
Trimethylamine N-Oxide (TMAO) Inducing Endothelial Injury: UPLC-MS/MS-Based Quantification and the Activation of Cathepsin B-Mediated NLRP3 Inflammasome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
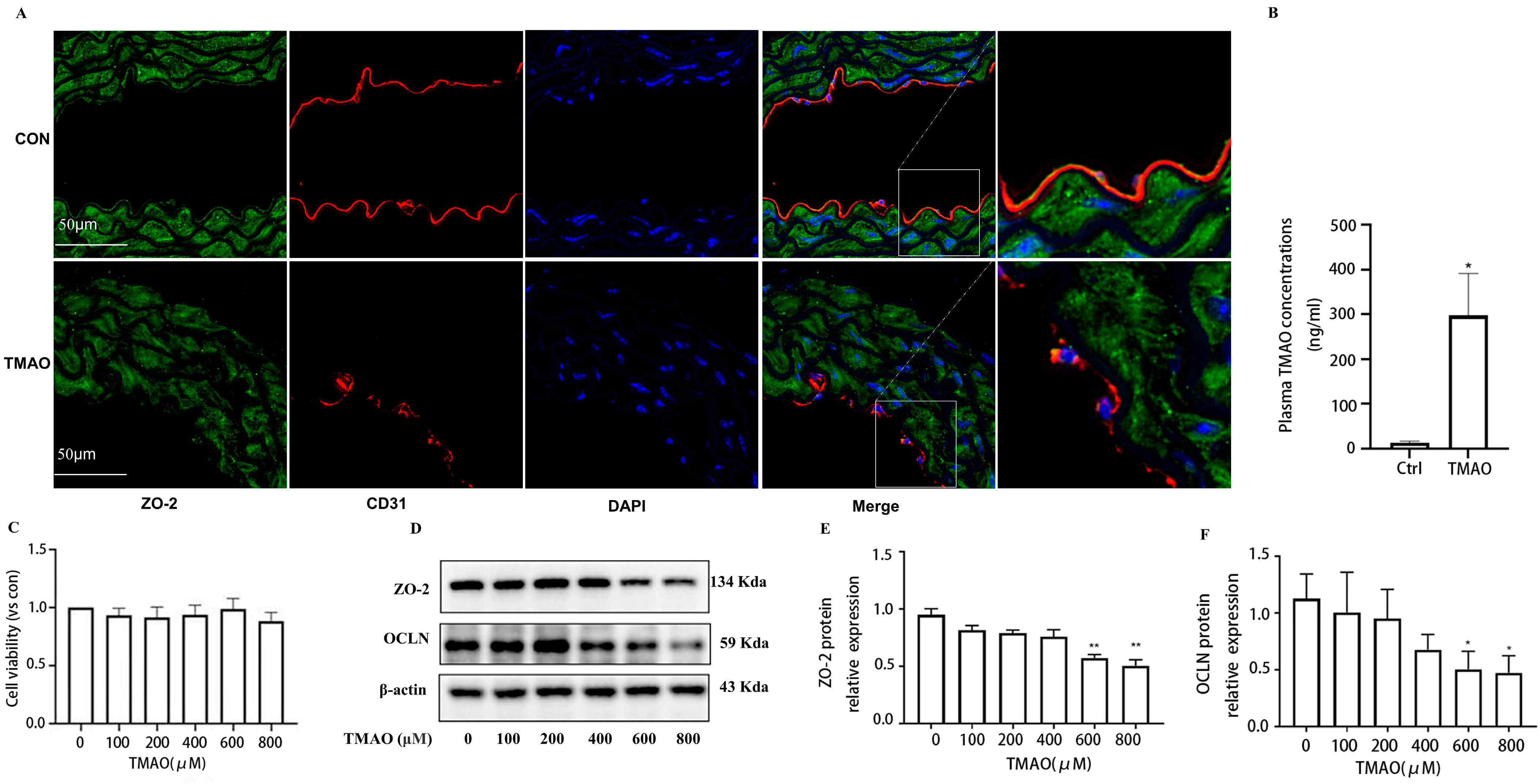
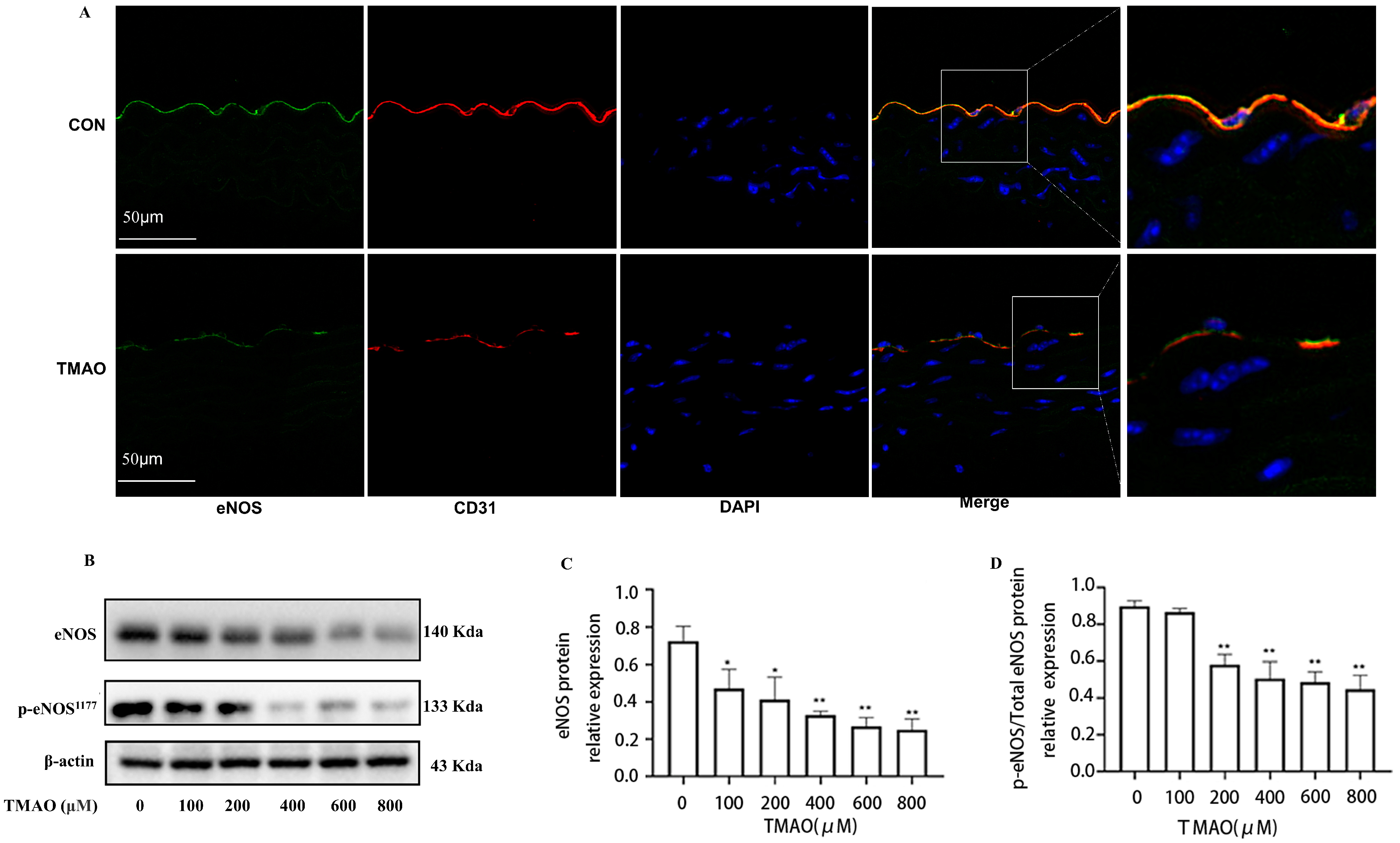
2.1. Quantification of Plasma TMAO Based on UPLC-MS/MS and Its Effects on Endothelial Injury
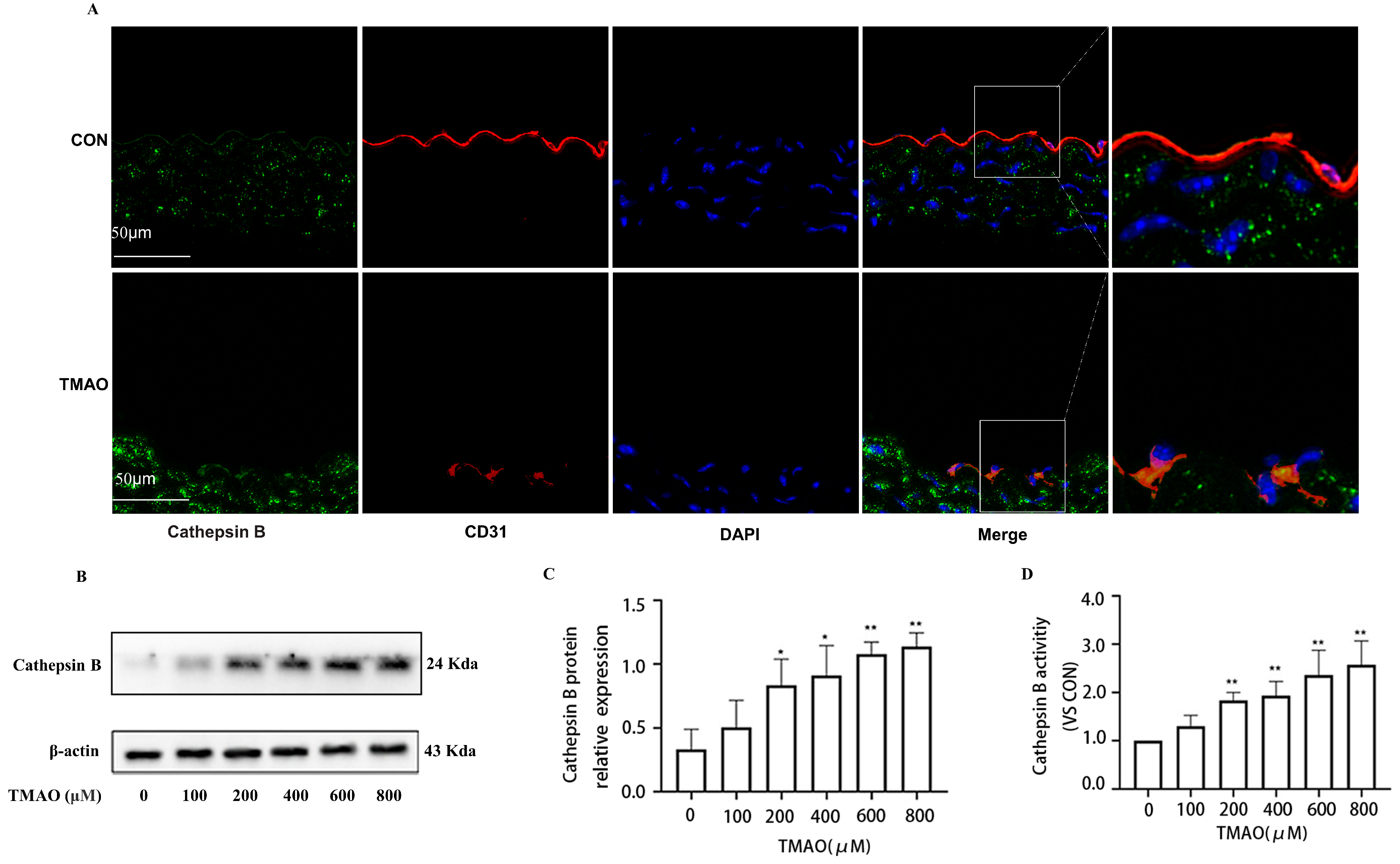
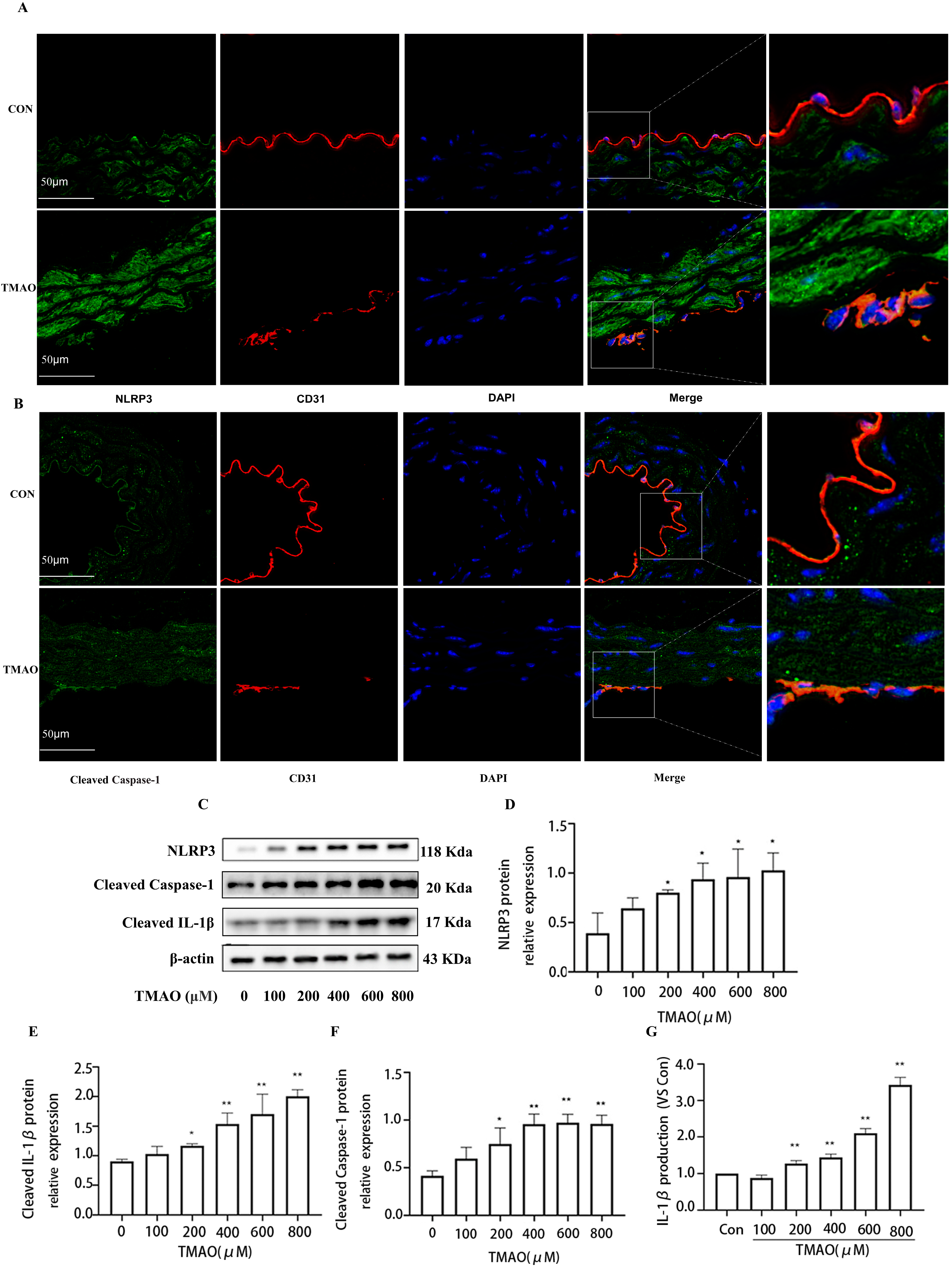
2.2. TMAO Upregulates the Expression of Cathepsin B In Vivo and In Vitro Accompanied by Increased Activity of Cathepsin B
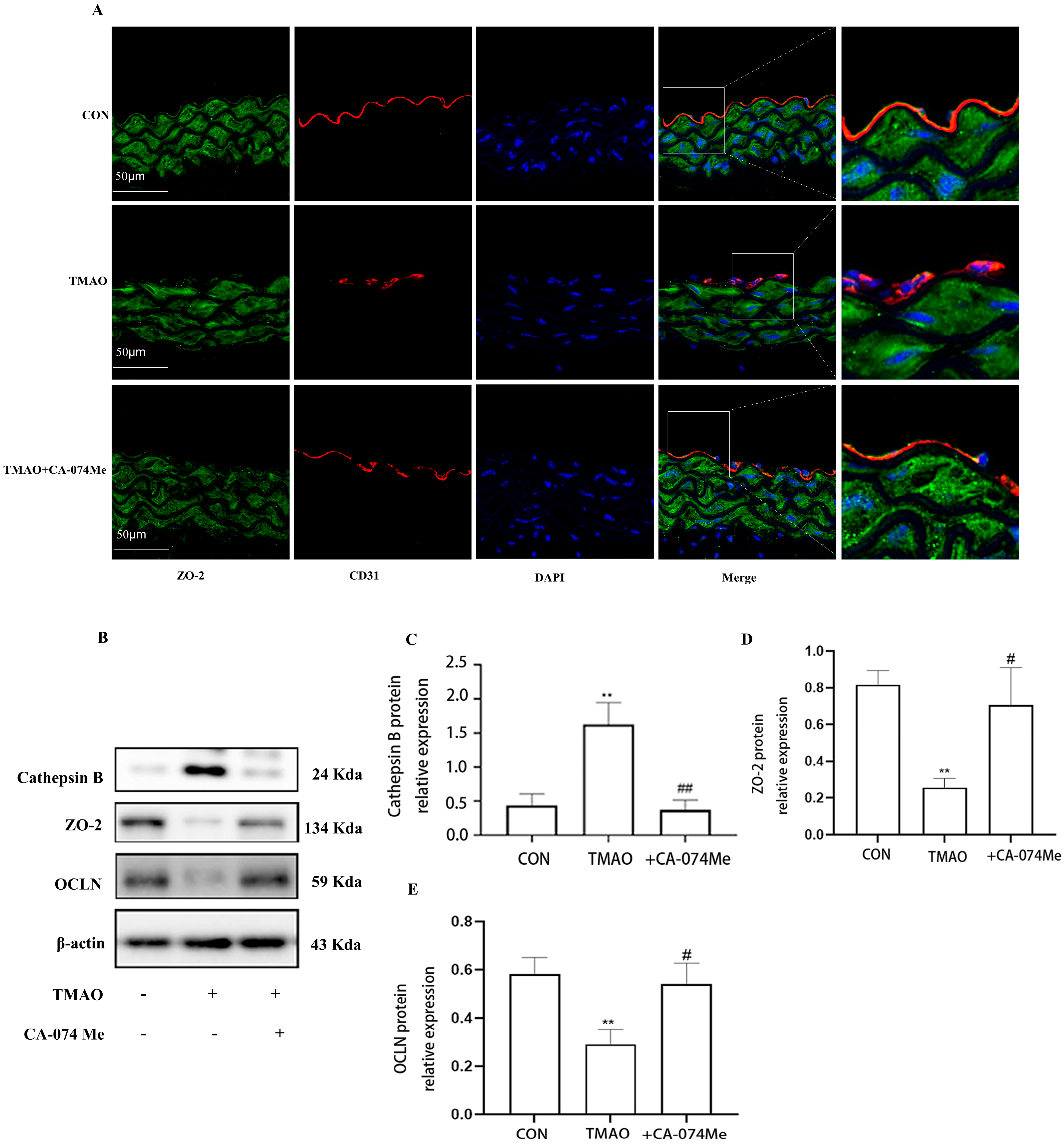
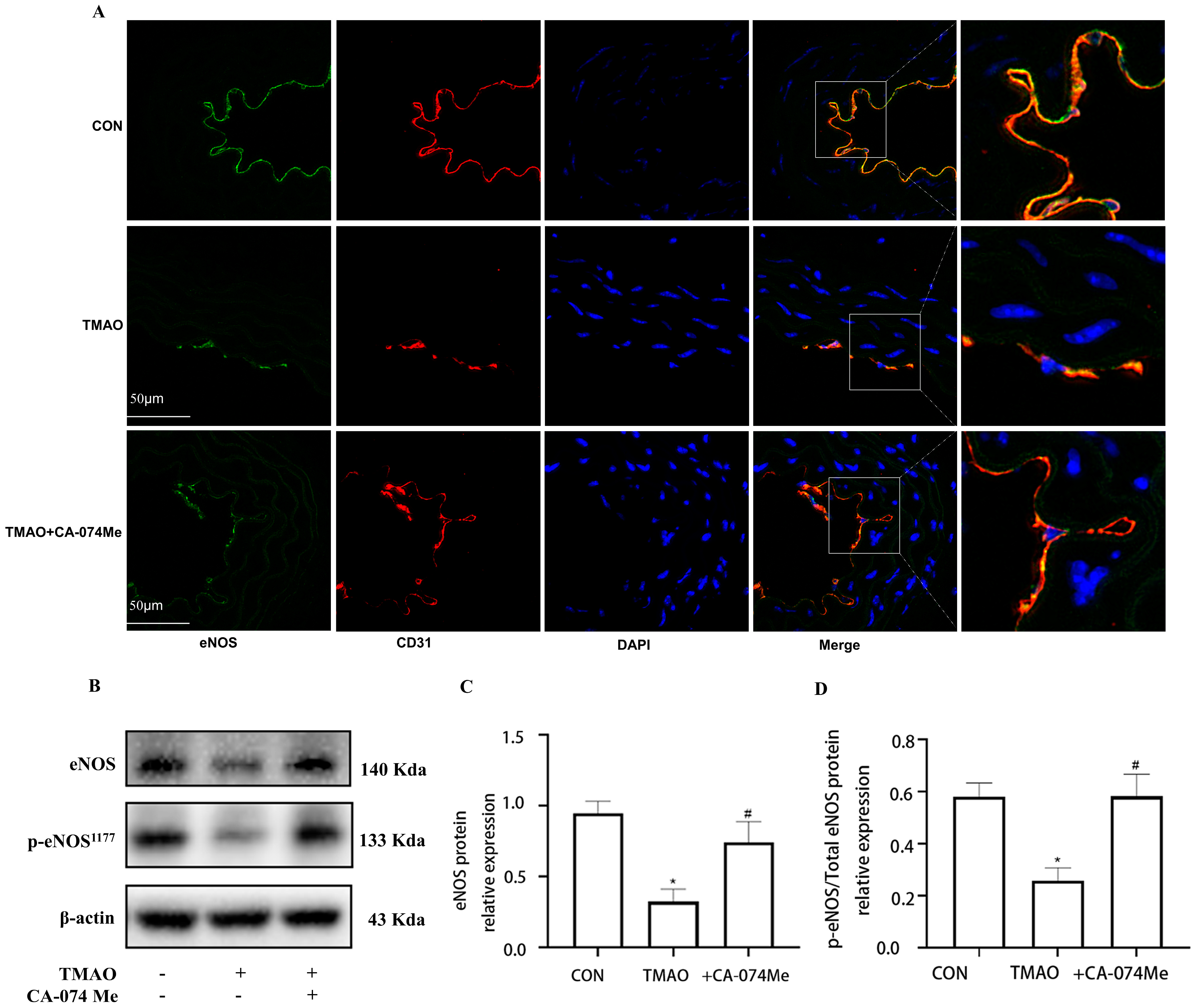
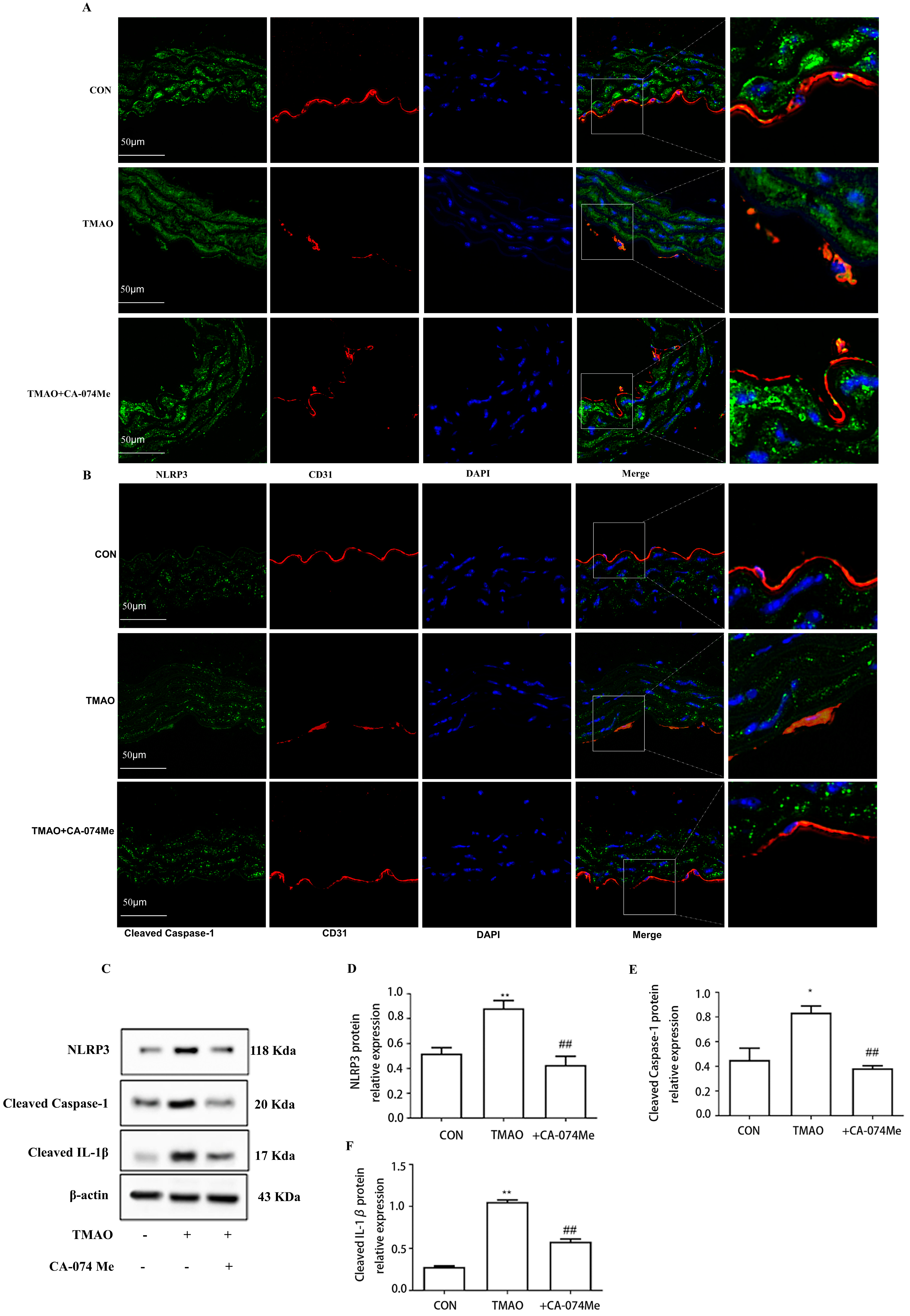
2.3. TMAO Causes Endothelial Injury via Cathepsin B
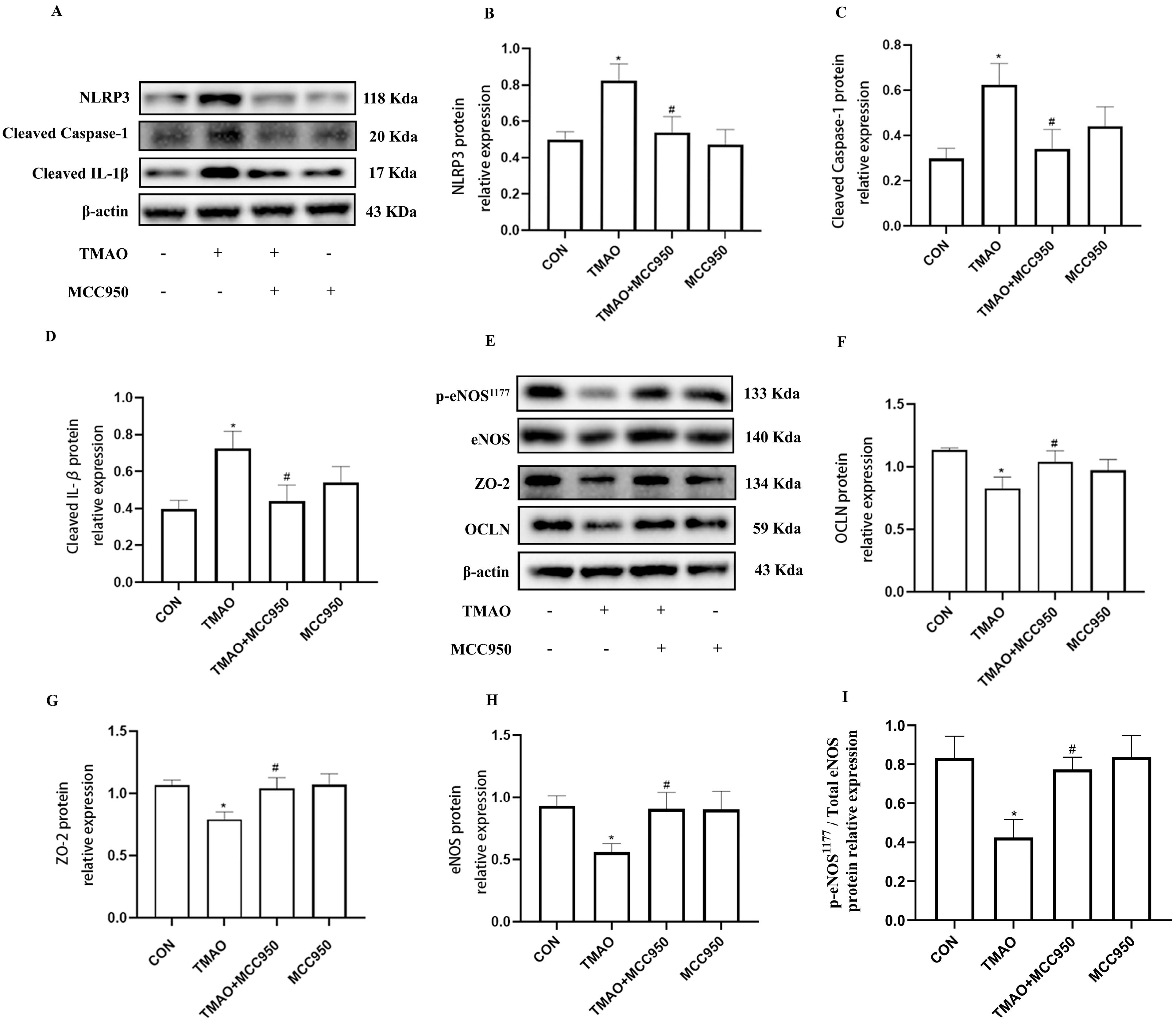
2.4. TMAO Causes Endothelial Injury via Cathepsin B/NLRP3 Inflammasome Axis
3. Materials and Methods
3.1. Reagents and Materials
3.2. Animals and Experimental Protocols
3.3. Ultra Performance Liquid Chromatography-Tandem Mass Spectrometry Quantification of TMAO
3.4. Cell Culture and Treatments
3.5. Cathepsin B Activity Assay
3.6. ELISA Assay
3.7. Immunofluorescence Detection
3.8. Endothelial Cell Viability Test
3.9. Protein Extraction and Western Blotting
3.10. Statistical Analyses
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chen, X.F.; Chen, X.; Tang, X. Short-chain fatty acid, acylation and cardiovascular diseases. Clin. Sci. 2020, 134, 657–676. [Google Scholar] [CrossRef]
- Verhaar, B.J.H.; Prodan, A.; Nieuwdorp, M.; Muller, M. Gut Microbiota in Hypertension and Atherosclerosis: A Review. Nutrients 2020, 12, 2982. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.; Weeks, T.L.; Hazen, S.L. Gut Microbiota and Cardiovascular Disease. Circ. Res. 2020, 127, 553–570. [Google Scholar] [CrossRef]
- Zhou, W.; Cheng, Y.; Zhu, P.; Nasser, M.I.; Zhang, X.; Zhao, M. Implication of Gut Microbiota in Cardiovascular Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 5394096. [Google Scholar] [CrossRef]
- Guasti, L.; Galliazzo, S.; Molaro, M.; Visconti, E.; Pennella, B.; Gaudio, G.V.; Lupi, A.; Grandi, A.M.; Squizzato, A. TMAO as a biomarker of cardiovascular events: A systematic review and meta-analysis. Intern. Emerg. Med. 2021, 16, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, S.; Asar, T.O.; Kumar, V.; Al-Abbasi, F.A.; Alhayyani, S.; Kamal, M.A.; Anwar, F. A cross-talk between gut microbiome, salt and hypertension. Biomed. Pharmacother. 2021, 134, 111156. [Google Scholar] [CrossRef]
- Suzuki, T.; Heaney, L.M.; Bhandari, S.S.; Jones, D.J.; Ng, L.L. Trimethylamine N-oxide and prognosis in acute heart failure. Heart 2016, 102, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Brunt, V.E.; Gioscia-Ryan, R.A.; Casso, A.G.; Van Dongen, N.S.; Ziemba, B.P.; Sapinsley, Z.J.; Richey, J.J.; Zigler, M.C.; Neilson, A.P.; Davy, K.P.; et al. Trimethylamine-N-Oxide Promotes Age-Related Vascular Oxidative Stress and Endothelial Dysfunction in Mice and Healthy Humans. Hypertension 2020, 76, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Lander, H.M.; Grant, A.M.; Albrecht, T.; Hill, T.; Peters, C.J. Endothelial cell permeability and adherens junction disruption induced by junin virus infection. Am. J. Trop. Med. Hyg. 2014, 90, 993–1002. [Google Scholar] [CrossRef]
- Giannotta, M.; Trani, M.; Dejana, E. VE-cadherin and endothelial adherens junctions: Active guardians of vascular integrity. Dev. Cell 2013, 26, 441–454. [Google Scholar] [CrossRef]
- Runkle, E.A.; Mu, D. Tight junction proteins: From barrier to tumorigenesis. Cancer Lett. 2013, 337, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, M.; Fleming, I. The eNOS signalosome and its link to endothelial dysfunction. Pflug. Arch. 2016, 468, 1125–1137. [Google Scholar] [CrossRef]
- Xi, H.; Zhang, Y.; Xu, Y.; Yang, W.Y.; Jiang, X.; Sha, X.; Cheng, X.; Wang, J.; Qin, X.; Yu, J.; et al. Caspase-1 Inflammasome Activation Mediates Homocysteine-Induced Pyrop-Apoptosis in Endothelial Cells. Circ. Res. 2016, 118, 1525–1539. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Fann, D.Y.; Lee, S.Y.; Manzanero, S.; Chunduri, P.; Sobey, C.G.; Arumugam, T.V. Pathogenesis of acute stroke and the role of inflammasomes. Ageing Res. Rev. 2013, 12, 941–966. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Yang, P.; Liu, X.; Lu, L.; Chen, Y.; Zhong, X.; Li, Z.; Liu, H.; Ou, C.; et al. Trimethylamine-N-Oxide Promotes Vascular Calcification Through Activation of NLRP3 (Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3) Inflammasome and NF-kappaB (Nuclear Factor kappaB) Signals. Arter. Thromb. Vasc. Biol. 2020, 40, 751–765. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Zhang, J.; Chen, H.; Zhuge, Y.; Chen, H.; Qian, F.; Zhou, K.; Niu, C.; Wang, F.; Qiu, H.; et al. Endothelial cell pyroptosis plays an important role in Kawasaki disease via HMGB1/RAGE/cathespin B signaling pathway and NLRP3 inflammasome activation. Cell Death Dis. 2019, 10, 778. [Google Scholar] [CrossRef] [PubMed]
- Castro-Ferreira, R.; Cardoso, R.; Leite-Moreira, A.; Mansilha, A. The Role of Endothelial Dysfunction and Inflammation in Chronic Venous Disease. Ann. Vasc Surg. 2018, 46, 380–393. [Google Scholar] [CrossRef]
- Chen, Y.; Li, X.; Boini, K.M.; Pitzer, A.L.; Gulbins, E.; Zhang, Y.; Li, P.L. Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim. Biophys. Acta 2015, 1853, 396–408. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Y.; Li, X.; Zhang, Y.; Gulbins, E.; Zhang, Y. Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget 2016, 7, 73229–73241. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, Y.; Zhang, Y.; Li, P.L.; Li, X. Contribution of cathepsin B-dependent Nlrp3 inflammasome activation to nicotine-induced endothelial barrier dysfunction. Eur. J. Pharmacol. 2019, 865, 172795. [Google Scholar] [CrossRef]
- Rox, K.; Rath, S.; Pieper, D.H.; Vital, M.; Bronstrup, M. A simplified LC-MS/MS method for the quantification of the cardiovascular disease biomarker trimethylamine-N-oxide and its precursors. J. Pharm. Anal. 2021, 11, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef]
- Duttaroy, A.K. Role of Gut Microbiota and Their Metabolites on Atherosclerosis, Hypertension and Human Blood Platelet Function: A Review. Nutrients 2021, 13, 144. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wu, Z.; Yan, J.; Liu, H.; Liu, Q.; Deng, Y.; Ou, C.; Chen, M. Gut microbe-derived metabolite trimethylamine N-oxide induces cardiac hypertrophy and fibrosis. Lab. Investig. 2019, 99, 346–357. [Google Scholar] [CrossRef]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Huang, L.; Tian, Y.; Cao, X.; Song, Y. Trimethylamine-N-Oxide Promotes High-Glucose-Induced Dysfunction and NLRP3 Inflammasome Activation in Retinal Microvascular Endothelial Cells. J. Ophthalmol. 2023, 28, 8224752. [Google Scholar] [CrossRef]
- O’Carroll, S.J.; Kho, D.T.; Wiltshire, R.; Nelson, V.; Rotimi, O.; Johnson, R.; Angel, C.E.; Graham, E.S. Pro-inflammatory TNFalpha and IL-1beta differentially regulate the inflammatory phenotype of brain microvascular endothelial cells. J. Neuroinflamm. 2015, 12, 131. [Google Scholar] [CrossRef]
- Subramaniam, S.; Boukhlouf, S.; Fletcher, C. A bacterial metabolite, trimethylamine N-oxide, disrupts the hemostasis balance in human primary endothelial cells but no coagulopathy in mice. Blood Coagul. Fibrinolysis 2019, 7, 324–330. [Google Scholar] [CrossRef]
- Bai, L.; Dai, J.; Xia, Y.; He, K.; Xue, H.; Guo, Q.; Tian, D.; Xiao, L.; Zhang, X.; Teng, X.; et al. Hydrogen Sulfide Ameliorated High Choline-Induced Cardiac Dysfunction by Inhibiting cGAS-STING-NLRP3 Inflammasome Pathway. Oxid. Med. Cell. Longev. 2022, 22, 1392896. [Google Scholar] [CrossRef]
- Florea, C.M.; Rosu, R.; Cismaru, G.; Moldovan, R.; Vlase, L.; Toma, V.; Decea, N.; Ancuta, B.; Filip, G.A. Chronic oral trimethyla-mine-N-oxide administration induces experimental incipient atherosclerosis in non-genetically modified mice. J. Physiol. Pharmacol. 2022, 5, 633–643. [Google Scholar]
- Takahashi, M. NLRP3 inflammasome as a key driver of vascular disease. Cardiovasc. Res. 2022, 118, 372–385. [Google Scholar] [CrossRef] [PubMed]
- Bai, B.; Yang, Y.; Wang, Q.; Li, M.; Tian, C.; Liu, Y.; Aung, L.H.H.; Li, P.F.; Yu, T.; Chu, X.M. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis. 2020, 11, 776. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lü, H.; Chen, S.; Xiang, H.; Liu, H.; Zhao, S. Trimethylamine oxide induces pyroptosis of vascular endothelial cells through ALDH2/ROS/NLRP3/GSDMD pathway. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2022, 9, 1171–1181. [Google Scholar]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Sutterwala, F.S.; Haasken, S.; Cassel, S.L. Mechanism of NLRP3 inflammasome activation. Ann. N. Y. Acad. Sci. 2014, 1319, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.T.; Lv, L.L.; Pan, M.M.; Wen, Y.; Wang, B.; Li, Z.L.; Wu, M.; Wang, F.M.; Crowley, S.D.; Liu, B.C. Hydroxychloroquine attenuates renal ischemia/reperfusion injury by inhibiting cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis. 2018, 9, 351. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Yang, C.; Wang, B.; Zhang, X.; Hu, T.; Gu, Y.; Li, J. Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. Biomed. Pharmacother. 2018, 97, 941–947. [Google Scholar] [CrossRef]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef]
- Dumas, M.E.; Rothwell, A.R.; Hoyles, L.; Aranias, T.; Chilloux, J.; Calderari, S.; Noll, E.M.; Péan, N.; Boulangé, C.L.; Blancher, C.; et al. Microbial-Host Co-metabolites Are Prodromal Markers Predicting Phenotypic Heterogeneity in Behavior, Obesity, and Impaired Glucose Tolerance. Cell Rep. 2017, 20, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Henderson, A.; Petriello, M.C.; Romano, K.A.; Gearing, M.; Miao, J.; Schell, M.; Sandoval-Espinola, W.J.; Tao, J.; Sha, B.; et al. Trimethylamine N-Oxide Binds and Activates PERK to Promote Metabolic Dysfunction. Cell Metab. 2019, 30, 1141–1151.e5. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, D.; Yu, W.; Liu, Y.; Jiang, Y.; Li, X.; Lv, J.; Li, Y. Trimethylamine N-Oxide (TMAO) Inducing Endothelial Injury: UPLC-MS/MS-Based Quantification and the Activation of Cathepsin B-Mediated NLRP3 Inflammasome. Molecules 2023, 28, 3817. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28093817
Lei D, Yu W, Liu Y, Jiang Y, Li X, Lv J, Li Y. Trimethylamine N-Oxide (TMAO) Inducing Endothelial Injury: UPLC-MS/MS-Based Quantification and the Activation of Cathepsin B-Mediated NLRP3 Inflammasome. Molecules. 2023; 28(9):3817. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28093817
Chicago/Turabian StyleLei, Dongyu, Wenbo Yu, Yi Liu, Yujie Jiang, Xiaohui Li, Jing Lv, and Ying Li. 2023. "Trimethylamine N-Oxide (TMAO) Inducing Endothelial Injury: UPLC-MS/MS-Based Quantification and the Activation of Cathepsin B-Mediated NLRP3 Inflammasome" Molecules 28, no. 9: 3817. https://0-doi-org.brum.beds.ac.uk/10.3390/molecules28093817