
Fluorinated Human Serum Albumin as Potential 19F Magnetic Resonance Imaging Probe

1
Institute of Chemical Biology and Fundamental Medicine SB RAS, 630090 Novosibirsk, Russia
2
Faculty of Natural sciences, Novosibirsk State University, 630090 Novosibirsk, Russia
*
Author to whom correspondence should be addressed.
Molecules 2023, 28(4), 1695; https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28041695
Submission received: 25 January 2023
/
Revised: 6 February 2023
/
Accepted: 9 February 2023
/
Published: 10 February 2023
(This article belongs to the Special Issue Fluorine in Imaging toward Biological and Medical Applications)
Abstract
:Fluorinated human serum albumin conjugates were prepared and tested as potential metal-free probes for 19F magnetic resonance imaging (MRI). Each protein molecule was modified by several fluorine-containing compounds via the N-substituted natural acylating reagent homocysteine thiolactone. Albumin conjugates retain the protein’s physical and biological properties, such as its 3D dimensional structure, aggregation ability, good solubility, proteolysis efficiency, biocompatibility, and low cytotoxicity. A dual-labeled with cyanine 7 fluorescence dye and fluorine reporter group albumin were synthesized for simultaneous fluorescence imaging and 19F MRI. The preliminary in vitro studies show the prospects of albumin carriers for multimodal imaging.

1. Introduction
Magnetic resonance imaging (MRI) is one of the most powerful noninvasive imaging techniques. 1H MRI allows real-time visualization of tissues, physiological processes, the diagnosis of diseases, monitoring the response to therapy and various biological events, and tracking pharmaceutical substances. MRI has unlimited diagnostic depth, high resolution, and excellent contrast abilities. However, in many cases, MRI requires a specific contrast agent (CA) to enhance the difference between the target tissue and the background [1,2,3,4]. The CA for 1H MRI is detected indirectly by their effect on the spin relaxation of water protons. Paramagnetic metal complexes of Gd and Mn, and iron oxide nanoparticles are among the primary-used CA [1,3,4,5,6,7]. However, recent concerns regarding low stability and potential toxicity of Gd-based and Fe3O4 nanoparticles CAs make their clinical use challenging [8,9,10,11,12,13,14]. Therefore, new metal-free technologies were developed [15]. Organic radical CAs have the same contrast mechanism as Gd chelates and are assembled from highly biocompatible species [16,17,18]. However, rapid radical reduction reactions in vivo and low relaxivity, both limit area progress. The earlier stages’ results of the proposed construction show high perspectives for these CAs in the future [16,19]. Another approach is a multinuclear MRI called nonproton MRI [20]. The use of multinuclear MRI has great potential in clinics due to the absence of tissue background in most cases [20]. Magnetically abundant nuclei such as 19F and 31P are the most popular for drug delivery and metabolism studies [20,21]. The 19F consists of 100% naturally occurring fluorine. It has a nuclear spin of 1/2 and a high gyromagnetic ratio, providing the most sensitive heteronuclear NMR measurements. Moreover, no background fluorine NMR signal in a human organism with optimal relaxation time possesses the ability to register 19F-containing molecules as “fire in the darkness.” Indeed, the 19F NMR chemical shifts span a high range of about ~300 ppm, which may be useful for multiple 19F imaging [22]. In recent years, 19F NMR/MRI has been used to monitor the biodegradation, biodistribution, and pharmacokinetics of fluorinated drugs, binding studies involving protein-DNA complexes, small molecule interactions with proteins, etc. [23,24,25,26,27,28,29,30]. Structural analysis of protein folding, DNA and RNA secondary structures, various macromolecule complexes, and their changes was performed using 19F NMR [28,30]. Recently, 19F electron nuclear double resonance (ENDOR) spectroscopy was evaluated for distance measurement between a spin probe and a fluorine label [31,32,33]. Fluorine-based tracer construction can be engineered to contain polymers [34,35] and nanoparticles [29,30,35,36,37,38] for cancer [35,39,40], Alzheimer’s disease, amyloid-β [41,42,43], neuro disorders [44], inflammation [45], and other various pathological conditions [26]. Improvements in 19F MRI equipment combined with the development of novel, sensitive probes will decrease the global disease burden.
Herein, we developed human serum albumin (HSA) based construction as a potential dual-modal CA for 19F MRI and fluorescence imaging. HSA is a major multifunctional protein in blood plasma [46,47,48]. It plays an important role in hormones, fatty acids, metal cations, and pharmaceutical transport [47,49,50,51,52,53,54]. HSA has attracted attention for therapy and diagnostics [10,49,50,51,52,53,54]. Albumin provides important features as a carrier platform: (1) high biocompatibility and lack of toxicity and immunogenicity; (2) a long half-life of 20 days in the bloodstream; (3) tumor accumulation by enhanced permeability and retention (EPR-effect) of macromolecules and receptor interaction; (4) robustness toward chemical modification; and (5) easily controlled surface chemistry [10,33,49,50,51,52,53,54,55,56,57]. Due to its properties, HSA has been utilized as a carrier for MRI probes and cancer treatment [53,55,56,57,58,59,60,61,62,63,64].
The maleimide-containing fluorescent dye Cy7 was chosen for Cys34 selective labeling. Conjugation or binding of drugs and dyes to albumin is a well-established technology [10,47,49,52,53,54]. Only one Cys34 of HSA has the free thiol group, which can be used for site-specific protein modification. On the other hand, the fluorine label should be multiplied to achieve enough 19F MRI sensitivity. We have suggested using a natural posttranslational modification of HSA to label lysine residues in proteins [65]. Homocysteine thiolactone (HTL) is a site-specific acylation compound [16,63,65] that can be used for the covalent conjugation of Lys residues, called N-homocysteinylation. The reaction involves only hyper-reactive Lys residues [16,63]. The predominant site is Lys525, and it has one of the lowest pKa among the albumin residues. Thiolactone chemistry provides a powerful tool to prepare fluorine- and spin-labeled albumin-based multimodal imaging probes and therapeutic agents [16,62,63,64]. HTL can be N-acylated by a fluorinated compound to produce the reagent for protein labeling. In the present work, we present the synthesis of N-fluorinated HTL compounds for HSA N-homocysteinylation. The acylation reagent synthesis, mechanism of formation, and kinetics studies were investigated. Two types of fluorinated albumin were tested as potential metal-free probes for 19F MRI. Dually labeled albumin with fluorescence dye (Cy5 or Cy7) and fluorine were synthesized to obtain a multimodal imaging probe. The conjugates were characterized by mass spectrometry (MALDI ToF MS) and 19F NMR. The biocompatibility was checked by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), circular dichroism (CD) spectroscopy, dynamic light scattering (DLS), and primary-used cytotoxicity test (MTT test) [66,67]. The trypsin proteolytic kinetics were applied to show the conjugates’ biodegradability in pseudo-tumor conditions. The results indicate the promise of fluorinated albumin to serve as a probe for 19F MRI.
2. Results and Discussion
2.1. Synthesis of N-Fluorinated Homocysteine Thiolactone Derivatives
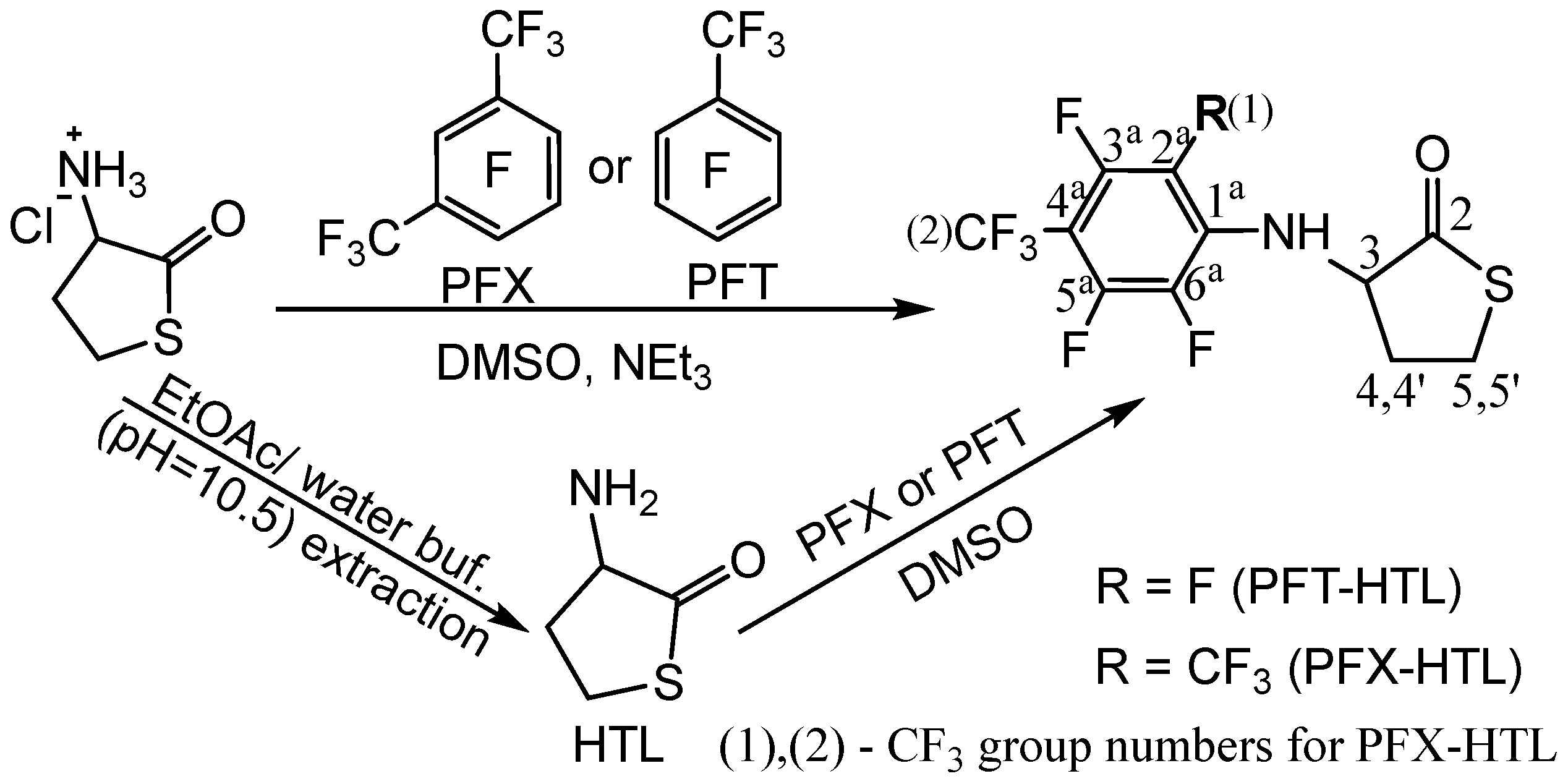
Homocysteine thiolactone (HTL) is a natural active cyclic thioester of nonprotein homocysteine amino acid (Hcy) [65]. HTL has a strained five-membered cycle, which may react with various nucleophiles [65,68]. HTL is a powerful synthetic tool for functional material production and protein labeling [16,69,70]. The amino group of HTL easily reacts with electrophilic reagents, which can be used for chemical modification. The synthesis of fluorinated HTL was performed according to one- or two-step procedures (Figure 1).
Nucleophilic aromatic substitution (SNAr) of fluorine in perfluoroarenes was used for stable bond formation with the HTL amino group. Dimethylsulfoxide (DMSO) solvent was utilized for HTL derivative synthesis. DMSO is an important solvent that dissolves both polar and nonpolar compounds and greatly increases the rate of nucleophilic substitution reactions. It exhibits several capabilities, such as high biocompatibility (nontoxic class 3 by the FDA), anti-inflammatory and bacteriostatic functions, and an antioxidant [71,72]. DMSO is widely used in life sciences and as an auxiliary solvent in the fabrication of polymers and drugs [73].
The one-stage synthetic pathway consists of an HTL hydrochloride reaction with perfluorotoluene (octafluorotoluene, PFT) or perfluoro-m-xylene (PFX) in the presence of a base (triethylamine) (Figure 1, top). The two-step procedure converts HTL hydrochloride to a free base of HTL with a subsequent reaction with perfluoroarenes (Figure 1, bottom). The free base of HTL synthesis looks simple. Nevertheless, HTL hydrochloride is chemically stable in bulk, and the free base of HTL is extremely reactive. The chemical preparation of an HTL-free base requires a thoughtful synthesis procedure. Due to its electrophilic-nucleophilic character, HTL is susceptible to condensation reactions (Figure 2) [64,74]. Self-condensation of two HTL molecules results in a dipeptide with a thiolactone ring, which may transform into a tripeptide by the intermolecular reaction with the 2,5-diketopiperazine compound (Figure 2). Diketopiperazines are the smallest cyclic peptides that attracted attention due to their ability to mimick peptidic pharmacophoric groups [75]. The wide spectrum of their biological properties is essential for modern medicinal chemistry [75]. The formation of 2,5-diketopiperazine was assumed by Vigneaud V. et al. in 1938 by elemental analysis [74]. The synthesis of fluorinated tripeptide and 2,5-diketopiperazine derivatives of homocysteine was previously confirmed in our laboratory by 1H NMR [64]. However, the detailed mechanism of compound formation was beyond the work [64].
The influence of various factors on a free base of HTL synthesis is presented in Table 1. Depending on the reaction time in the alkali solution, a HTL-free base or self-condensation product may be obtained (Table 1). To overcome HTL stability limitations, as low a time as possible in alkali solution and a 0.26 M and lower HTL concentration are required (see Section 3.3.1). However, if the goal of the procedure is to synthesize homocysteine peptides, a higher concentration of HTL and a 10–20 min time frame should be used.
High-time (30 min) in an alkali solution results in homocysteine 2,5-diketopiperazine disulfide polymer precipitation (Figure 2), as demonstrated by polymer reduction with dithiothreitol (DTT) and 1H NMR analysis. By comparing different organic solvents and considering their boiling point, miscibility with water, toxicity, and hazardousness, ethyl acetate turned out to be the solvent of choice. The amino group of HTL has extremely low pKa of 6.67 [76], 7.1 [77], and herein 6.56 ± 0.03 (25 °C) and 6.40 ± 0.02 (37 °C) (Supplementary Materials). Due to the almost neutral HTL charge in alkali solution, HTL is easily and with good efficiency (87%) extracted in ethyl acetate solution. Possible side reactions like HTL self-condensation could be suppressed by performing the organic solvent evaporation at as low a temperature as possible (self-cooling). The HTL was obtained at a moderate rate of 96%. In the case of a 0.26 M HTL concentration and 10 min in alkali with subsequent extraction and evaporation, the HTL yield is reduced. The major reaction byproduct is 2,5-diketopiperazine of homocysteine. Using a higher HTL concentration of 0.4 M, homocysteine tripeptide can be synthesized in greater quantities. It should be mentioned that the free base of HTL is not stable even when the temperature is below zero (Table 1). This compound should be used as quickly as possible for further synthesis.
The intrinsic instability of the HTL-free base requires highly reactive electrophiles for efficient reaction with the amino group; examples include acid halides, activated esters, and anhydrides [58,69]. Herein, we show the possibility of obtaining N-substituted derivatives of perfluoroarene. The fluorinated thiolactones were derived from the corresponding perfulorotoluene (PFT), perfluoro-m-xylene (PFX), perfluoro-m-xylene and perfluoro-o-xylene mixture. The SNAr is generally held to proceed either stepwise (Meisenheimer complexes) or by a concerted mechanism, which depends on the structural features of the reactants [78,79]. In a perfluoropolyaromatic compound, substitution may occur at one position preferentially. According to experimental data and theoretical calculations [80], the main SNAr sites for PFT and PFX are ortho- and para-positions towards the CF3 group [79,81]. Previously, we found that the reaction of HTL with PFT occurs in para position. Using a 1.4-fold excess of HTL, the PFT-HTL was found to be a major product with a 57% yield (Figure 3). In a side reaction, S-fluorinated 2,5-diketopiperazine and tripeptide derivatives were obtained (Figure 3 and Figure S1) [64]. Nevertheless, the initial HTL-free base doesn’t contain a sufficient amount of self-condensation products to indicate their synthesis in DMSO solution during the reaction. Moreover, the amino group of HTL reacts with PFT much slower than the SH group of homocysteine derivatives, as shown by 19F NMR. About 5 min is enough for SH nucleophiles to yield a SNAr reaction, in comparison toNH2 that requires several hours. High excess of HTL results in a PFT quick substitution in para- and then ortho-position, which can easily be observed by CF3 signal changes from triplet to duplet state by 19F NMR. Instead of PFT, the PFX compound reacts with HTL free base much quicker, with a PFX-HTL yield of ~ 93%. The sub-products are S-fluorinated derivatives of 2,5-diketopiperazine (6%) and tripeptide (1%). However, perfluoro-m-xylene is not a commercially available product. Its precursor is a perfluoro-m-xylene and perfluoro-o-xylene mixture, which reacts in the same way as HTL. Perfluoro-o-xylene stays intact in the reaction conditions. The fluorinated products of the reaction mixture were found to be poorly soluble in water at such high concentrations. It permitted us to precipitate them with water. The physicochemical characteristics of compounds are presented in the experimental part and supplementary materials.
The one-step synthetic pathway, using a system of DMSO, HTL hydrochloride, perfluoroarene, and triethylamine deprotection of the hydrochloride salt in various compound concentrations and conditions, is presented in Table 2. The simple reaction setup, use of fewer reactants, high yield, and fewer purification steps were considered essential factors. However, the reaction time is a major limitation of the one-step procedure. The increase in temperature allows the product to be obtained in ordinary time. Furthermore, a high base excess increases HTL self-condensation and N-substitution by triethylamine impurities, such as perfluoroarenes. In this way, we optimized the triethylamine excess and purified the base before synthesis (Table 2). Another possibility to reduce byproducts is using an expensive, sterically shielded N-ethylisopropylamine.
To evaluate a reaction mechanism (Figure 4) between perfluoroarenes and HTL, various ratios of compounds were investigated by 19F NMR (Figure 5 and Figure 6). Higher concentrations (0.1 M) of perfluoroarenes were used for the reaction to proceed in a short time (Table S1). To identify the rate constant k2, the reactant steady state approximation was used. The kinetics curve in the perfluoroarene and HTL ratio~1:1 is well described by the mechanism one. The k2 value is 0.43 ± 0.02 M−1s−1 and 0.016 ± 0.001 M−1s−1 for PFX and PFT, respectively. However, for at least a 2-fold excess of HTL per PFX, the mechanism changes to number two (with HTL catalysis).
2.2. Synthesis and Characterization of Fluorinated HSA Conjugates
There are only a few works that handle covalent modification of HSA by fluorine-containing compounds for 19F MRI [39,40,58,64,82]. Yu et al. [40] and Zhu et al. [39] have selectively modified albumin on Cys34 residue. However, line broadening and signal splitting occurred during the work, hindering the 19F MRI application [40]. Recently [39], fluorinated tags with six CF3 groups (two perfluoro-tert-butyl moieties) were conjugated to Cys34, generating a low width 19F peak. However, only one molecule is possible to conjugate with albumin by such a procedure. In the present work, we present the multiple lysine labeling of albumin. N-fluorinated HTL was used as a site-specific acylation reagent. Previously, we used PFT HTL to synthesize albumin with 3.1 ± 0.1 fluorinated tags [64]. N-Homocysteinylation of HSA by PFT-HTL and PFX-HTL was performed under physiological-like conditions (PBS, 37 °C, pH 7.4) (Figure 7). Unreacted low-MW compounds were removed using Centricon concentrators by standard procedure. The yields of fluorinated HSA conjugates were 90–95%. For comparison, N-homocysteinylated HSA was synthesized. The mixture of S-substituted derivatives of PFT and PFT-HTL reacts with albumin in the same manner as pure PFT-HTL. S-substituted derivatives of PFT precipitate in water solutions and do not influence the reaction.
The protein modification was proven by MALDI-ToF MS, UV spectroscopy, and 19F NMR. PFT-HSA and PFX-HSA contain 3.9 ± 0.1 and 4.9 ± 0.1 fluorinated residues, respectively. The 19F NMR spectra of PFT-HSA and PFX-HSA are presented in Figure 8. Multiple albumin labeling results in broad 19F signals. In the case of PFX-HSA, two CF3 groups of perfluoroxylene rings showed one split, but showed much more intense 19F signals than the PFT-HSA conjugate.
Protein over-labeling usually leads to significant changes in conformation, protein damage, loss of function, and precipitation. Wrong albumin modification may lead to protein oligomer formation and amyloidal transformation [65]. Therefore, we precisely controlled the protein labeling. Fluorinated albumin conjugates were characterized by gel electrophoresis (SDS-PAGE), circular dichroism (CD), and dynamic light scattering (DLS) (Table 3).
HSA solutions always contain some dimers, oligomers, and noncovalent, reversible dimer fractions (SDS PAGE Figure S2, Table 3) [83]. However, the amount of oligomer fraction is extremely high in the Hcy-HSA sample. N-homocysteinylation of HSA by natural HTL causes protein conformational changes and oligomer formation (Table 3, Hcy-HSA sample) [65,84]. On the contrary, the reaction with PFT-HTL and PFX-HTL leads only to slight changes in α-helical and β-sheet content and inhibits aggregation (Table 3, PFT-HSA and PFX-HSA samples). Despite the higher modification degree, the PFX-HSA conjugate better reproduces the properties of the native protein (Table 3, cf. HSA and PFX-HSA). The prevalent protein fraction is a monomer (~6 nm) in PFT-HSA and PFX-HSA samples, which is shown by number and volume modes by DLS (Figures S3 and S4, Table 3). The presence in the samples of a small number of large-sized particles can be seen in intensity mode, which is in good correlation with the native HSA sample (Figures S5 and S6).
2.3. Cytotoxicity of HSA Conjugates
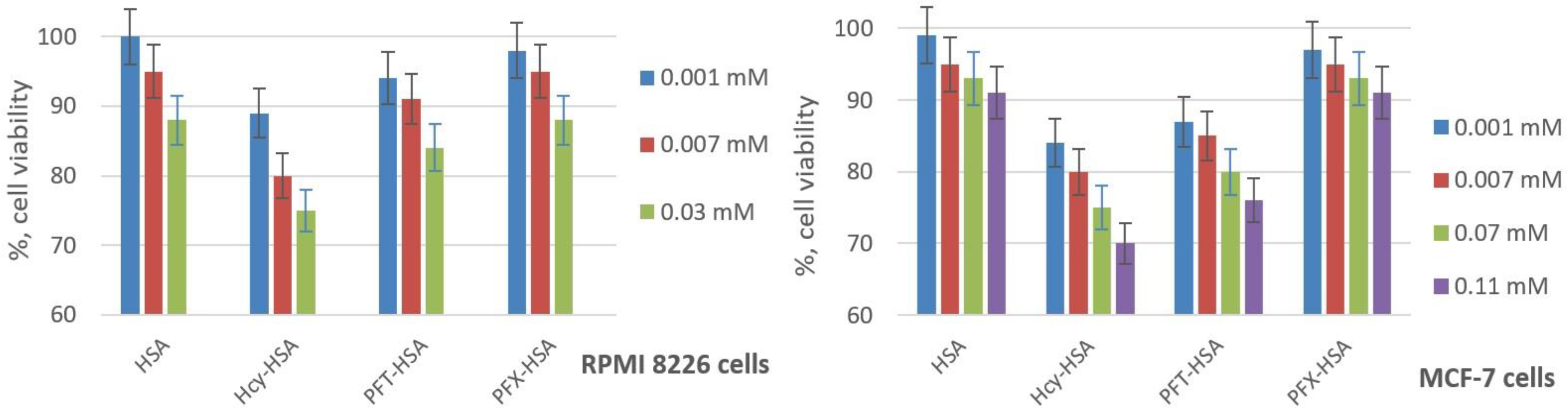
The ability of fluorinated HSA conjugates to cause cell damage was investigated by the widely-applied 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test using breast cancer MCF-7 and human multiple myeloma RPMI 8226 cells [66,67]. During the exponential growth phase, cell cultures were treated for 72 h, with the amounts of the HSA conjugates in the range expected for MRI applications. The fluorinated HSA conjugates show no significant cell viability difference from native HSA (Figure 9), which indicates their high biocompatibility. On the contrary, the incubation of the cells with N-homocysteinylated HSA (Hcy-HSA) results in a reduction in cell viability [85]. The cell viability in RPMI cell lines is slightly higher for PFX-HSA than for PFT-HSA, and significantly higher for MCF-7 cells, which displays better possible prospects of perfluoroxylene-based albumin conjugation.
2.4. Dual-Labeled Albumin Conjugates Synthesis
HSA has only one free SH group, Cys34, which can be used for site-specific modification [39,64]. However, the SH group of Cys34 may be in reduced form (in healthy patients, ~70%), oxidized as a disulfide (~25%), or a sulfinic or sulfonic acid (~5%). Ellman’s test indicated that the starting HSA in the present study contained 0.28 ± 0.05 sulfhydryl groups per protein molecule. Therefore, the albumin was treated with dithiothreitol (DTT) to convert the Cys-34 into free thiol used for labeling.
To synthesize dual-modified protein, we first obtained a fluorescently labeled fluorinated HSA (Figure 10). In order to visualize the cell uptake, HSA was labeled with Cy5 dye, which was shown to exhibit strong fluorescence for efficient identification of the molecule by flow cytometry [64]. The Cy7 is an optimal residue for further in vivo fluorescence imaging experiments [63]. Fluorescent dyes Cy7 or Cy5 were coupled to the single Cys-34 residue of albumin using the maleimide derivative of the dye via a Michael addition reaction. The HSA-Cy7 conjugate was modified by an N-homocysteinylation reaction using PFT-HTL or PFX-HTL compounds in PBS at 37 °C. PFT-HSA-Cy7 and PFX-HSA-Cy7 were characterized by different methods such as 19F NMR, MALDI ToF MS, UV, etc. (see experimental part). The cytotoxicity studies on RPMI and MCF-7 cell lines show the same good cell viability shown in Figure 9. The incubation of cells with PFT-HSA-Cy and PFX-HSA-Cy conjugates did not reveal any significant reduction in cell viability. The flow cytometry analysis shows almost the same cell internalization (~80%) for HSA-Cy5, PFT-HSA-Cy5, and PFX-HSA-Cy5 conjugates (Figure S7). The obtained data suggest that albumin modification does not influence cell penetration, and excellent cell viability studies are not related to low accumulation efficacy. Generally, our initial results have demonstrated the potential of PFT-HSA-Cy and PFX-HSA-Cy to serve as biocompatible optical and 19F MR imaging agents. 19F MRI is suitable for preoperative imaging studies. Fluorescence imaging is a powerful intraoperative tool for tumor margin demarcation [86,87]. Finally, it is important to incorporate multimodal imaging to precisely excise tumors.
2.5. Trypsin Degradation of Fluorinated HSA Conjugates
A trypsin digest of the HSA and its conjugates was carried out under physiological-like conditions at pH 7.4 (PBS buffer). The reaction was monitored by SDS-PAGE. As shown in Figure 11, N-homocysteinylated HSA is degraded by trypsin slower than unmodified HSA, which is in good correlation with previously published data [88]. On the contrary, N-homocysteinylated albumin by PFT-HTL or PFX-HTL shows higher proteolysis susceptibility, which provides efficient biodegradation of fluorinated conjugates in the organism. PFT-HSA and PFX-HSA show the same tryptic digest efficiency. A similar pattern of susceptibilities to proteolysis was observed in PFT-HSA-Cy and PFX-HSA-Cy. As proteolysis proceeded, new peptide bands appeared with smaller molecular weights below HSA (Figure S8). HSA is degraded specifically into certain large fragments (~55 kDa) and much slower than the lower MW fragments ~45, 40, and 35 kDa.
Proteases are enzymes that catalyze the hydrolysis of the peptide bonds in proteins. A number of the proteinases are secreted and active in the lysosomal compartment and extracellular matrix, where protease activity and concentration are highest. Many tumors have been shown to have elevated levels of proteases at an early stage [89]. Interestingly, persistent proteolysis has been observed in spreading tumors and metastasis [90]. The detection of a number of cancers may potentially benefit from protease imaging, including prostate, colon, brain, etc. [89,90,91,92]. Under the proteolysis condition in tumor tissue, heavy protein albumin (66.5 kDa) will decrease molecular weight, which highly influences the correlation time of fluorinated residues and 19F NMR peak width. Trypsinolysis generates a fast-motional regime for the fluorinated residues, and causes T1 and T2 relaxation time changes. The possible enzymatic hydrolysis of fluorinated albumin may serve as a stimuli-responsive 19F MRI probe [26]. However, such a proposition requires further research. An effective protease-activated switch system for nitroxide-labeled albumin and 1H MRI was evaluated [90,93]. Protein digestion switches from a stable, low-relaxivity state to one of high-relaxivity.
3. Materials and Methods
3.1. Chemicals
5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB), DL-homocysteine thiolactone (HTL) hydrochloride, and all solvents and other reagents, unless stated differently, were purchased from Sigma (St. Louis, MO, USA) at the highest available grade and used without purification. MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay kit was purchased from Invitrogen (Waltham, MA, USA). Centricon concentrators with a 3 kDa molecular weight cut-off were purchased from Millipore. Perfluoro-m-xylene (PFX) was prepared and kindly provided by V.E. Platonov (Novosibirsk Institute of Organic Chemistry, SB RAS). Cyanine dye derivatives were obtained from Lumiprobe (Moscow, Russia). Sequencing-grade modified trypsin for MALDI ToF peptide analysis was purchased from Promega (Madison, WI, USA) (cat. no. V5111). Trypsin for protein trypsinolysis was obtained from Gibco (Carlsbad, CA, USA) (cat. no. 15090046). The human serum albumin (HSA) used in this study was purchased from Sigma–Aldrich (St. Louis, MO, USA) (cat. no. A3782). MS (MALDI ToF) m/z HSA 66.48 kDa. Ellman’s test (SH group content) indicated that HSA in this study contained 0.28 ± 0.05 sulfhydryl groups per protein molecule.
3.2. Physicochemical Characterization
The number of thiol groups per albumin molecule was determined using the Ellman’s method, as described in the literature [94], or on the Thermo Scientific website with 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB) at pH 8 at 412 nm (ε = 1.4 × 104 M−1cm−1), using a UV-1800 spectrometer with unmodified HSA as a control. DTNB produces a measurable, yellow-colored product when it reacts with free SH groups.
Electronic absorption spectra were recorded on a UV-1800 spectrometer (Shimadzu, Tokyo, Japan). The concentrations of HSA solutions were determined by absorption at 278 nm, pH 7.4 (PBS), using the molar extinction coefficient ε = 3.7 × 104 M−1cm−1. The concentrations of HTL solutions were determined by absorption at 240 nm using the molar extinction coefficient ε (PBS, pH 7.4; or 0.25 M carbonate buffer, pH 10.5) = (5.0 ± 0.1) × 103 M−1cm−1.
Column chromatography was performed on silica gel 60 (0.063–0.200 mm) using CHCl3/(CH3)2CO 1:2.5 (V:V) and CHCl3 for elution.
SDS-PAGE. Human serum albumin conjugates were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis using 7% PAAG under Laemmli conditions with subsequent Coomassie Brilliant Blue staining. Quantitative data were obtained by digitizing the gel using GelPro Analyzer software 3.0.
NMR spectra were recorded at 25 °C in 5 mm NMR sample tubes. 1H, 13C, and 19F NMR spectra were recorded on an AV-300 NMR spectrometer (Bruker, Rheinstetten, Germany) at 300.13, 75.43, and 282.37 MHz, respectively. 19F NMR spectra for kinetics studies were recorded on a Spinsolve 80 NMR spectrometer (Magritek, Aachen, Germany) at 80 MHz. The chemical shifts were expressed in parts per million, ppm (δ). All 1H chemical shifts were calculated relative to the residual 1H NMR signal of the deuterated NMR solvent. C6F6 (δ = 0.00 ppm) was used as the external reference for chemical shifts in the 19F NMR spectra. The spin-spin coupling constants (J) are reported in hertz (Hz), and spin multiples are given as s (singlet), d (doublet), t (triplet), q (quartet), and m (multiplet). Abbreviation br. means broad. The coupling constants (J) in the difficult case were calculated by simulation of the spin system using the Spinworks freeware software package 4.0.5.
Circular dichroism (CD) data were collected at 25 °C with a JASCO J-600 spectrophotometer with a time constant of 4 s and a bandwidth of 1 nm, using a 0.01 cm path length quartz cell. All CD spectra were obtained from 190 to 240 nm, and the final spectrum was obtained as an average of 20 spectra. To determine the percentages of α-helices, β-sheets, and the disordered structures, we minimized the difference between the theoretical and experimental curves. The theoretical curves were calculated as a linear combination of the basis spectra of various components of the secondary structures taken from the CCA+ software [95]. For recording the CD spectra, samples consisted of 5 µM albumin in PBS. Changes in the secondary structure of the protein were examined by deconvolution of CD spectra to determine the α-helix and β-sheet content.
ESI MS was performed on a mass spectrometer, the LC/MSD Trap XCT (Agilent, Santa Clara, CA, USA).
MALDI-ToF mass spectra of proteins were recorded on a Bruker Autoflex Speed (Bruker, Germany) MALDI-TOF mass spectrometer in a positive linear mode. A smartbeam-II laser was used with 2,5-dihydroxyacetophenone (2,5-DHAP) as the matrix. Protein samples were desalted by ZipTip C4 pipette tips. A 2 µL protein sample solution was mixed with 2 µL of 2% TFA (trifluoroacetic acid). For the latter solution, 2 µL of the matrix (2,5-DHAP) was added. The mixture was pipetted up and down until the crystallization started. Approximately 1 μL of the resulting solution was deposited on the 384-grit steel target plate and allowed to dry before being introduced into the mass spectrometer. Mass spectra were obtained by averaging 2500–3000 laser shots. External calibration was provided by the [M+H]+ peak of human serum albumin at m/z 66,467. MALDI ToF data were deconvoluted using the software package mMass 5.5.0 [96,97,98].
Dynamic light scattering (DLS) measurements were carried out on a Malvern Zetasizer Nano device (Malvern Instruments, Worcestershire, UK) at 25 °C. The protein samples (50 µM) were prepared in PBS buffer, and measurements of their size were conducted.
3.3. Synthesis of S- and N-Fluorinated Derivatives of Homocysteine Thiolactone
3.3.1. Synthesis of Homocysteine Thiolactone (HTL) Free Base
DL-homocysteine thiolactone hydrochloride (20.0 mg, 0.130 mmol) was stirred with 0.5 mL of preliminarily cooled 4 °C 0.25 M sodium carbonate buffer (pH 10.5) for 30 s. The alkali solution of HTL was quickly shaken with preliminary cooled to 4 °C ethyl acetate (13 mL), followed by organic solvent separation into a flask. The solvent was immediately removed in vacuo for 5–7 min. Evaporation was performed without a water bath. Under such conditions, the organic solvent HTL solution is self-cooled. After stopping the evaporation due to excessive cooling, the flask with the solution is lowered into a water bath (25 °C) until fogging disappears, and the procedure is repeated. The product was directly used for the next step owing to its instability even at temperatures below zero. The extraction efficiency was estimated by measuring the electronic absorption of the diluted water solutions of the given HTL solution before and after extraction (λmax 240 nm; ε = (5.0 ± 0.1) × 103 M−1cm−1). The efficiency of extraction was 87%. The thiolactone cycle retention rate during the extraction stage was 98%. The thiolactone cycle retention rate after extraction and evaporation was ~96%. The remaining 4% of compounds are 2,5-diketopiperazine of homocysteine (3%) and homocysteine tripeptide (1%).
3.3.2. Reaction of HTL Free Base with Perfluorotoluene (PFT) and Perfluoro-m-xylene (PFX) (The Method № 1)
The synthetic procedure for PFT-HTL and PFX-HTL was adapted from Chubarov et al. [58,64]. For this synthesis, a free base of HTL obtained by a procedure in Section 3.3.1 was used. HTL (15.21 mg, 0.13 mmol) was quickly dissolved in 0.6 mL DMSO, and PFT (6.38 µL, 21.24 mg, 0.09 mmol) or 1,4- and 1,3-perfluoroxylene mixture (PFX) (11.22 µL, 0.12 mmol; 0.09 mmol of 1,3-PFX), or 1,3-PFX (8.1 µL, 0.09 mmol) was added. The mixtures were allowed to react at 25 °C for 14 h. After the reaction was completed, 0.4 mL of water was added to the solution, yielding a white precipitate. The mixture was centrifuged for 2 min at 6000× g, and the supernatant was discarded. The washing procedure was repeated at least one more time. The target compounds were separated by flash chromatography on silica gel 60 (0.063–0.200 mm) using CHCl3/(CH3)2CO 1:2.5 (V:V) and CHCl3 for elution. The PFX-HTL compound was not required for flash chromatography.
3.3.3. Reaction of HTL Hydrochloride with Perfluorotoluene (PFT) and Perfluoro-m-xylene (PFX) in the Presence of a Triethylamine (The Method № 2)
The PFT (4.25 µL, 14.1 mg, 0.060 mmol) or 1,4- and 1,3-perfluoroxylene mixture (PFX) (8.09 µL, 0.09 mmol; 0.065 mmol of 1,3-PFX) and 13.5 µL of fresh anhydrous trimethylamine (refluxed under p-toluenesulfonyl chloride) were added to DL-HTL hydrochloride (10.00 mg, 0.065 mmol) dissolved in 6.5 mL of DMSO. The mixtures were allowed to react at 75 °C for 70 h. After cooling to ambient temperature, the reaction mixture was evaporated under reduced pressure using a liquid-nitrogen-cooled trap. After that, 0.4 mL of water was added to the solutions. The mixture was centrifuged for 2 min at 6000× g, and the supernatant was discarded. The procedure was repeated at least one more time. The target compound PFT-HTL was separated by flash chromatography on silica gel 60 (0.063–0.200 mm) using CHCl3/(CH3)2CO 1:2.5 (V:V) and CHCl3 for elution. The PFX-HTL compound was not required for flash chromatography. Perfluoro-o-xylene stays intact in the reaction conditions.
3.3.4. N-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenyl) Homocysteine Thiolactone (PFT-HTL)
UV-vis (CH3CN): λmax 240 nm, ε = (1.3 ± 0.1) × 104 M−1cm−1; 256 nm, ε = (2.1 ± 0.1) × 104 M−1cm−1. 19F NMR (DMSO-d6): δ 108.91 (t, 3F, CF3), 17.46 (m, 2F, F-3a, F-5a), 5.02 (m, 2F, F-2a, F-6a), JCF3,3a = JCF3,5a = 20.2, J2a,3a = J5a,6a = 19.0, J3a,6a = J2a,5a = 5.0, JNH,2a = JNH,6a = 2.5, coupling constants (J) were calculated by simulation of the spin system using SpinWorks freeware software package. 1H NMR (DMSO-d6): δ 6.97 (br.d, 1H, NH), 4.75 (m, 1H, H-3), 3.42 (m, 1H, H-5′), 3.30 (m, 1H, H-5), 2.57 (m, 1H, H-4′), 2.33 (m, 1H, H-4), J3,4 = 12.8, J3,4′ = 7.0, J3,5 = 0.5, J3,NH = 8.8, J4,4′ = 12.2, J4,5 = 7.0, J4,5′ = 12.2, J4′,5 = 1.3, J4′,5′ = 5.5, J5,5′ = 11.3, JNH,2a = JNH,6a = 2.5, J4,NH = 2.0, J4′,NH = 1.0. 13C NMR (CD3CN): δ 204.6 (C-2), 146.4 (C-2a,6a), 144.0 (C-3a,5a), 123.4 (C-1a), 109.0 (CF3), 108.5 (C-4a), 64.3 (C-3), 30.3 (C-4,4′ or 5,5′), 28.0 (C-5,5′ or 4,4′). ESI MS (m/z): calc. for C11H5NOSF7 [M-H]: 331.998, found 331.997. The yield of PFT-HTL was 57 % (method № 1), and 94 % (method № 2).
3.3.5. N-(2,3,5-Trifluoro-4,6-bis(trifluoromethyl)phenyl) Homocysteine Thiolactone (PFX-HTL)
UV–vis (CH3CN): λmax 255 nm ε = (1.3 ± 0.1) × 104 M−1cm−1; 296 nm ε = (6.0 ± 0.1) × 103 M−1cm−1; 325 nm ε = (1.9 ± 0.1) × 103 M−1cm−1. 19F NMR (CD3CN): 108.78 (d, 3F, CF3(1)), 108.17 (t, 3F, CF3(2)), 45.18 (m, 1F, F-3a), 31.60 (m, 1F, F-5a), 7.91 (m, 1F, F-6a), JCF3(1),3a = 30.9, JCF3(1),5a = 3.6, JCF3(2),3a = JCF3(2),5a = 22.3, J3a,5a = 2.6, J3a,6a = 10.0, J5a,6a = 18.0, JNH,6a = 2.0. 1H NMR (DMSO-d6): 6.52 (br. d, 1H, NH), 4.71 (m, 1H, H-3), 3.43 (m, 1H, H-5′), 3.31 (m, 1H, H-5), 2.76 (m, 1H, H-4′), 2.26 (m, 1H, H-4), J3,4 = 12.8, J3,4′ = 7.0, J3,5 = 0.5, J3,NH = 8.4, J4,4′ = 12.2, J4,5 = 7.0, J4,5′ = 12.2, J4′,5 = 1.3, J4′,5 ’= 5.3, J5,5 ’= 11.5, J4,NH = 2.0, J4′,NH = 1.0. 13C NMR (CD3CN): 207.2 (CO), 155.0 (C-3a), 141.6 (C-5a), 140.7 (C-6a), 138.3 (C-1a), 125.8 (CF3), 124.4 (CF3), 123.1 (C-4a), 121.6 (C-2a), 66.2 (C-3), 33.4 (C-5,5′), 28.0 (C-4,4′). ESI MS negative mode (m/z) calculated for C12H5NOSF9 [M-H]-: 381.995, found 381.896. The yield of PFX-HTL was 93 % (method № 1) and 99 % (method № 2).
3.4. Synthesis of N-homocysteinylated Albumin (Hcy-HSA)
The synthetic procedure for Hcy-HSA was adapted from Chubarov et al. [16]. Briefly, to 0.5 mL of a 0.53 mM solution of HSA in PBS (15 mM KH2PO4, 145 mM NaCl, pH 7.4 with 0.2 mM EDTA), a solution of a 6-fold molar excess of D,L-homocysteine thiolactone hydrochloride dissolved in 0.04 mL PBS. The reaction mixture was incubated at 37°C for 18 h with stirring at 500 rpm. The excess reagents were removed by filtration using Centricon concentrators (3 kDa molecular weight cutoff; Amicon Centriprep YM30, Millipore) according to the procedure described by the manufacturer, with PBS as the eluent. The yield of Hcy-HSA was ~90%. Hcy-HSA UV–vis (PBS, pH 7.4): λmax 278. MALDI TOF MS m/z 66.86 kDa (~3.2 ± 0.1 Hcy residues). The number of SH groups per protein molecule was analyzed by Ellman’s assay, which is 3.37 ± 0.05 (excluding albumin-free SH group 3.09 ± 0.05).
3.5. Synthesis of HSA-Cy5 and HSA-Cy7
Commercially available HSA contains 0.1–0.7 free SH groups per protein, depending on the preparation method and plasma grade. According to our Ellmans’ test results, albumin A3782 contains 0.28 SH groups per protein molecule (see Materials subsection). To enlarge albumin modification by fluorescence dye, the reduction reaction by dithiothreitol (DTT) was performed [99]. Under this condition, no intramolecular disulfides (protein S-S bridges) were broken [99]. Using the Ellmans’ test, it was found that reduced albumin (HSA-SH) contains 0.9 ± 0.1 sulfhydryl group per protein molecule.
The synthetic procedure was adapted from Chubarov et al. [64]. Briefly, to 1 mL of a 1.0 mM solution of HSA-SH in PBS (15 mM KH2PO4, 145 mM NaCl, pH 7.4), add a solution of a 3-fold molar excess of sulfo-Cy5 maleimide or a 1.2-fold molar excess of a Cy7 maleimide derivative dissolved in 0.1 mL DMSO. During this process, the solution was stirred vigorously. The reaction mixture was incubated at 37 °C for 12 h with stirring at 500 rpm. The excess reagents were removed by filtration using Centricon concentrators (3 kDa molecular weight cutoff; Amicon Centriprep YM30, Millipore) according to the procedure described by the manufacturer, with PBS as the eluent. The yield of HSA-Cy5 and HSA-Cy7 was ~95%. HSA-Cy5 UV–vis (PBS, pH 7.4): λmax 278, 652 nm (ε652 = (2.3 ± 0.1) × 105 M−1cm−1, ~0.85 sulfo-Cy5 residues). HSA-Cy7 UV–vis (PBS, pH 7.4): λmax 278, 762 nm (ε762 = (1.8 ± 0.1) × 105 M−1cm−1, ~0.90 Cy7 residues).
3.6. Synthesis of Fluorinated Albumin Conjugates (PFT-HSA, PFX-HSA, PFT-HSA-Cy5, PFX-HSA-Cy5, PFT-HSA-Cy7, PFX-HSA-Cy7)
The synthetic procedure was adapted from Chubarov et al. [58,64]. Briefly, to 0.4 mL of the 1.15 mM solution of HSA or HSA-Cy in PBS (15 mM KH2PO4, pH 7.4, 145 mM NaCl), the solution of 20-fold molar excess of N-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenyl) homocysteine thiolactone (PFT-HTL) or -(3,5,6-trifluoro-2,4-bis(trifluoromethyl)phenyl) homocysteine thiolactone (PFX-HTL) in 150 µL DMSO was added. The reaction mixture was incubated at 37 °C for 20 h with stirring at 300 rpm. The excess reagents were removed by filtration using Centricon concentrators (3 kDa molecular weight cutoff; Amicon Centriprep YM30, Millipore) according to the procedure described by the manufacturer, with PBS as the eluent.
PFT-HSA. The yield was ~90%. UV-vis (PBS, pH 7.4): λmax 256 ε256 = (9.7 ± 0.1) × 104 M−1cm−1. 19F NMR (PBS, pH 7.4): δ 110.7–106.1 (br. m., max 108.9 ppm, CF3), 10.0–20.0 (br. m., max 16.6 ppm, F-3, F-5), −5.0–5.0 (br. m., max 0.0 ppm, F-2, F-6). MALDI TOF MS m/z 67.8 kDa (~3.95 ± 0.05 PFT-Hcy residues). The average amount of PFT-Hcy residue per protein molecule by UV and MALDI TOF MS is 3.9 ± 0.1.
PFT-HSA-Cy5. The yield was ~92%. UV-vis (PBS, pH 7.4): λmax 256, 652 nm ε652 = (2.3 ± 0.1) × 105 M−1cm−1. MALDI TOF MS m/z 68.5 kDa.
PFT-HSA-Cy7. The yield was ~90%. UV-vis (PBS, pH 7.4): λmax 256, 762 nm ε762 = (1.8 ± 0.1) × 105 M−1cm−1. MALDI TOF MS m/z 68.4 kDa.
PFX-HSA. The yield was ~95%. UV-vis (PBS, pH 7.4): λmax 256, 278, 300 ε256 = (9.7 ± 0.1) × 104 M−1cm−1. 19F NMR (PBS, pH 7.4): δ 109.0 (br. m., CF3(1), CF3(2)), 47.0 (br. m., F-3), 33.8 (br. m., F-5), 4.0 (br. m., F-6). MALDI TOF MS m/z 68.4 kDa (~5.0 ± 0.1 PFX residues). The average amount of PFX-Hcy residue per protein molecule by UV and MALDI TOF MS is 4.9 ± 0.1.
PFX-HSA-Cy5. The yield was ~90%. UV-vis (PBS, pH 7.4): λmax 256, 278, 300, 652 nm ε652 = (2.3 ± 0.1) × 105 M−1cm−1. MALDI TOF MS m/z 69.1 kDa.
PFX-HSA-Cy7. The yield was ~90%. UV-vis (PBS, pH 7.4): λmax 256, 278, 300, 762 nm ε762 = (1.8 ± 0.1) × 105 M−1cm−1. MALDI TOF MS m/z 69.0 kDa.
3.7. Synthesis of PFT-HSA Using the Mixture of S- and N-Substituted Derivatives of PFT
To 0.4 mL of the 1.15 mM solution of HSA in PBS, the solution of a 20-fold molar excess of PFT-HTL with various amounts of S-substituted derivatives of PFT in 150 µL DMSO was added. The reaction mixture was incubated at 37 °C for 20 h with stirring at 300 rpm. The excess reagents were removed by filtration using Centricon concentrators (3 kDa molecular weight cutoff; Amicon Centriprep YM30, Millipore) according to the procedure described by the manufacturer, with PBS as the eluent.
3.8. Susceptibility of Albumin Conjugates to Proteolysis
The solution of HSA and HSA conjugates 50 µL of 1 mM were digested with 2.5 µL trypsin («Gibco» cat. no. 15090046) at an enzyme-substrate ratio of 1:100 in PBS buffer (pH 7.4) for 0.5–6 h at 37 °C. Digest aliquots of 3.75 µL were diluted with 71.25 µL of PBS. The latter solution aliquot (7.5 µL) was mixed 1:1 with SDS-PAGE sample buffer (50 mM tris hydrochloride pH 6.8, 1% SDS, 10% glycerol, and 0.025% bromophenol blue with or without 0.1% DTT), denatured for 5 min at 95 °C, and subjected to SDS-PAGE on 10% gels. All samples were analyzed by SDS-PAGE on the same day. Protein bands were visualized by staining with Coomassie Brilliant Blue. Quantitative data were obtained by digitizing the gel using GelPro Analyzer software.
3.9. Cell Culture and Toxicity Assay
Cell Culture. Tumor cell lines from human mammary adenocarcinoma MCF-7 and human glioblastoma T98G were cultured in IMDM and EMEM medium, respectively, supplemented with 10% fetal bovine serum (FBS) (Invitrogen), penicillin (100 units/mL), and streptomycin (100 μg/mL) at 37 °C with 5% CO2 in a humid atmosphere.
The inhibition of cell proliferation was determined using a colorimetric assay based on the cleavage of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) by mitochondrial dehydrogenases in viable cells, leading to formazan formation [67]. Briefly, exponentially growing cells were plated in a 96-well plate (2000 cells per well). After overnight incubation, the cells were treated with media containing albumin conjugates. The solutions of conjugates were applied into the media for 72 h at 37 °C. A 10 μL aliquot of MTT solution (25 mg/mL in PBS) was added to each well, and the plates were incubated at 37 °C for 3 h. The medium was removed, and the dark blue crystals of formazan were dissolved in 0.1 mL of isopropanol. The absorbance at 570 nm (peak) and 620 nm (baseline) was read using a microplate reader, Multiscan EX (Thermo Electron Corporation). The results were expressed as a percentage of the control values. All values in the present study are given as the mean ± standard deviation (SD) values, and all measurements were repeated three times.
4. Conclusions
Human serum albumin (HSA) is an attractive protein for targeted delivery. Recently, various albumin-binding prodrugs and probes or covalent constructions have also been developed for cancer treatment [10,46,54,100]. The fluorinated HSA has great potential for 19F MRI diagnostics and therapy [39,58,63,64]. Herein, a new family of fluorinated albumin conjugates using perfluo-m-xylene moieties was synthesized. These conjugates retain important biocompatible properties of albumin, such as high cell viability and cell internalization. Sparing perfluo-m-xylene conjugation through HTL natural acylation reagent, leads to the preservation of HSA physical properties, such as 3D dimensional structure and oligomerization susceptibility. The combination of good biodegradation in cancer tissue and the long blood half-life of albumin can provide a possible enzyme-activatable 19F MRI probe, which warrants further investigation. In conclusion, the fluorinated albumin species have a high potential for 19F MRI tumor diagnostics. Nevertheless, further optimization of fluorine tags is required to prove a single low-width 19F peak. An extended biomedical study on in vivo models is indispensable for clinical potential examination.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/molecules28041695/s1, Figure S1: Structures of nonfluorinated and S-fluorinated 2,5-diketopiperazine and tripeptide derivatives; Table S1: The ingredients and reaction conditions of the reaction between HTL and perfluoroarenes for mechanism studies; Figure S2: SDS–PAGE of PFT-HSA (line 1), PFX-HSA (line 2), HSA (line 3), and Hcy-HSA (line 4) conjugates; Figure S3: DLS size distribution of A—PFT-HSA; B—PFX-HSA by number; Figure S4: DLS size distribution of A—PFT-HSA; B—PFX-HSA by volume; Figure S5: DLS size distribution of A—PFT-HSA; B—PFX-HSA by intensity; Figure S6: DLS size distribution of HSA by intensity; Figure S7: Cellular uptake of fluorescently labeled albumin conjugates; Figure S8: Typical albumin proteolysis SDS-PAGE data.
Author Contributions
Conceptualization, A.S.C.; data curation, A.S.C. and D.E.M.; investigation, A.S.C. and D.E.M.; writing—original draft preparation, A.S.C. and D.E.M.; supervision, A.S.C.; project administration, A.S.C.; funding acquisition, A.S.C.; All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation (grant no. 21-74-00120) and the protein proteolysis studies was supported by the Russian state-funded project for ICBFM SB RAS (grant number 121112900217-3).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from authors.
References
- Xiao, Y.-D.; Paudel, R.; Liu, J.; Ma, C.; Zhang, Z.-S.; Zhou, S.-K. MRI contrast agents: Classification and application (Review). Int. J. Mol. Med. 2016, 38, 1319–1326. [Google Scholar] [CrossRef]
- Angelovski, G. What We Can Really Do with Bioresponsive MRI Contrast Agents. Angew. Chem. Int. Ed. 2016, 55, 7038–7046. [Google Scholar] [CrossRef]
- Wahsner, J.; Gale, E.M.; Rodríguez-Rodríguez, A.; Caravan, P. Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers. Chem. Rev. 2018, 119, 957–1057. [Google Scholar] [CrossRef]
- Geraldes, C.F.G.C.; Peters, J.A. MRI Contrast Agents in Glycobiology. Molecules 2022, 27, 8297. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Meade, T.J. Molecular Magnetic Resonance Imaging with Gd(III)-Based Contrast Agents: Challenges and Key Advances. J. Am. Chem. Soc. 2019, 141, 17025–17041. [Google Scholar] [CrossRef]
- Kim, H.-K.; Lee, G.H.; Chang, Y. Gadolinium as an MRI contrast agent. Future Med. Chem. 2018, 10, 639–661. [Google Scholar] [CrossRef]
- Caspani, S.; Magalhães, R.; Araújo, J.P.; Sousa, C.T. Magnetic Nanomaterials as Contrast Agents for MRI. Materials 2020, 13, 2586. [Google Scholar] [CrossRef]
- Petrov, K.D.; Chubarov, A.S. Magnetite Nanoparticles for Biomedical Applications. Encyclopedia 2022, 2, 1811–1828. [Google Scholar] [CrossRef]
- Popova, V.; Dmitrienko, E.; Chubarov, A. Magnetic Nanocomposites and Imprinted Polymers for Biomedical Applications of Nucleic Acids. Magnetochemistry 2023, 9, 12. [Google Scholar] [CrossRef]
- Chubarov, A.S. Serum Albumin for Magnetic Nanoparticles Coating. Magnetochemistry 2022, 8, 13. [Google Scholar] [CrossRef]
- Thomsen, H.S.; Morcos, S.K.; Almén, T.; Bellin, M.-F.; Bertolotto, M.; Bongartz, G.; Clement, O.; Leander, P.; Heinz-Peer, G.; Reimer, P.; et al. Nephrogenic systemic fibrosis and gadolinium-based contrast media: Updated ESUR Contrast Medium Safety Committee guidelines. Eur. Radiol. 2013, 23, 307–318. [Google Scholar] [CrossRef]
- Pasquini, L.; Napolitano, A.; Visconti, E.; Longo, D.; Romano, A.; Tomà, P.; Espagnet, M.C.R. Gadolinium-Based Contrast Agent-Related Toxicities. CNS Drugs 2018, 32, 229–240. [Google Scholar] [CrossRef]
- Ramalho, J.; Ramalho, M. Gadolinium Deposition and Chronic Toxicity. Magn. Reson. Imaging Clin. N. Am. 2017, 25, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Malikova, H.; Holesta, M. Gadolinium Contrast Agents—Are they Really Safe? J. Vasc. Access 2017, 18 (Suppl. S2), S1–S7. [Google Scholar] [CrossRef] [PubMed]
- Akakuru, O.U.; Iqbal, M.Z.; Saeed, M.; Liu, C.; Paunesku, T.; Woloschak, G.; Hosmane, N.S.; Wu, A. The Transition from Metal-Based to Metal-Free Contrast Agents for T1 Magnetic Resonance Imaging Enhancement. Bioconjug. Chem. 2019, 30, 2264–2286. [Google Scholar] [CrossRef]
- Dobrynin, S.; Kutseikin, S.; Morozov, D.; Krumkacheva, O.; Spitsyna, A.; Gatilov, Y.; Silnikov, V.; Angelovski, G.; Bowman, M.K.; Kirilyuk, I.; et al. Human Serum Albumin Labelled with Sterically-Hindered Nitroxides as Potential MRI Contrast Agents. Molecules 2020, 25, 1709. [Google Scholar] [CrossRef]
- Nguyen, H.V.-T.; Detappe, A.; Harvey, P.; Gallagher, N.; Mathieu, C.; Agius, M.P.; Zavidij, O.; Wang, W.; Jiang, Y.; Rajca, A.; et al. Pro-organic radical contrast agents (“pro-ORCAs”) for real-time MRI of pro-drug activation in biological systems. Polym. Chem. 2020, 11, 4768–4779. [Google Scholar] [CrossRef]
- Nguyen, H.V.-T.; Detappe, A.; Gallagher, N.M.; Zhang, H.; Harvey, P.; Yan, C.; Mathieu, C.; Golder, M.R.; Jiang, Y.; Ottaviani, M.F.; et al. Triply Loaded Nitroxide Brush-Arm Star Polymers Enable Metal-Free Millimetric Tumor Detection by Magnetic Resonance Imaging. ACS Nano 2018, 12, 11343–11354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lloveras, V.; Lope-Piedrafita, S.; Calero-Pérez, P.; Wu, S.; Candiota, A.P.; Vidal-Gancedo, J. Metal-Free Radical Dendrimers as MRI Contrast Agents for Glioblastoma Diagnosis: Ex Vivo and In Vivo Approaches. Biomacromolecules 2022, 23, 2767–2777. [Google Scholar] [CrossRef]
- Bartusik-Aebisher, D.; Bober, Z.; Zalejska-Fiolka, J.; Kawczyk-Krupka, A.; Aebisher, D. Multinuclear MRI in Drug Discovery. Molecules 2022, 27, 6493. [Google Scholar] [CrossRef]
- Godovikova, T.S.; Lisitskiy, V.A.; Antonova, N.M.; Popova, T.V.; Zakharova, O.D.; Chubarov, A.S.; Koptyug, I.V.; Sagdeev, R.Z.; Kaptein, R.; Akulov, A.E.; et al. Ligand-Directed Acid-Sensitive Amidophosphate 5-Trifluoromethyl-2′-Deoxyuridine Conjugate as a Potential Theranostic Agent. Bioconjug. Chem. 2013, 24, 780–795. [Google Scholar] [CrossRef] [PubMed]
- Dawid, J.; Krawczyk, T. F-19 MRI Probes for Multimodal Imaging. Chem. A Eur. J. 2022, 28, e202102556. [Google Scholar] [CrossRef]
- Chen, H.; Viel, S.; Ziarelli, F.; Peng, L. 19F NMR: A valuable tool for studying biological events. Chem. Soc. Rev. 2013, 42, 7971–7982. [Google Scholar] [CrossRef] [PubMed]
- Dahanayake, J.N.; Kasireddy, C.; Karnes, J.P.; Verma, R.; Steinert, R.M.; Hildebrandt, D.; Hull, O.A.; Ellis, J.M.; Mitchell-Koch, K.R. Progress in Our Understanding of 19F Chemical Shifts. In Annual Reports on NMR Spectroscopy, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 93, pp. 281–365. [Google Scholar]
- Ruiz-Cabello, J.; Barnett, B.P.; Bottomley, P.A.; Bulte, J.W. Fluorine (19F) MRS and MRI in biomedicine. NMR Biomed. 2011, 24, 114–129. [Google Scholar] [CrossRef]
- Cho, M.H.; Shin, S.H.; Park, S.H.; Kadayakkara, D.K.; Kim, D.; Choi, Y. Targeted, Stimuli-Responsive, and Theranostic 19F Magnetic Resonance Imaging Probes. Bioconjug. Chem. 2019, 30, 2502–2518. [Google Scholar] [CrossRef] [PubMed]
- Tirotta, I.; Dichiarante, V.; Pigliacelli, C.; Cavallo, G.; Terraneo, G.; Bombelli, F.B.; Metrangolo, P.; Resnati, G. 19F Magnetic Resonance Imaging (MRI): From Design of Materials to Clinical Applications. Chem. Rev. 2015, 115, 1106–1129. [Google Scholar] [CrossRef]
- Cobb, S.L.; Murphy, C.D. 19F NMR applications in chemical biology. J. Fluor. Chem. 2009, 130, 132–143. [Google Scholar] [CrossRef]
- Qin, L.; Sheridan, C.; Gao, J. Synthesis of tetrafluorinated aromatic amino acids with distinct signatures in 19F NMR. Org. Lett. 2012, 14, 528–531. [Google Scholar] [CrossRef]
- Zhang, C.; Yan, K.; Fu, C.; Peng, H.; Hawker, C.J.; Whittaker, A.K. Biological Utility of Fluorinated Compounds: From Materials Design to Molecular Imaging, Therapeutics and Environmental Remediation. Chem. Rev. 2021, 122, 167–208. [Google Scholar] [CrossRef]
- Meyer, A.; Dechert, S.; Dey, S.; Höbartner, C.; Bennati, M. Measurement of Angstrom to Nanometer Molecular Distances with 19F Nuclear Spins by EPR/ENDOR Spectroscopy. Angew. Chem. Int. Ed. 2020, 59, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Kehl, A.; Hiller, M.; Hecker, F.; Tkach, I.; Dechert, S.; Bennati, M.; Meyer, A. Resolution of chemical shift anisotropy in 19F ENDOR spectroscopy at 263 GHz/9.4 T. J. Magn. Reson. 2021, 333, 107091. [Google Scholar] [CrossRef]
- Asanbaeva, N.B.; Sukhanov, A.A.; Diveikina, A.A.; Rogozhnikova, O.Y.; Trukhin, D.V.; Tormyshev, V.M.; Chubarov, A.S.; Maryasov, A.G.; Genaev, A.M.; Shernyukov, A.V.; et al. Application of W-band 19F electron nuclear double resonance (ENDOR) spectroscopy to distance measurement using a trityl spin probe and a fluorine label. Phys. Chem. Chem. Phys. 2022, 24, 5982–6001. [Google Scholar] [CrossRef]
- Wu, T.; Li, A.; Chen, K.; Peng, X.; Zhang, J.; Jiang, M.; Chen, S.; Zheng, X.; Zhou, X.; Jiang, Z.-X. Perfluoro-tert-butanol: A cornerstone for high performance fluorine-19 magnetic resonance imaging. Chem. Commun. 2021, 57, 7743–7757. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cui, J.; Li, C.; Zhou, H.; Chang, J.; Aras, O.; An, F. 19F MRI Nanotheranostics for Cancer Management: Progress and Prospects. Chemmedchem 2022, 17, e202100701. [Google Scholar] [CrossRef] [PubMed]
- Mali, A.; Kaijzel, E.L.; Lamb, H.J.; Cruz, L.J. 19F-nanoparticles: Platform for in vivo delivery of fluorinated biomaterials for 19F-MRI. J. Control. Release 2021, 338, 870–889. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.M.; Gigliobianco, M.R.; Firouzabadi, B.M.; Censi, R.; Di Martino, P. Nanotechnology as a Versatile Tool for 19F-MRI Agent’s Formulation: A Glimpse into the Use of Perfluorinated and Fluorinated Compounds in Nanoparticles. Pharmaceutics 2022, 14, 382. [Google Scholar] [CrossRef]
- Wu, L.; Liu, F.; Liu, S.; Xu, X.; Liu, Z.; Sun, X. Perfluorocarbons-Based 19F Magnetic Resonance Imaging in Biomedicine. Int. J. Nanomed. 2020, 15, 7377–7395. [Google Scholar] [CrossRef]
- Zhu, L.; Li, Y.; Jiang, M.; Ke, C.; Long, H.; Qiu, M.; Zhang, L.; Ye, C.; Zhou, X.; Jiang, Z.-X.; et al. Self-Assembly of Precisely Fluorinated Albumin for Dual Imaging-Guided Synergistic Chemo–Photothermal–Photodynamic Cancer Therapy. ACS Appl. Mater. Interfaces 2023, 15, 2665–2678. [Google Scholar] [CrossRef]
- Yue, X.; Feng, Y.; Yu, Y.B. Synthesis and characterization of fluorinated conjugates of albumin. J. Fluor. Chem. 2013, 152, 173–181. [Google Scholar] [CrossRef]
- Yeo, S.K.; Shepelytskyi, Y.; Grynko, V.; Albert, M.S. Molecular Imaging of Fluorinated Probes for Tau Protein and Amyloid-β Detection. Molecules 2020, 25, 3413. [Google Scholar] [CrossRef]
- Yanagisawa, D.; Ibrahim, N.F.; Taguchi, H.; Morikawa, S.; Tomiyama, T.; Tooyama, I. Fluorine-19 Magnetic Resonance Imaging for Detection of Amyloid β Oligomers Using a Keto Form of Curcumin Derivative in a Mouse Model of Alzheimer’s Disease. Molecules 2021, 26, 1362. [Google Scholar] [CrossRef] [PubMed]
- Tooyama, I.; Yanagisawa, D.; Taguchi, H.; Kato, T.; Hirao, K.; Shirai, N.; Sogabe, T.; Ibrahim, N.F.; Inubushi, T.; Morikawa, S. Amyloid imaging using fluorine-19 magnetic resonance imaging (19F-MRI). Ageing Res. Rev. 2016, 30, 85–94. [Google Scholar] [CrossRef]
- Modo, M. 19F Magnetic Resonance Imaging and Spectroscopy in Neuroscience. Neuroscience 2021, 474, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Bouvain, P.; Temme, S.; Flögel, U. Hot spot 19F magnetic resonance imaging of inflammation. WIREs Nanomed. Nanobiotechnol. 2020, 12, e1639. [Google Scholar] [CrossRef]
- De Simone, G.; di Masi, A.; Ascenzi, P. Serum Albumin: A Multifaced Enzyme. Int. J. Mol. Sci. 2021, 22, 10086. [Google Scholar] [CrossRef]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef] [PubMed]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Unraveling the mysteries of serum albumin—More than just a serum protein. Front. Physiol. 2014, 5, 299. [Google Scholar] [CrossRef]
- Belinskaia, D.A.; Voronina, P.A.; Shmurak, V.I.; Jenkins, R.O.; Goncharov, N.V. Serum Albumin in Health and Disease: Esterase, Antioxidant, Transporting and Signaling Properties. Int. J. Mol. Sci. 2021, 22, 10318. [Google Scholar] [CrossRef]
- Nishi, K.; Yamasaki, K.; Otagiri, M. Serum Albumin, Lipid and Drug Binding. Vertebr. Invertebr. Respir. Proteins Lipoproteins Other Body Fluid Proteins 2020, 94, 383–397. [Google Scholar]
- van der Vusse, G.J. Albumin as Fatty Acid Transporter. Drug Metab. Pharmacokinet. 2009, 24, 300–307. [Google Scholar] [CrossRef]
- Otagiri, M.; Giam Chuang, V.T. Albumin in Medicine: Pathological and Clinical Applications; Springer: Singapore, 2016; pp. 1–277. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Kragh-Hansen, U. Human Serum Albumin: A Multifunctional Protein. In Albumin in Medicine: Pathological and Clinical Applications; Springer: Singapore, 2016; pp. 1–24. [Google Scholar]
- Sockolosky, J.T.; Szoka, F.C. The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy. Adv. Drug Deliv. Rev. 2015, 91, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Sleep, D.; Cameron, J.; Evans, L.R. Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta BBA Gen. Subj. 2013, 1830, 5526–5534. [Google Scholar] [CrossRef]
- Larsen, M.T.; Kuhlmann, M.; Hvam, M.L.; Howard, K.A. Albumin-based drug delivery: Harnessing nature to cure disease. Mol. Cell. Ther. 2016, 4, 3. [Google Scholar] [CrossRef]
- Chubarov, A.S.; Shakirov, M.M.; Koptyug, I.V.; Sagdeev, R.Z.; Knorre, D.G.; Godovikova, T.S. Synthesis and characterization of fluorinated homocysteine derivatives as potential molecular probes for 19F magnetic resonance spectroscopy and imaging. Bioorg. Med. Chem. Lett. 2011, 21, 4050–4053. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, X. Simple bioconjugate chemistry serves great clinical advances: Albumin as a versatile platform for diagnosis and precision therapy. Chem. Soc. Rev. 2016, 45, 1432–1456. [Google Scholar] [CrossRef] [PubMed]
- Bern, M.; Sand, K.M.K.; Nilsen, J.; Sandlie, I.; Andersen, J.T. The role of albumin receptors in regulation of albumin homeostasis: Implications for drug delivery. J. Control. Release 2015, 211, 144–162. [Google Scholar] [CrossRef]
- Sikder, S.; Gote, V.; Alshamrani, M.; Sicotte, J.; Pal, D. Long-term delivery of protein and peptide therapeutics for cancer therapies. Expert Opin. Drug Deliv. 2019, 16, 1113–1131. [Google Scholar] [CrossRef]
- Popova, T.V.; Khan, H.; Chubarov, A.S.; Lisitskiy, V.A.; Antonova, N.M.; Akulov, A.E.; Shevelev, O.B.; Zavjalov, E.L.; Silnikov, V.N.; Ahmad, S.; et al. Biotin-decorated anti-cancer nucleotide theranostic conjugate of human serum albumin: Where the seed meets the soil? Bioorg. Med. Chem. Lett. 2018, 28, 260–264. [Google Scholar] [CrossRef]
- Lisitskiy, V.A.; Khan, H.; Popova, T.V.; Chubarov, A.S.; Zakharova, O.D.; Akulov, A.E.; Shevelev, O.B.; Zavjalov, E.L.; Koptyug, I.V.; Moshkin, M.P.; et al. Multifunctional human serum albumin-therapeutic nucleotide conjugate with redox and pH-sensitive drug release mechanism for cancer theranostics. Bioorg. Med. Chem. Lett. 2017, 27, 3925–3930. [Google Scholar] [CrossRef]
- Chubarov, A.S.; Zakharova, O.D.; Koval, O.A.; Romaschenko, A.V.; Akulov, A.E.; Zavjalov, E.L.; Razumov, I.A.; Koptyug, I.V.; Knorre, D.G.; Godovikova, T.S. Design of protein homocystamides with enhanced tumor uptake properties for 19F magnetic resonance imaging. Bioorg. Med. Chem. 2015, 23, 6943–6954. [Google Scholar] [CrossRef]
- Chubarov, A.S. Homocysteine Thiolactone: Biology and Chemistry. Encyclopedia 2021, 1, 445–459. [Google Scholar] [CrossRef]
- Präbst, K.; Engelhardt, H.; Ringgeler, S.; Hübner, H. Basic Colorimetric Proliferation Assays: MTT, WST, and Resazurin. In Cell Viability Assays; Gilbert, D.F., Friedrich, O., Eds.; Springer: New York, NY, USA, 2017; Volume 1601, pp. 1–17. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Espeel, P.; Du Prez, F.E. One-pot multi-step reactions based on thiolactone chemistry: A powerful synthetic tool in polymer science. Eur. Polym. J. 2015, 62, 247–272. [Google Scholar] [CrossRef]
- Espeel, P.; Prez, F.E. One-Pot Double Modification of Polymers Based on Thiolactone Chemistry. Adv. Polym. Sci. 2015, 269, 105–132. [Google Scholar] [CrossRef]
- Frank, D.; Espeel, P.; Claessens, S.; Mes, E.; Du Prez, F.E. Synthesis of thiolactone building blocks as potential precursors for sustainable functional materials. Tetrahedron 2016, 72, 6616–6625. [Google Scholar] [CrossRef]
- Cui, H.-L. Recent Advances in DMSO-Based Direct Synthesis of Heterocycles. Molecules 2022, 27, 8480. [Google Scholar] [CrossRef]
- Bisconti, M.; Grosjean, P.; Arcolia, V.; Simon, J.-F.; Hennebert, E. Influence of Two Widely Used Solvents, Ethanol and Dimethyl Sulfoxide, on Human Sperm Parameters. Int. J. Mol. Sci. 2022, 24, 505. [Google Scholar] [CrossRef]
- Kuroda, K.; Komori, T.; Ishibashi, K.; Uto, T.; Kobayashi, I.; Kadokawa, R.; Kato, Y.; Ninomiya, K.; Takahashi, K.; Hirata, E. Non-aqueous, zwitterionic solvent as an alternative for dimethyl sulfoxide in the life sciences. Commun. Chem. 2020, 3, 163. [Google Scholar] [CrossRef]
- Vigneaud, V.; Patterson, W.I.; Hunt, M. Opeining of the Ring of the Thiolactone of Homocysteine. J. Biol. Chem 1938, 126, 217–231. [Google Scholar] [CrossRef]
- Martins, M.B.; Carvalho, I. Diketopiperazines: Biological activity and synthesis. Tetrahedron 2007, 63, 9923–9932. [Google Scholar] [CrossRef]
- Jakubowski, H. Mechanism of the Condensation of Homocysteine Thiolactone with Aldehydes. Chem. A Eur. J. 2006, 12, 8039–8043. [Google Scholar] [CrossRef]
- Garel, J.; Tawfik, D.S. Mechanism of Hydrolysis and Aminolysis of Homocysteine Thiolactone. Chem. A Eur. J. 2006, 12, 4144–4152. [Google Scholar] [CrossRef] [PubMed]
- Kwan, E.E.; Zeng, Y.; Besser, H.A.; Jacobsen, E.N. Concerted nucleophilic aromatic substitutions. Nat. Chem. 2018, 10, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.D.; Martin, P.A.; Sandford, G.; Williams, D.L.H. Mechanisms of reactions of halogenated compounds: Part 7. Effects of fluorine and other groups as substituents on nucleophilic aromatic substitution. J. Fluor. Chem. 2008, 129, 998–1002. [Google Scholar] [CrossRef]
- Muir, M.; Baker, J. A simple calculational model for predicting the site for nucleophilic substitution in aromatic perfluorocarbons. J. Fluor. Chem. 2005, 126, 727–738. [Google Scholar] [CrossRef]
- Kovtonyuk, V.N.; Gatilov, Y.V.; Nikul’Shin, P.V.; Bredikhin, R.A. Synthesis of Polyfluorinated Thia- and Oxathiacalixarenes Based on Perfluoro-m-xylene. Molecules 2021, 26, 526. [Google Scholar] [CrossRef] [PubMed]
- Mehta, V.D.; Kulkarni, P.V.; Mason, R.P.; Constantinescu, A.; Antich, P.P. Fluorinated Proteins as Potential 19F Magnetic Resonance Imaging and Spectroscopy Agents. Bioconjug. Chem. 1994, 5, 257–261. [Google Scholar] [CrossRef]
- Chubarov, A.; Spitsyna, A.; Krumkacheva, O.; Mitin, D.; Suvorov, D.; Tormyshev, V.; Fedin, M.; Bowman, M.K.; Bagryanskaya, E. Reversible Dimerization of Human Serum Albumin. Molecules 2021, 26, 108. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine Modification in Protein Structure/Function and Human Disease. Physiol. Rev. 2019, 99, 555–604. [Google Scholar] [CrossRef]
- Jakubowski, H. Molecular basis of homocysteine toxicity in humans. Cell. Mol. Life Sci. 2004, 61, 470–487. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Du, Y.; Ye, J.; Kou, D.; Qiu, J.; Wang, J.; Tian, J.; Chen, X. Intraoperative Imaging-Guided Cancer Surgery: From Current Fluorescence Molecular Imaging Methods to Future Multi-Modality Imaging Technology. Theranostics 2014, 4, 1072–1084. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Xu, A.; Yu, Y.; Fu, C.; Liang, G. Biomedical Applications of Fluorescent and Magnetic Resonance Imaging Dual-Modality Probes. Chembiochem 2019, 20, 499–510. [Google Scholar] [CrossRef]
- Głowacki, R.; Jakubowski, H. Cross-talk between Cys34 and Lysine Residues in Human Serum Albumin Revealed by N-Homocysteinylation. J. Biol. Chem. 2004, 279, 10864–10871. [Google Scholar] [CrossRef]
- McIntyre, J.O.; Matrisian, L.M. Molecular imaging of proteolytic activity in cancer. J. Cell. Biochem. 2003, 90, 1087–1097. [Google Scholar] [CrossRef]
- Mellet, P.; Massot, P.; Madelin, G.; Marque, S.R.A.; Harté, E.; Franconi, J.-M.; Thiaudière, E. New Concepts in Molecular Imaging: Non-Invasive MRI Spotting of Proteolysis Using an Overhauser Effect Switch. PLoS ONE 2009, 4, e5244. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hong, H.; Zhang, Y.; Cai, W. Molecular Imaging of Proteases in Cancer. Cancer Growth Metastasis 2009, 2, CGM.S2814. [Google Scholar] [CrossRef]
- Mahmood, U.; Weissleder, R. Near-infrared optical imaging of proteases in cancer. Mol. Cancer Ther. 2003, 2, 489–496. [Google Scholar]
- Koonjoo, N.; Parzy, E.; Massot, P.; Lepetit-Coiffé, M.; Marque, S.R.A.; Franconi, J.-M.; Thiaudiere, E.; Mellet, P. In vivo Overhauser-enhanced MRI of proteolytic activity. Contrast Media Mol. Imaging 2014, 9, 363–371. [Google Scholar] [CrossRef]
- Janatova, J.; Fuller, J.K.; Hunter, M.J. The Heterogeneity of Bovine Albumin with Respect to Sulfhydryl and Dimer Content. J. Biol. Chem. 1968, 243, 3612–3622. [Google Scholar] [CrossRef]
- Perczel, A.; Hollósi, M.; Tusn’dy, G.; Fasman, G.D. Convex constraint analysis: A natural deconvolution of circular dichroism curves of proteins. Protein Eng. Des. Sel. 1991, 4, 669–679. [Google Scholar] [CrossRef]
- Strohalm, M.; Kavan, D.; Novák, P.; Volný, M.; Havlíček, V. mMass 3: A Cross-Platform Software Environment for Precise Analysis of Mass Spectrometric Data. Anal. Chem. 2010, 82, 4648–4651. [Google Scholar] [CrossRef] [PubMed]
- Niedermeyer, T.H.J.; Strohalm, M. mMass as a Software Tool for the Annotation of Cyclic Peptide Tandem Mass Spectra. PLoS ONE 2012, 7, e44913. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.; Attalla, M.I. N-Nitrosopiperazines Form at High PH in Post-Combustion Capture Solutions Containing Piperazine: A Low-Energy Collisional Behaviour Study. Rapid Commun. Mass Spectrom. 2010, 24, 3567–3577. [Google Scholar] [CrossRef]
- Tormyshev, V.M.; Chubarov, A.S.; Krumkacheva, O.A.; Trukhin, D.V.; Rogozhnikova, O.Y.; Spitsyna, A.S.; Kuzhelev, A.A.; Koval, V.; Fedin, M.V.; Godovikova, T.S.; et al. Methanethiosulfonate Derivative of OX063 Trityl: A Promising and Efficient Reagent for Side-Directed Spin Labeling of Proteins. Chem. A Eur. J. 2020, 26, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Ishima, Y.; Maruyama, T.; Otagiri, M.; Chuang, V.T.G.; Ishida, T. The New Delivery Strategy of Albumin Carrier Utilizing the Interaction with Albumin Receptors. Chem. Pharm. Bull. 2022, 70, 330–333. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Synthesis of the fluorinated N-substituted HTL derivatives.

Figure 2.
HTL reactions in alkali solution.

Figure 3.
HTL-free base reaction with PFT and PFX in various ratios. The PFT:HTL ratios are 1:1 (line 1), 1:1.4 (line 2), and 1:5 (line 3). Line 4 PFX:HTL ratio is 1:1.4.
Figure 3.
HTL-free base reaction with PFT and PFX in various ratios. The PFT:HTL ratios are 1:1 (line 1), 1:1.4 (line 2), and 1:5 (line 3). Line 4 PFX:HTL ratio is 1:1.4.

Figure 4.
The possible mechanisms of reaction between PFT or PFX and HTL with (1) and without (2) basic catalysis by HTL excess. R = F or CF3.
Figure 4.
The possible mechanisms of reaction between PFT or PFX and HTL with (1) and without (2) basic catalysis by HTL excess. R = F or CF3.

Figure 5.
Typical 19F NMR spectra for PFX and HTL reaction in DMSO at 0 (bottom), 40 (center), and 163 (top) min. The reagent ratio PFX:HTL:NEt3 equals to 1:1.25:1.65.
Figure 5.
Typical 19F NMR spectra for PFX and HTL reaction in DMSO at 0 (bottom), 40 (center), and 163 (top) min. The reagent ratio PFX:HTL:NEt3 equals to 1:1.25:1.65.

Figure 6.
Typical 19F NMR spectra of the PFX and HTL reaction in DMSO in the presence of triethylamine in the CF3 groups’ shift diapason. The PFX:HTL:NEt3 ratio is 1:2.5:3.3.
Figure 6.
Typical 19F NMR spectra of the PFX and HTL reaction in DMSO in the presence of triethylamine in the CF3 groups’ shift diapason. The PFX:HTL:NEt3 ratio is 1:2.5:3.3.

Figure 7.
Synthetic route to PFT-HSA (n = 3.9 ± 0.1) and PFX-HSA (n = 4.9 ± 0.1) conjugates. Framework (shown schematically as a heart-like structure)—human serum albumin (HSA).
Figure 7.
Synthetic route to PFT-HSA (n = 3.9 ± 0.1) and PFX-HSA (n = 4.9 ± 0.1) conjugates. Framework (shown schematically as a heart-like structure)—human serum albumin (HSA).

Figure 8.
19F NMR spectra (300 MHz) of PFT-HSA (top) and PFX-HSA (bottom) conjugates in PBS at 37 °C.
Figure 8.
19F NMR spectra (300 MHz) of PFT-HSA (top) and PFX-HSA (bottom) conjugates in PBS at 37 °C.

Figure 9.
Effects of HSA conjugates on the viability of MCF-7 and RPMI 8226 cell lines. Cell viability was normalized using cells treated with PBS buffer as a 100% viability control. Hcy-HSA—N-homocysteinylated HSA containing 3 Hcy residues.
Figure 9.
Effects of HSA conjugates on the viability of MCF-7 and RPMI 8226 cell lines. Cell viability was normalized using cells treated with PBS buffer as a 100% viability control. Hcy-HSA—N-homocysteinylated HSA containing 3 Hcy residues.

Figure 10.
Synthetic route to PFT-HSA-Cy and PFX-HSA-Cy conjugates. Framework (shown schematically as a heart-like structure)—human serum albumin (HSA).
Figure 10.
Synthetic route to PFT-HSA-Cy and PFX-HSA-Cy conjugates. Framework (shown schematically as a heart-like structure)—human serum albumin (HSA).

Figure 11.
Susceptibility of HSA conjugates to tryptic proteolysis (enzyme/substrate ratio of 1:100). Samples were analyzed by SDS-PAGE. Albumin bands were visualized by Coomassie blue staining. Quantitative data were obtained by digitizing the gel (SDS-PAGE) using GelPro Analyzer software (Media Cybernetics). The bands in the gel lower than 66.5 kDa (HSA monomer MW) were marked as digested albumin (Figure S8).
Figure 11.
Susceptibility of HSA conjugates to tryptic proteolysis (enzyme/substrate ratio of 1:100). Samples were analyzed by SDS-PAGE. Albumin bands were visualized by Coomassie blue staining. Quantitative data were obtained by digitizing the gel (SDS-PAGE) using GelPro Analyzer software (Media Cybernetics). The bands in the gel lower than 66.5 kDa (HSA monomer MW) were marked as digested albumin (Figure S8).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
A brief summary of HTL free base synthesis results.
| Synthesis Step | Conditions | Compounds Yield, % | |||
|---|---|---|---|---|---|
| CHTL, M | Time, min | HTL | 2,5-Diketopiperazine | Peptide | |
| Extraction from aqueous solution (pH 10.5) a | 0.40 a | 0.5 a | 91 | 9 | |
| 0.26 | 0.5 | 98 | 1–2 | ||
| 0.20 | 0.5 | 98 | 1–2 | ||
| 0.40 | 10 | 80 | 20 | ||
| 0.26 | 10 | 90 | 10 | ||
| 0.26 | 30 | 60 | 40 b | ||
| Evaporation in vacuo | CHTL = 0.26 M, | ||||
| self-cooling c | 0.5 min | 96 d | 3 | 1 | |
| water bath 25 °C | 0.5 min | 87 | 11 | 2 | |
| water bath 40 °C | 0.5 min | 73 | 24 | 3 | |
| CHTL = 0.26 M, water bath 25 °C | 10 min | 81 | 15 | 5 | |
| CHTL = 0.40 M, water bath 25 °C | 0.5 min | 77 | 15 | 8 | |
| CHTL = 0.40 M, water bath 25 °C | 10 min | 65 | 20 | 15 | |
| Compound storage in solid state | without storage | 96 d | 3 | 1 | |
| −20 °C | 1 day | 94 | 5 | 1 | |
| −20 °C | 1 week | 89 | 9 | 2 | |
| 4 °C | 1 day | 88 | 11 | 1 | |
| 25 °C | 1 day | 75 | 13 | 2 | |
a HTL extraction from 0.25 M sodium carbonate buffer (pH 10.5) using cooled ethyl acetate (4 °C). CHTL and time are presented for aqueous buffer incubation. The HTL compound quantity was estimated by calculation of adsorption by UV spectroscopy of the water solution aliquots on the wavelength 240 nm (HTL, ε240 = (5.0 ± 0.1) × 103 M−1cm−1); b White precipitate of homocysteine 2,5-diketopiperazine -S-S- polymer formation. The compound is not soluble in water, or ordinary solvents. The structure was defined using a DTT compound for S-S bond reduction with subsequent analysis by 1H NMR (DMSO-d6, 300 MHz); c Evaporation of ethyl acetate without a water bath. Under such conditions, the organic solvent HTL solution is self-cooled. After stopping the evaporation due to excessive cooling, the flask with the solution is lowered into a water bath (25 °C) until fogging disappears, and the procedure is repeated; d The ratio was calculated by 1H NMR (DMSO-d6, 300 MHz).
Table 2.
The ingredients, reaction conditions, and products of the reaction between HTL and perfluoroarenes in the presence of triethylamine in DMSO.
Table 2.
The ingredients, reaction conditions, and products of the reaction between HTL and perfluoroarenes in the presence of triethylamine in DMSO.
| Conditions | T, °C | Time, h | Compounds Ratio per Perfluoroarene by 19F NMR, % | |||
|---|---|---|---|---|---|---|
 |  |  | Initial PFT or PFX | |||
| PFT 0.01 M, HTL 0.02 M, NEt3 0.04 M | 25 | 20 | 35 | 0 | 0 | 65 |
| 45 | 60 | 5 | 3 | 32 | ||
| 120 | 83 | 11 | 6 | 0 | ||
| PFT 0.02 M, HTL 0.02 M, NEt3 0.04 M | 25 | 20 | 31 | 0 | 0 | 69 |
| 45 | 50 | 2.5 | 0.5 | 47 | ||
| 70 | 61 | 3 | 1 | 35 | ||
| 120 | 83 | 5.5 | 1.5 | 10 | ||
| 150 | 92.5 | 6 | 1.5 | 0 | ||
| PFT 0.01 M, HTL 0.02 M, NEt3 0.08 M | 25 | 20 | 30 | 0 | 0 | 70 |
| 45 | 50 | 3.5 | 1.5 | 45 | ||
| 150 | 87 | 8 | 5 | 0 | ||
| PFT 0.02 M, HTL 0.02 M, NEt3 0.04 M | 50 | 20 | 72 | 2 | 1 | 25 |
| 45 | 88 | 3 | 1.5 | 7.5 | ||
| 70 | 90 | 6.5 | 3.5 | 0 | ||
| PFT 0.009 M, HTL 0.01 M, NEt3 0.013 M | 75 | 20 | 63 | 1 | 0.5 | 35 |
| 45 | 84 | 3 | 1 | 12 | ||
| 70 | 94 | 5 | 1 | 0 | ||
| PFX 0.009 M, HTL 0.01 M, NEt3 0.013 M | 75 | 15 | ~100 | ~0 | 0 | 0 |
Table 3.
Fluorinated HSA characteristics.
| HSA Type | CD Analysis | DLS Analysis | SDS-PAGE Analysis | |||
|---|---|---|---|---|---|---|
| n | α-Helix, % | β-Sheet, % | Hydrodynamic Diameter, nm | Oligomer, % | Monomer, % | |
| HSA | - | 60.0 | 4.5 | 6.1 ± 0.6 1 | 7.0 | 93.0 |
| PFT-HSA | 3.9 ± 0.1 | 56.4 | 6.8 | 6.2 ± 0.8 | 8.7 | 91.3 |
| PFX-HSA | 4.9 ± 0.1 | 58.2 | 5.0 | 6.0 ± 0.9 | 7.5 | 92.5 |
| Hcy-HSA | 2.9 ± 0.1 | 50.0 | 10.0 | n.d. | 83.0 | 17.0 |
1 DLS data of 50 µM HSA conjugates in PBS by Number mode.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mitin, D.E.; Chubarov, A.S. Fluorinated Human Serum Albumin as Potential 19F Magnetic Resonance Imaging Probe. Molecules 2023, 28, 1695. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28041695
AMA Style
Mitin DE, Chubarov AS. Fluorinated Human Serum Albumin as Potential 19F Magnetic Resonance Imaging Probe. Molecules. 2023; 28(4):1695. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28041695
Chicago/Turabian StyleMitin, Dmitry E., and Alexey S. Chubarov. 2023. "Fluorinated Human Serum Albumin as Potential 19F Magnetic Resonance Imaging Probe" Molecules 28, no. 4: 1695. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28041695