 | 0 | Yes | High |
| 17 | [SB195] | 348.32 | 7 | 2 | 66.91 | 2.45 | −2.28 | 0 | Yes | High |
| 18 | [SB196] | 366.31 | 8 | 2 | 66.91 | 2.66 | −2.27 | 0 | Yes | High |
| 19 | [SB203] | 360.33 | 6 | 1 | 58.12 | 2.67 | −3.1 | 0 | Yes | High |
| 20 | [SB204] | 351.32 | 7 | 1 | 64.11 | 2.99 | −3.36 | 0 | Yes | High |
| 21 | [SB205] | 337.3 | 7 | 1 | 64.11 | 2.7 | −2.99 | 0 | Yes | High |
| 22 | [SB206] | 392.37 | 7 | 1 | 75.19 | 2.72 | −3.21 | 0 | Yes | High |
| 23 | [SB208] | 346.31 | 6 | 2 | 66.91 | 2.54 | −3.14 | 0 | Yes | High |
| 24 | [SB209] | 348.32 | 6 | 2 | 66.91 | 2.63 | −3.1 | 0 | Yes | High |
| 25 | [SB210] | 351.32 | 7 | 1 | 64.11 | 3.08 | −3.08 | 0 | Yes | High |
| 26 | [SB213] | 337.3 | 7 | 1 | 64.11 | 2.71 | −2.7 | 0 | Yes | High |
| 27 | [SB220] | 350.34 | 7 | 2 | 66.91 | 2.59 | −2.96 | 0 | Yes | High |
| 28 | [SB234] | 353.3 | 8 | 1 | 73.34 | 2.08 | −2.15 | 0 | Yes | High |
| 29 | [SB242] | 378.35 | 7 | 1 | 75.19 | 2.39 | −2.66 | 0 | Yes | High |
| 30 | [SB247] | 380.36 | 8 | 1 | 67.35 | 2.34 | −2.59 | 0 | Yes | High |
| 31 | [SB262] | 350.3 | 8 | 1 | 76.47 | 2.6 | −3.23 | 0 | Yes | High |
| 32 | [SB282] | 390.4 | 7 | 1 | 58.12 | 3.28 | −3.72 | 0 | Yes | High |
| 33 | [SB283] | 426.43 | 7 | 2 | 66.91 | 3.73 | −4.29 | 0 | Yes | High |
| 34 | [SB306] | 390.4 | 7 | 1 | 58.12 | 3.26 | −3.83 | 0 | Yes | High |
| 35 | [SB335] | 368.33 | 8 | 2 | 66.91 | 2.72 | −2.9 | 0 | Yes | High |
| 36 | [SB339] | 376.38 | 7 | 1 | 58.12 | 2.95 | −3.28 | 0 | Yes | High |
| 37 | [SB340] | 379.38 | 7 | 1 | 64.11 | 3.53 | −4.09 | 0 | Yes | High |
| 38 | [SB342] | 392.37 | 7 | 1 | 75.19 | 2.68 | −3.24 | 0 | Yes | High |
| 39 | [SB353] | 348.32 | 7 | 2 | 66.91 | 2.3 | −2.4 | 0 | Yes | High |
| 40 | [SB357] | 391.39 | 7 | 1 | 64.11 | 3.56 | −4.11 | 0 | Yes | High |
| 41 | [SB362] | 406.4 | 8 | 2 | 78.35 | 2.62 | −3.13 | 0 | Yes | High |
| 42 | [SB363] | 377.36 | 7 | 1 | 64.11 | 3.41 | −3.54 | 0 | Yes | High |
| 43 | [SB364] | 376.38 | 7 | 2 | 66.91 | 2.93 | −3.31 | 0 | Yes | High |
| 44 | [SB367] | 379.33 | 8 | 1 | 73.34 | 2.55 | −2.95 | 0 | Yes | High |
| 45 | [SB375] | 390.4 | 7 | 2 | 66.91 | 3.24 | −3.8 | 0 | Yes | High |
| 46 | [SB381] | 379.33 | 8 | 1 | 73.34 | 2.61 | −2.65 | 0 | Yes | High |
| 47 | [SB382] | 393.36 | 8 | 1 | 73.34 | 2.78 | −3.14 | 0 | Yes | High |
| 48 | [SB395] | 404.43 | 7 | 2 | 66.91 | 3.62 | −4.16 | 0 | Yes | High |
| 49 | [SB523] | 390.4 | 7 | 1 | 58.12 | 3.26 | −3.54 | 0 | Yes | High |
| 50 | [SB856] | 376.38 | 7 | 2 | 66.91 | 3.01 | −3.49 | 0 | Yes | High |
| S. No. | Compound ID | Docking Score (kcal/mol) | Interactions |
|---|---|---|---|
| 1 | Elenbecestat | −42.82 | Asp32, Asp228, Ser35, Ile 118, Leu30, Gly13, Tyr14, Gly34, Ser229, Thr232 |
| 2 | SB6 | −33.54 | Asp32, Tyr71, Gly230, Val166, Trp115, Thr231, Thr232, Arg235 |
| 3 | SB12 | −43.35 | Asp32, Asp228, Thr329, Arg235, Gly230, Thr232, Val166, Thr231, Trp115, Gly34, Lys224, Thr329, Tyr198, Val332, Leu30, Ile226 |
| 4 | SB19 | −37.70 | Asp32, Asp228, Thr232, Arg235, Gly34, Gly230, Thr231, Val166, |
| 5 | SB20 | −34.53 | Asp32, Asp228, Gly34, Gly230, Thr231, Arg235, Thr232, Val166, |
| 6 | SB21 | −43.06 | Asp32, Asp228, Arg235, Val332, Gly230, Gly34, Thr329, Lys224, Thr231, Val166, Thr232 |
| 7 | SB22 | −38.46 | Asp32, Asp228, Gly34, Tyr71, Trp115, Gly230, Thr231, Thr232, Val166 |
| 8 | SB35 | −41.27 | Asp32, Asp228, Thr232, Thr231, Gly230, Val332, Arg235, Gly34, Trp115, Val166 |
| 9 | SB37 | −38.47 | Asp32, Asp228, Gly34, Arg235, Thr232, Thr231, Gly230, Val166 |
| 10 | SB38 | −38.04 | Asp32, Asp228, Val166, Gly34, Tyr71, Trp115, Gly230, Thr231, Thr232 |
| 11 | SB41 | −37.06 | Asp32, Val166, Thr232, Thr231, Tyr71 |
| 12 | SB42 | −35.51 | Asp32, Asp228, Tyr71, Gly34, Gly230, Lys224, Val332, Trp115, Val166, Thr329, Thr231, Thr232 |
| 13 | SB63 | −41.42 | Asp32, Asp228, Gly34, Trp115, Gly230, Val166, Thr231, Thr232 |
| 14 | SB92 | −43.31 | Asp32, Asp228, Gly34, Arg235, Thr232, Thr231, Gly230, Val166, Trp115 |
| 15 | SB139 | −36.55 | Asp32, Asp228, Tyr71, Gly34, Gly230, Val332, Trp115, Val166, Thr231, Thr232 |
| 16 | SB153 | −32.568 | Asp32, Asp228, Gly230, Thr231, Thr232, Trp115, Tyr71, Val332, Lys224, Thr329, Arg235 |
| 17 | SB171 | −33.11 | Asp32, Asp228, Trp115, Tyr71, Thr231, Thr232, Gly230, Gly34 |
| 18 | SB195 | −41.40 | Asp32, Asp228, Gly34, Gly230, Thr231, Arg235, Thr232, Val166, Trp115, Tyr71 |
| 19 | SB196 | −37.88 | Asp32, Asp228, Gly34, Trp115, Gly230, Val166, Thr231, Thr232 |
| 20 | SB203 | −38.86 | Asp32, Gly34, Tyr71, Trp115, Gly230, Thr231, Thr232, Val166 |
| 21 | SB204 | −42.01 | Asp32, Asp228, Gly34, Gly230, Thr231, Thr232, Val166, Trp115 |
| 22 | SB205 | −34.14 | Asp32, Asp228, Gly34, Val332, Thr231, Thr232, Gly230, Trp115, Tyr71, Val166 |
| 23 | SB206 | −37.43 | Asp32, Asp228, Gly34, Gly230, Thr231, Thr232, Thr329, Trp115, Lys224, Val332, Arg235 |
| 24 | SB208 | −30.56 | Asp32, Asp228, Gly34, Gly230, Thr231, Tyr71 |
| 25 | SB209 | −31.31 | Asp32, Asp228, Tyr71, Gly34, Trp115, Gly230, Val166, Thr232, Thr231 |
| 26 | SB210 | −40.6 | Asp32, Trp115, Thr231, Thr232, Val166, Gly34, Tyr71, Gly230 |
| 27 | SB213 | −33.89 | Asp32, Asp228, Tyr71, Gly34, Val332, Thr231, Thr232, Gly230, Trp115 |
| 28 | SB220 | −40.46 | Asp32, Asp228, Gly34, Gly230, Tyr71, Trp115, Val166, Thr232, Thr231 |
| 29 | SB234 | −33.02 | Asp32, Asp228, Tyr71, Gly34, Gly230, Thr231, Thr232, Trp115 |
| 30 | SB242 | −37.75 | Asp32, Arg235, Tyr71, Gly230, Thr231, Thr232, Val166, Trp115 |
| 31 | SB247 | −36.85 | Asp228, Gly230, Val166, Trp115, Thr231, Thr232, Thr329, Arg235, Val332 |
| 32 | SB262 | −40.15 | Asp32, Trp115, Thr231, Thr232, Val166, Gly34, Tyr71, Gly230 |
| 33 | SB283 | −43.30 | Asp32, Asp228, Trp115, Tyr71, Thr231, Thr232, Gly230, Gly34, Val166 |
| 34 | SB306 | −44.35 | Asp32, Asp228, Gly34, Tyr71, Trp115, Gly230, Thr231, Thr232, Val166 |
| 35 | SB335 | −33.29 | Asp32, Asp228, Gly34, Gly230, Val332, Arg235, Lys224, Thr329, Thr231, Thr232, Trp115, Tyr71 |
| 36 | SB339 | −41.84 | Asp32, Asp228, Gly34, Tyr71, Trp115, Gly230, Val166, Thr231, Thr232 |
| 37 | SB340 | −39.28 | Asp32, Asp228, Tyr71, Gly34, Gly230, Thr231, Thr232, Trp115, Val166 |
| 38 | SB342 | −38.96 | Asp32, Asp228, Val166, Gly34, Tyr71, Trp115, Gly230, Thr231, Thr232, Arg235, Thr329 Lys224, Val332 |
| 39 | SB353 | −39.54 | Asp32, Asp228, Trp115, Gly34, Gly230, Tyr71, Val166, Thr231, Thr232 |
| 40 | SB357 | −39.79 | Asp32, Asp228, Val166, Gly34, Gly230, Tyr71, Trp115 |
| 41 | SB362 | −41.43 | Asp32, Asp228, Thr329, Thr231, Thr232, Lys224, Val332, Gly34, Gly230, Tyr71 |
| 42 | SB363 | −42.46 | Asp32, Asp228, Val332, Thr231, Thr232, Thr329, Lys224, Gly230, Gly34, Tyr71 |
| 43 | SB364 | −35.77 | Asp32, Asp228, Thr231, Thr232, Gly34, Gly230, Val332, Tyr71, Thr329, Lys224 |
| 44 | SB367 | −41.62 | Asp32, Asp228, Gly34, Tyr71, Thr231, Gly230, Thr232, Val332, Lys224, Thr329 |
| 45 | SB375 | −38.84 | Asp32, Asp228, Gly34, Gly230, Thr231, Thr329, Tyr71, Val332, Lys224 |
| 46 | SB381 | −41.17 | Asp32, Asp228, Gly34, Gly230, Tyr71, Thr231, Val332, Thr329, Lys224 |
| 47 | SB382 | −39.06 | Asp32, Asp228, Lys224, Thr329, Val332, Gly34, Gly230, Thr231, Tyr71 |
| 48 | SB395 | −33.28 | Asp32, Asp228, Val332, Gly230, Gly34, Thr231, Tyr71, Thr329, Lys224 |
| 49 | SB523 | −40.50 | Asp32, Asp228, Gly34, Gly230, Lys224, Thr329, Val332, Thr231, Tyr71 |
| 50 | SB856 | −40.44 | Asp32, Asp228, Lys224, Thr329, Val332, Gly34, Gly230, Thr231, Tyr71 |
| Molecule Name | Clinical and Pharmacology Consideration | Clinical Trial Status | Clinical Trial ID |
|---|---|---|---|
| Lanabecestat (AZD3293, LY3314814) | Reduce CSF Aβ levels | Phase 3 trials terminated due to less efficacy | Clinicaltrials.gov ID: NCT02245737 |
| (JNJ-54861911) Atabecestat | Reduce CSF Amyloid-β levels | Phase 2 or 3 trials were terminated due to liver toxicity | Clinicaltrials.gov ID: NCT02569398 |
| CNP520 | Reduce CSF Aβ levels | Phase II/III completed | Clinicaltrials.gov ID: NCT02576639 |
| (E2609) Elenbecestat | Decreased Amyloid-β levels in cerebrospinal fluid and plasma Less decline in functional cognition. | Currently in Phase III clinical trials but side effects like dermatitis, respiratory tract infection, abnormal nightmares and dreams, headache, falls, and diarrhea but safe for liver | Clinicaltrials.gov ID: NCT03036280, NCT02956486 |
| (MK-8931) Verubecestat | Decreased CNS amyloid-β levels in animals and patients of AD | Phase 3 trials were withdrawn due to lack of efficacy as well as dermatological and behavioral side effects | Clinicaltrials.gov ID: NCT01953601 |
| LY2886721 | Potent BACE-1 inhibitor | Phase 1/2 terminated due to abnormal liver biochemical toxicity. | Clinicaltrials.gov ID: NCT01561430 |
| LY320262 | BACE-1 inhibitor | Phase II but no clinical efficacy as of now | Clinicaltrials.gov ID: NCT02323334 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhatia, S.; Singh, M.; Sharma, P.; Mujwar, S.; Singh, V.; Mishra, K.K.; Singh, T.G.; Singh, T.; Ahmad, S.F. Scaffold Morphing and In Silico Design of Potential BACE-1 (β-Secretase) Inhibitors: A Hope for a Newer Dawn in Anti-Alzheimer Therapeutics. Molecules 2023, 28, 6032. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28166032
Bhatia S, Singh M, Sharma P, Mujwar S, Singh V, Mishra KK, Singh TG, Singh T, Ahmad SF. Scaffold Morphing and In Silico Design of Potential BACE-1 (β-Secretase) Inhibitors: A Hope for a Newer Dawn in Anti-Alzheimer Therapeutics. Molecules. 2023; 28(16):6032. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28166032
Chicago/Turabian StyleBhatia, Shiveena, Manjinder Singh, Pratibha Sharma, Somdutt Mujwar, Varinder Singh, Krishna Kumar Mishra, Thakur Gurjeet Singh, Tanveer Singh, and Sheikh Fayaz Ahmad. 2023. "Scaffold Morphing and In Silico Design of Potential BACE-1 (β-Secretase) Inhibitors: A Hope for a Newer Dawn in Anti-Alzheimer Therapeutics" Molecules 28, no. 16: 6032. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28166032