
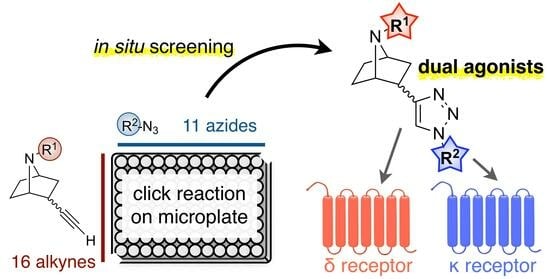
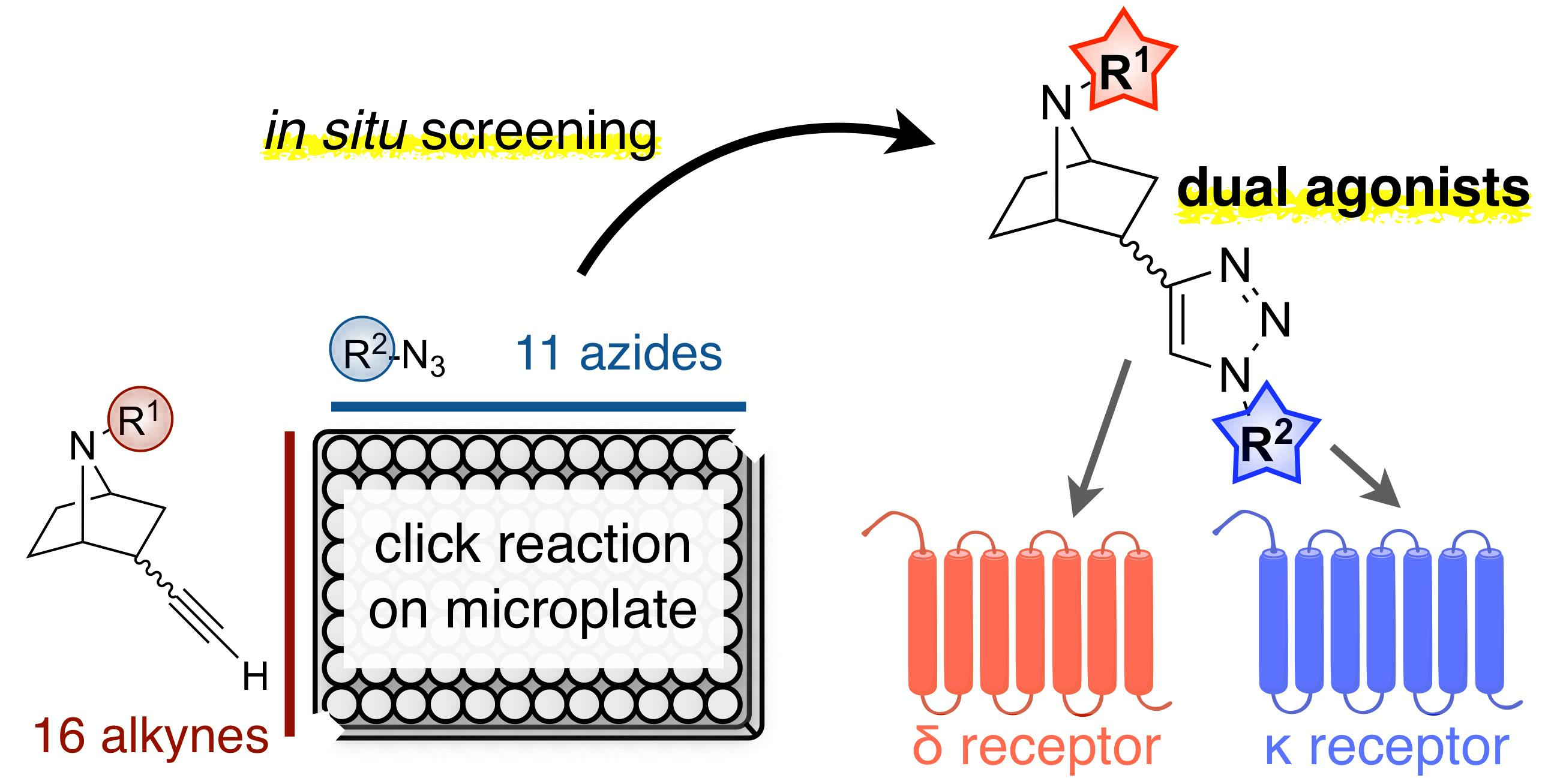
Discovery of 7-Azanorbornane-Based Dual Agonists for the Delta and Kappa Opioid Receptors through an In Situ Screening Protocol
 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Construction of the Compound Library by Click Reactions and a Screening Assay without Compound Purification
2.2. Validation of the Activities after Purification of the Compounds
3. Discussion
3.1. Applicability of the In Situ Screening Protocol for the Opioid Receptors
3.2. Preliminary Structure-Activity Relationship Information of the Obtained Agonists
4. Materials and Methods
4.1. Organic Synthesis
4.1.1. General Remarks
4.1.2. Preparation of Alkynes 1–7
4.1.3. Click Reaction in Microplates
4.1.4. Synthesis of the Triazoles
4.2. Biological Evaluation
4.2.1. Cell Culture
4.2.2. CellKeyTM Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Hernández-López, L.; von Baeckmann, C.; Martínez-Esaín, J.; Cortés- Martínez, A.; Faraudo, J.; Caules, C.; Parella, T.; Maspoch, D.; Carné-Sánchez, A. (Bio)Functionalisation of Metal-Organic Polyhedra by using Click Chemistry. Chem. Eur. J. 2023, e202301945. [Google Scholar] [CrossRef]
- Shioi, R.; Karaki, F.; Yoshioka, H.; Noguchi-Yachide, T.; Ishikawa, M.; Dodo, K.; Hashimoto, Y.; Sodeoka, M.; Ohgane, K. Image-based screen capturing misfolding status of Niemann-Pick type C1 identifies potential candidates for chaperone drugs. PLoS ONE 2020, 15, e0243746. [Google Scholar] [CrossRef]
- Peng, B.; Thorsell, A.-G.; Karlberg, T.; Schüler, H.; Yao, S.Q. Small Molecule Microarray Based Discovery of PARP14 Inhibitors. Angew. Chem. Int. Ed. 2017, 56, 248–253. [Google Scholar] [CrossRef]
- Bhardwaj, A.; Kaur, J.; Wuest, M.; Wuest, F. In situ click chemistry generation of cyclooxygenase-2 inhibitors. Nat. Commun. 2017, 8, 1. [Google Scholar] [CrossRef]
- Nemati, F.; Bischoff-Kont, I.; Salehi, P.; Nejad-Ebrahimi, S.; Mohebbi, M.; Bararjanian, M.; Hadian, N.; Hassanpour, Z.; Jung, Y.; Schaerlaekens, S.; et al. Identification of novel anti-cancer agents by the synthesis and cellular screening of a noscapine-based library. Bioorg. Chem. 2021, 115, 105135. [Google Scholar] [CrossRef]
- Jana, B.; Iram, S.; Thomas, J.; Liekens, S.; Dehaen, W. Synthesis and anticancer activity of novel aza-artemisinin derivatives. Bioorg. Med. Chem. 2017, 25, 3671–3676. [Google Scholar] [CrossRef]
- Srinivasan, R.; Li, J.; Ng, S.L.; Kalesh, K.A.; Yao, S.Q. Methods of using click chemistry in the discovery of enzyme inhibitors. Nat. Protoc. 2007, 2, 2655–2664. [Google Scholar] [CrossRef]
- Duval, R.; Kolb, S.; Braud, E.; Genest, D.; Garbay, C. Rapid Discovery of Triazolobenzylidene-Thiazolopyrimidines (TBTP) as CDC25 Phosphatase Inhibitors by Parallel Click Chemistry and in Situ Screening. J. Comb. Chem. 2009, 11, 947–950. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ota, Y.; Ri, M.; Bando, M.; Gotoh, A.; Itoh, Y.; Tsumoto, H.; Tatum, P.R.; Mizukami, T.; Nakagawa, H.; et al. Rapid Discovery of Highly Potent and Selective Inhibitors of Histone Deacetylase 8 Using Click Chemistry to Generate Candidate Libraries. J. Med. Chem. 2012, 55, 9562–9575. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Hao, X.; Jing, L.; Sun, L.; Cherukupalli, S.; Liu, S.; Wu, G.; Xu, S.; Zhang, X.; Shi, X.; et al. Discovery of potent and selective Cdc25 phosphatase inhibitors via rapid assembly and in situ screening of Quinonoid-focused libraries. Bioorg. Chem. 2021, 115, 105254. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, V.; Martínez-Bailén, M.; Carmona, A.T.; Mészáros, Z.; Kulik, N.; Slámová, K.; Křen, V.; Bojarová, P.; Robina, I.; Moreno-Vargas, A.J. Discovery of human hexosaminidase inhibitors by in situ screening of a library of mono-and divalent pyrrolidine iminosugars. Bioorg. Chem. 2022, 120, 105650. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Zhang, C.-J.; Wu, J.; Yao, Q. Fused Bicyclic Caspase-1 Inhibitors Assembled by Copper-Free Strain-Promoted Alkyne–Azide Cycloaddition (SPAAC). Chem. Eur. J. 2017, 23, 360–369. [Google Scholar] [CrossRef]
- Xin, Y.; Liu, S.; Liu, Y.; Qian, Z.; Liu, H.; Zhang, B.; Guo, T.; Thompson, G.J.; Stevens, R.C.; Sharpless, K.B.; et al. Affinity selection of double-click triazole libraries for rapid discovery of allosteric modulators for GLP-1 receptor. Proc. Natl. Acad. Sci. USA 2023, 120, e2220767120. [Google Scholar] [CrossRef]
- Blackman, M.L.; Royzen, M.; Fox, J.M. Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels−Alder Reactivity. J. Am. Chem. Soc. 2008, 130, 13518–13519. [Google Scholar] [CrossRef]
- Devaraj, N.K.; Weissleder, R.; Hilderbrand, S.A. Tetrazine-based cycloadditions: Application to pretargeted live cell imaging. Bioconjugate Chem. 2008, 19, 2297–2299. [Google Scholar] [CrossRef]
- Agard, N.J.; Prescher, J.A.; Bertozzi, C.R. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. [Google Scholar] [CrossRef]
- Wong, I.L.K.; Zhu, X.; Chan, K.-F.; Law, M.C.; Lo, A.M.Y.; Hu, X.; Chow, L.M.C.; Chan, T.H. Discovery of Novel Flavonoid Dimers To Reverse Multidrug Resistance Protein 1 (MRP1, ABCC1) Mediated Drug Resistance in Cancers Using a High Throughput Platform with "Click Chemistry". J. Med. Chem. 2018, 61, 9931–9951. [Google Scholar] [CrossRef]
- Rillahan, C.D.; Schwartz, E.; Rademacher, C.; McBride, R.; Rangarajan, J.; Fokin, V.V.; Paulson, J.C. On-chip synthesis and screening of a sialoside library yields a high affinity ligand for Siglec-7. ACS Chem. Biol. 2013, 8, 1417–1422. [Google Scholar] [CrossRef]
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Karaki, F.; Umemoto, S.; Ashizawa, K.; Oki, T.; Sato, N.; Ogino, T.; Ishibashi, N.; Someya, R.; Miyano, K.; Hirayama, S.; et al. A New Lead Identification Strategy: Screening an sp3 -rich and Lead-like Compound Library Composed of 7-Azanorbornane Derivatives. ChemMedChem 2019, 14, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Lovering, F.; Bikker, J.; Humblet, C. Escape from flatland: Increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. [Google Scholar] [CrossRef]
- Miyano, K.; Sudo, Y.; Yokoyama, A.; Hisaoka-Nakashima, K.; Morioka, N.; Takebayashi, M.; Nakata, Y.; Higami, Y.; Uezono, Y. History of the G Protein–Coupled Receptor (GPCR) Assays From Traditional to a State-of-the-Art Biosensor Assay. J. Pharmacol. Sci. 2014, 126, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Granier, S.; Manglik, A.; Kruse, A.C.; Kobilka, T.S.; Thian, F.S.; Weis, W.I.; Kobilka, B.K. Structure of the δ-opioid receptor bound to naltrindole. Nature 2012, 485, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into µ-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 2018, 172, 55–67. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Karaki, F.; Oki, T.; Sakao, Y.; Sato, N.; Hirayama, S.; Miyano, K.; Uezono, Y.; Fujii, H. Identification of a Putative β-Arrestin Superagonist of the Growth Hormone Secretagogue Receptor (GHSR). ChemMedChem 2021, 16, 3463–3476. [Google Scholar] [CrossRef]
- Portoghese, P.S.; Sultana, M.; Takemori, A.E. Naltrindole, a highly selective and potent non-peptide δ opioid receptor antagonist. Eur. J. Pharmacol. 1988, 146, 185–186. [Google Scholar] [CrossRef] [PubMed]
- Asghar, J.; Latif, L.; Alexander, S.P.H.; Kendall, D.A. Development of a novel cell-based, In-Cell Western/ERK assay system for the high-throughput screening of agonists acting on the delta-opioid receptor. Front. Pharmacol. 2022, 13, 933356. [Google Scholar] [CrossRef] [PubMed]
- Portoghese, P.S.; Lipkowski, A.W.; Takemori, A.E. Binaltorphimine and nor-binaltorphimine, potent and selective k-opioid receptor antagonists. Life Sci. 1987, 40, 1287–1292. [Google Scholar] [CrossRef]
- Yadav, V.D.; Kumar, L.; Kumari, P.; Kumar, S.; Singh, M.; Siddiqi, M.; Yadav, P.N.; Batra, S. Synthesis and Assessment of Fused β-Carboline Derivatives as Kappa Opioid Receptor Agonists. ChemMedChem 2021, 16, 1917–1926. [Google Scholar] [CrossRef]
- Wang, X.; Huang, B.; Liu, X.; Zhan, P. Discovery of bioactive molecules from CuAAC click-chemistry-based combinatorial libraries. Drug Discov. Today 2016, 21, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Váradi, A.; Marrone, G.F.; Eans, S.O.; Ganno, M.L.; Subrath, J.J.; Le Rouzic, V.; Hunkele, A.; Pasternak, G.W.; McLaughlin, J.P.; Majumdar, S. Synthesis and characterization of a dual kappa-delta opioid receptor agonist analgesic blocking cocaine reward behavior. ACS. Chem. Neurosci. 2015, 6, 1813–1824. [Google Scholar] [CrossRef]
- Uenohara, Y.; Tsumura, S.; Hirayama, S.; Higashi, E.; Watanabe, Y.; Gouda, H.; Nagase, H.; Fujii, H. Morphinan derivatives with an oxabicyclo[3.2.1]octane structure as dual agonists toward δ and κ opioid. Bioorg. Med. Chem. 2022, 53, 116552. [Google Scholar] [CrossRef] [PubMed]
- Nadin, A.; Hattotuwagama, C.; Churcher, I. Lead-Oriented Synthesis: A New Opportunity for Synthetic Chemistry. Angew. Chem. Int. Ed. 2012, 51, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Matthes, H.W.; Maldonado, R.; Simonin, F.; Valverde, O.; Slowe, S.; Kitchen, I.; Befort, K.; Dierich, A.; Le Meur, M.; Dollé, P.; et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the µ-opioid-receptor gene. Nature 1996, 383, 819–823. [Google Scholar] [CrossRef]
- Ochandarena, N.E.; Niehaus, J.K.; Tassou, A.; Scherrer, G. Cell-type specific molecular architecture for mu opioid receptor function in pain and addiction circuits. Neuropharmacology 2023, 238, 109597. [Google Scholar] [CrossRef]
- Atigari, D.V.; Paton, K.F.; Uprety, R.; Váradi, A.; Alder, A.F.; Scouller, B.; Miller, J.H.; Majumdar, S.; Kivell, B.M. The mixed kappa and delta opioid receptor agonist, MP1104, attenuates chemotherapy-induced neuropathic pain. Neuropharmacology 2021, 185, 108445. [Google Scholar] [CrossRef] [PubMed]
- Ngai, M.H.; Yang, P.-Y.; Liu, K.; Shen, Y.; Wenk, M.R.; Yao, S.Q.; Lear, M.J. Click-based synthesis and proteomic profiling of lipstatin analogues. Chem. Commun. 2010, 46, 8335–8337. [Google Scholar] [CrossRef] [PubMed]
- Fotea, C.; D’Silva, C. Adhesion enhancement of chromium tanned heavy-duty leather (Salz leather) under extreme conditions using photoreagents as surface primers. Int. J. Adhes. Adhes. 2005, 25, 442–449. [Google Scholar] [CrossRef]
- Pirali, T.; Gatti, S.; Brisco, R.D.; Tacchi, S.; Zaninetti, R.; Brunelli, E.; Massarotti, A.; Sorba, G.; Canonico, P.L.; Moro, L.; et al. Estrogenic Analogues Synthesized by Click Chemistry. ChemMedChem 2007, 2, 437–440. [Google Scholar] [CrossRef]
- Tran, A.T.; Cergol, K.M.; Britton, W.J.; Bokhari, S.A.I.; Ibrahim, M.; Lapthorn, A.J.; Payne, R.J. Rapid assembly of potent type II dehydroquinase inhibitorsvia “Click” chemistry. Med. Chem. Comm. 2010, 1, 271–275. [Google Scholar] [CrossRef]
- Kwok, S.W.; Fotsing, J.R.; Fraser, R.J.; Rodionov, V.O.; Fokin, V.V. Transition-Metal-Free Catalytic Synthesis of 1,5-Diaryl-1,2,3-triazoles. Org. Lett. 2010, 12, 4217–4219. [Google Scholar] [CrossRef]
- Piccinno, M.; Aragay, G.; Mihan, F.Y.; Ballester, P.; Cort, A.D. Unexpected Emission Properties of a 1,8-Naphthalimide Unit Covalently Appended to a Zn–Salophen. Eur. J. Inorg. Chem. 2015, 2664–2670. [Google Scholar] [CrossRef]
- Yamamoto, K.; Bruun, T.; Kim, J.Y.; Zhang, L.; Lautens, M. A New Multicomponent Multicatalyst Reaction (MC)2R: Chemoselective Cycloaddition and Latent Catalyst Activation for the Synthesis of Fully Substituted 1,2,3-Triazoles. Org. Lett. 2016, 18, 2644–2647. [Google Scholar] [CrossRef]
- Rogers, S.A.; Melander, C. Construction and Screening of a 2-Aminoimidazole Library Identifies a Small Molecule Capable of Inhibiting and Dispersing Bacterial Biofilms across Order, Class, and Phylum. Angew Chem. Int. Ed. 2008, 47, 5229–5231. [Google Scholar] [CrossRef]
- Berry, M.T.; Castrejon, D.; Hein, J.E. Oxidative Esterification of Aldehydes Using Mesoionic 1,2,3-Triazolyl Carbene Organocatalysts. Org. Lett. 2014, 16, 3676–3679. [Google Scholar] [CrossRef]
- Jeong, W.; Lee, J.H.; Kim, J.; Lee, W.J.; Seong, J.-H.; Park, J.; Rhee, Y.H. A Ru-catalyzed one-pot synthesis of homopropargylic amines from alkyl azides under photolytic conditions. RSC Adv. 2014, 4, 20632–20635. [Google Scholar] [CrossRef]
- Meguro, Y.; Miyano, K.; Hirayama, S.; Yoshida, Y.; Ishibashi, N.; Ogino, T.; Fujii, Y.; Manabe, S.; Eto, M.; Nonaka, M.; et al. Neuropeptide oxytocin enhances μ opioid receptor signaling as a positive allosteric modulator. J. Pharmacol. Sci. 2018, 137, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Miyano, K.; Yoshida, Y.; Hirayama, S.; Takahashi, H.; Ono, H.; Meguro, Y.; Manabe, S.; Komatsu, A.; Nonaka, M.; Mizuguchi, T.; et al. Oxytocin Is a Positive Allosteric Modulator of κ-Opioid Receptors but Not δ-Opioid Receptors in the G Protein Signaling Pathway. Cells 2021, 10, 2651. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karaki, F.; Takamori, T.; Kawakami, K.; Sakurai, S.; Hidaka, K.; Ishii, K.; Oki, T.; Sato, N.; Atsumi, N.; Ashizawa, K.; et al. Discovery of 7-Azanorbornane-Based Dual Agonists for the Delta and Kappa Opioid Receptors through an In Situ Screening Protocol. Molecules 2023, 28, 6925. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28196925
Karaki F, Takamori T, Kawakami K, Sakurai S, Hidaka K, Ishii K, Oki T, Sato N, Atsumi N, Ashizawa K, et al. Discovery of 7-Azanorbornane-Based Dual Agonists for the Delta and Kappa Opioid Receptors through an In Situ Screening Protocol. Molecules. 2023; 28(19):6925. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28196925
Chicago/Turabian StyleKaraki, Fumika, Taro Takamori, Koumei Kawakami, Sae Sakurai, Kyoko Hidaka, Kei Ishii, Tomoya Oki, Noriko Sato, Nao Atsumi, Karin Ashizawa, and et al. 2023. "Discovery of 7-Azanorbornane-Based Dual Agonists for the Delta and Kappa Opioid Receptors through an In Situ Screening Protocol" Molecules 28, no. 19: 6925. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28196925