Radioligands for Tropomyosin Receptor Kinase (Trk) Positron Emission Tomography Imaging

 , , ,
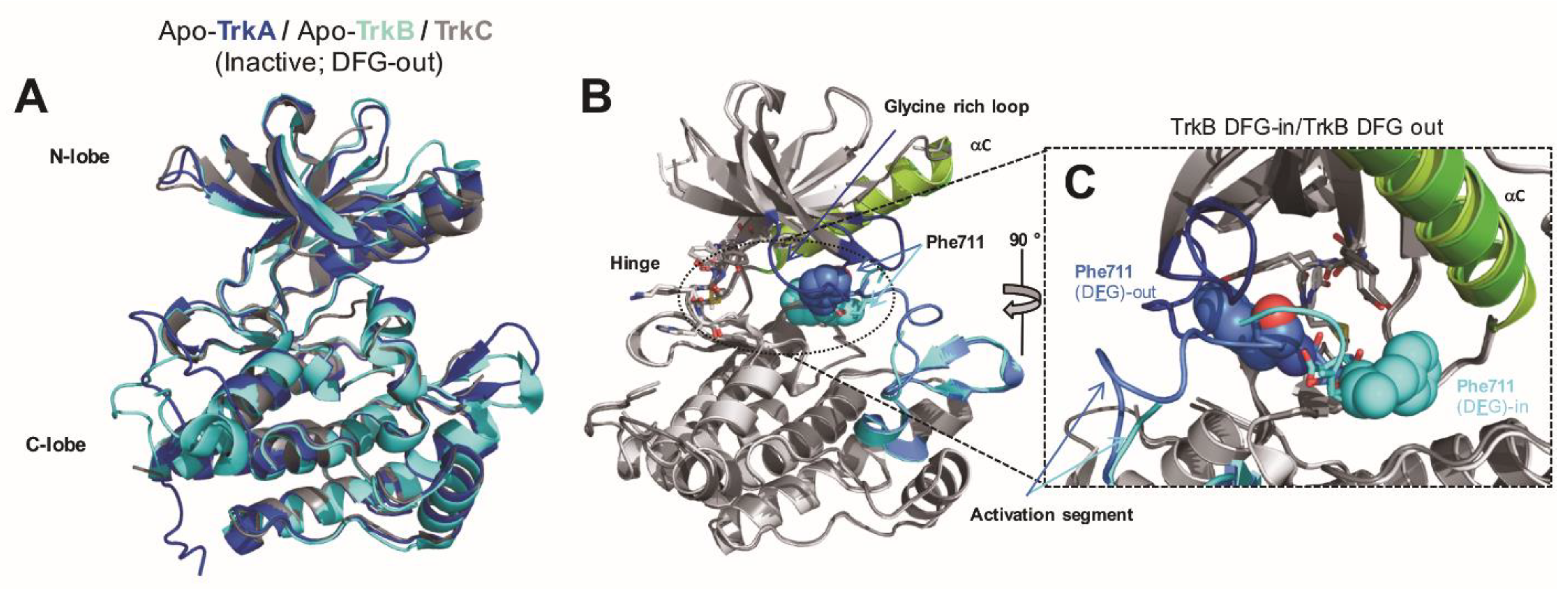
, , , 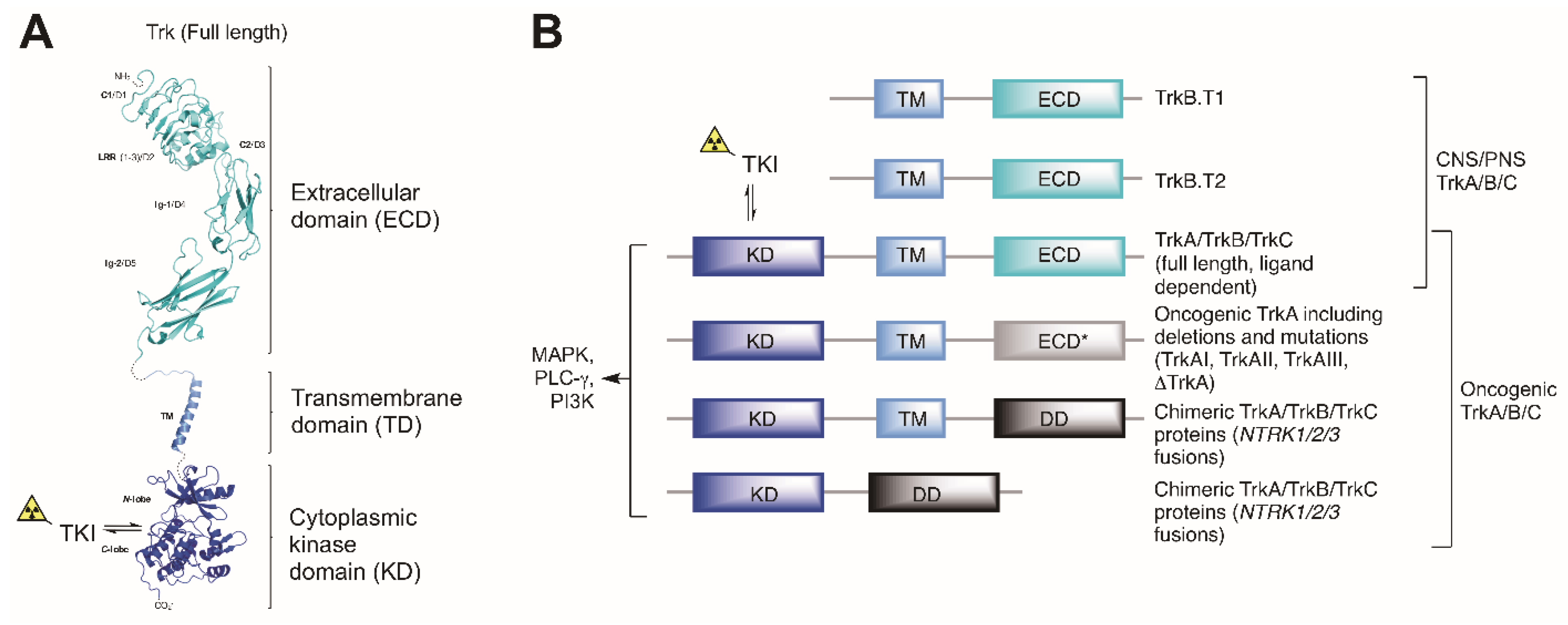{kind=link}
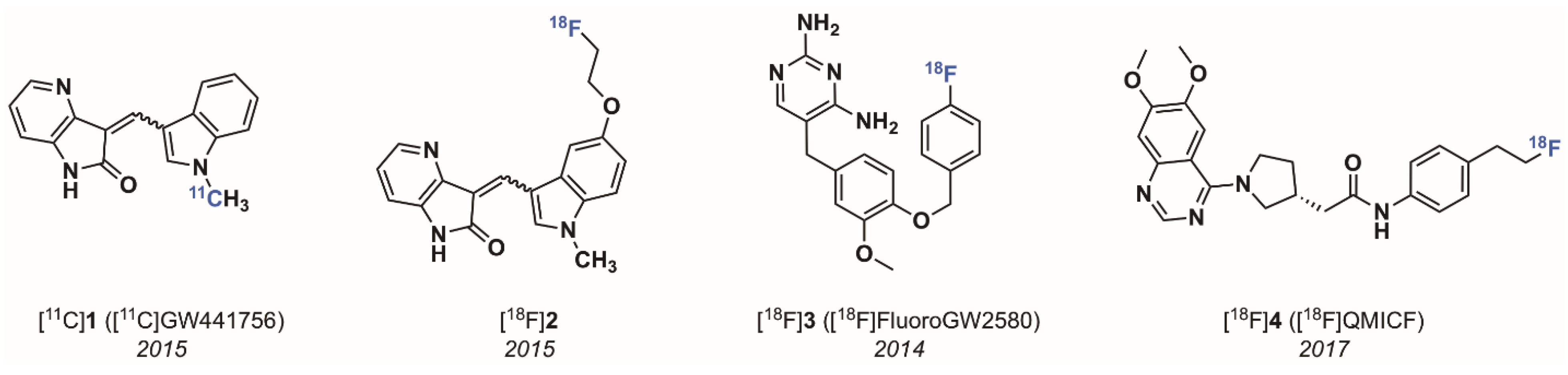
{kind=link}
{kind=link}
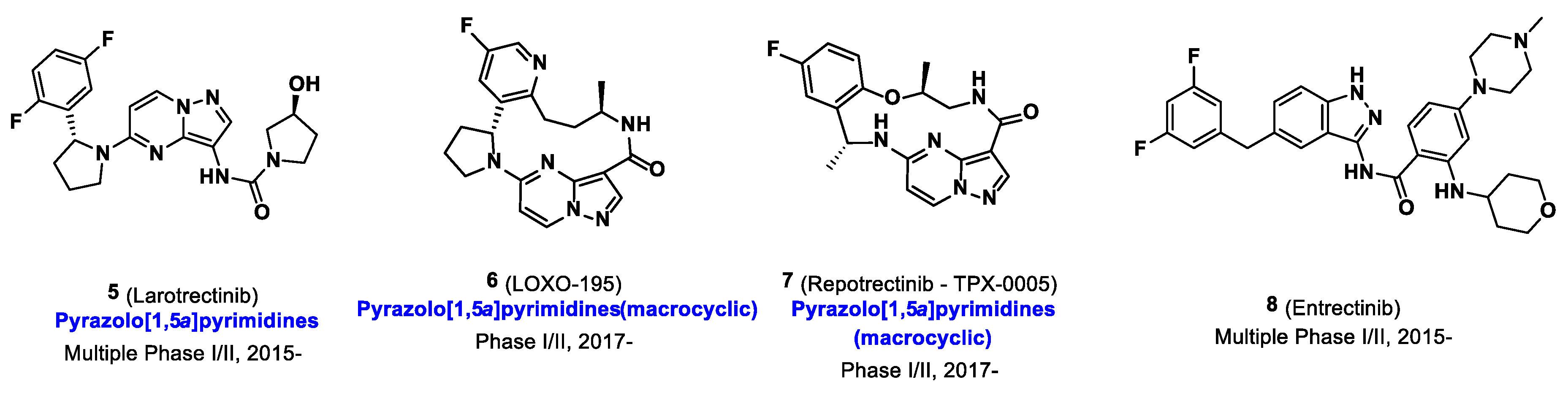{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Development of Trk Radioligands for PET Imaging
2.1. Binding Site Considerations
2.1.1. The 4-Aza-2-oxindole Radioligands
2.1.2. 2,4-Diaminopyridmidine and Quinazoline-Based Radioligands
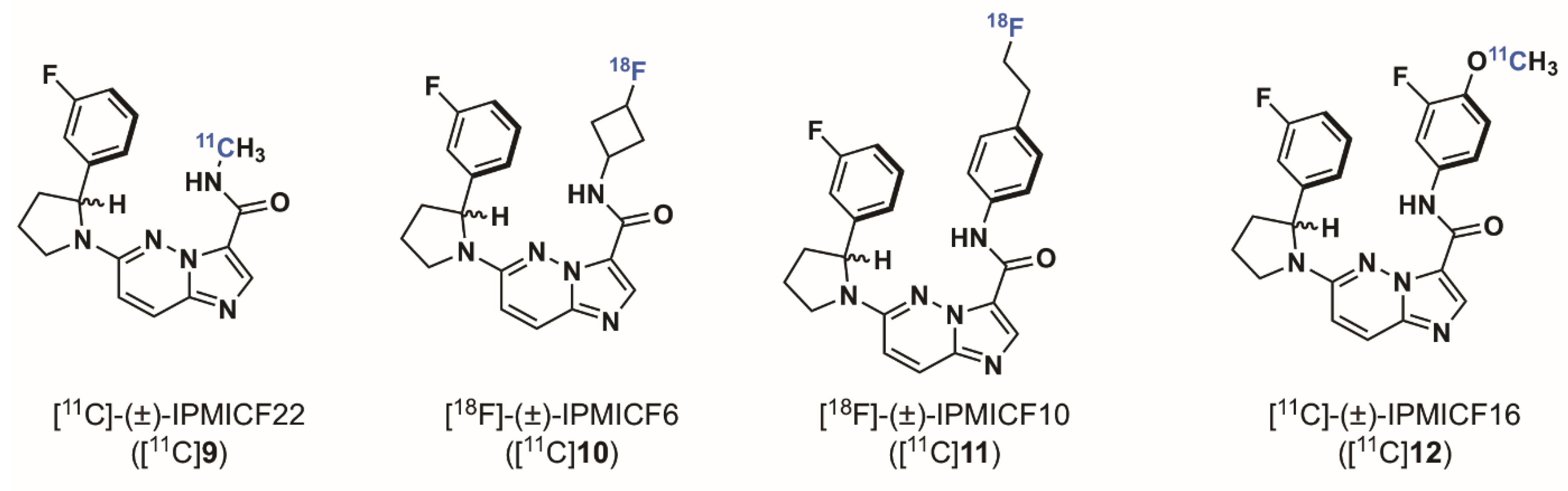
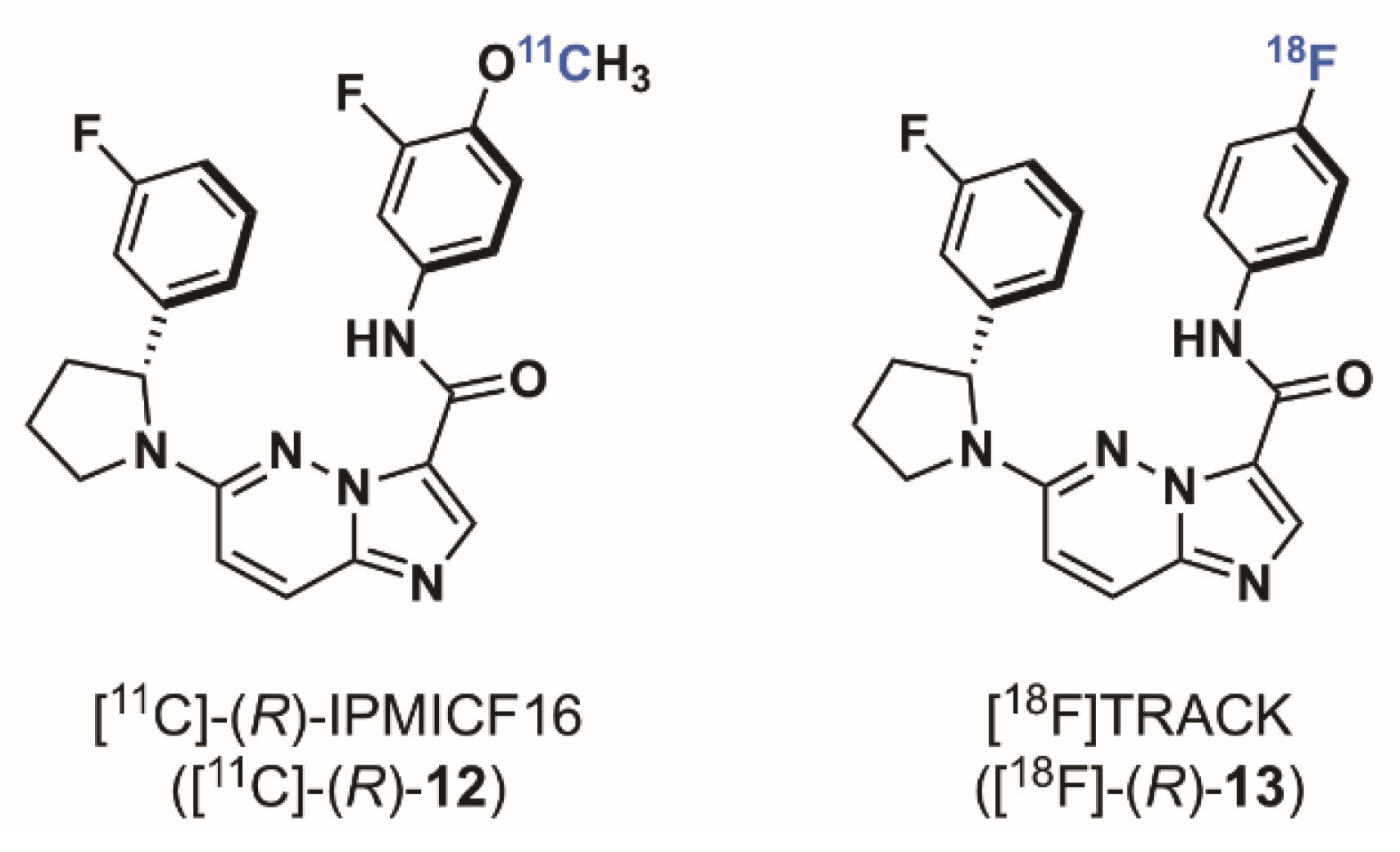
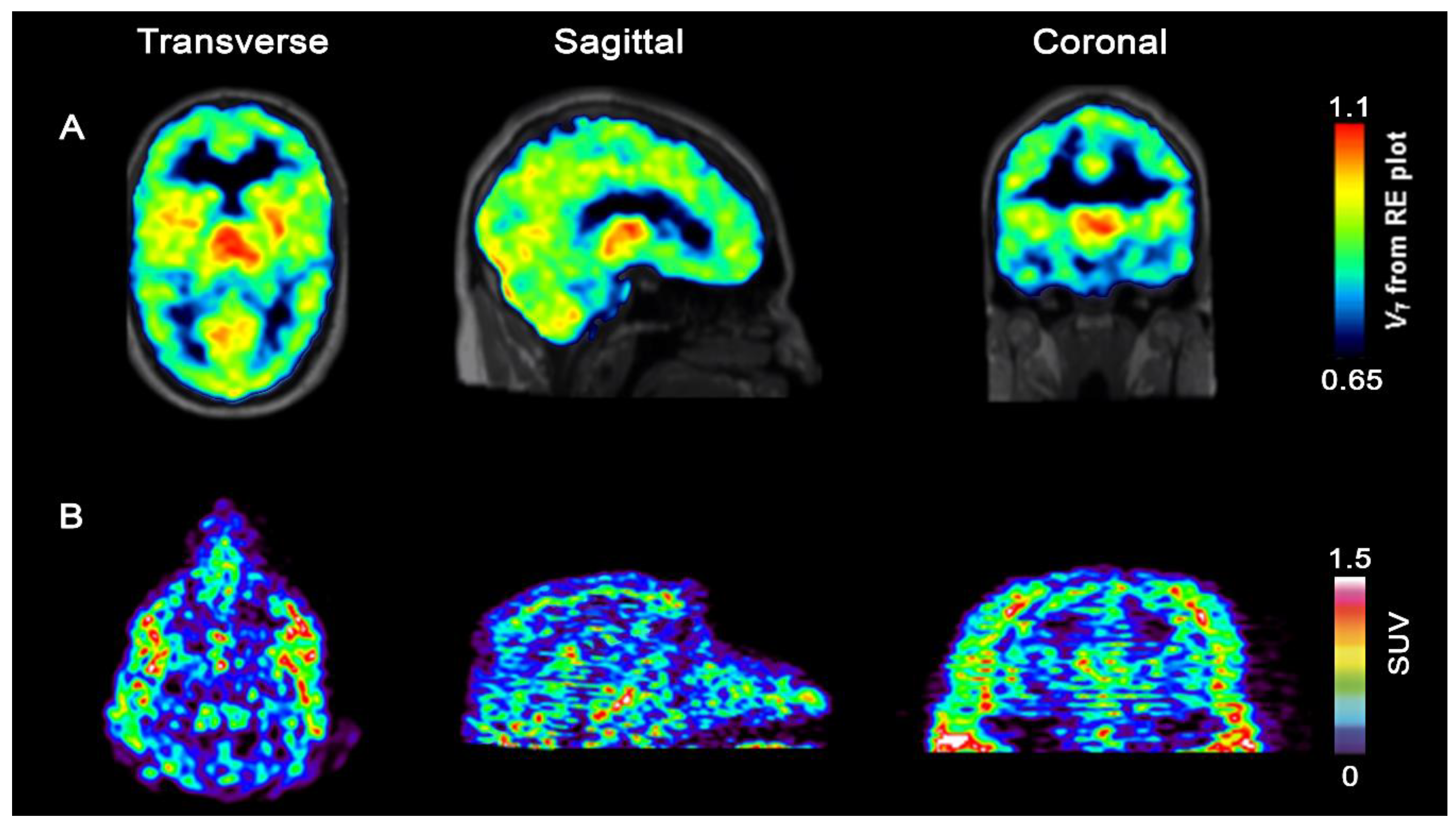
2.1.3. Imidazo[1,2-b]pyridazine-Based Radioligands
3. Conclusions
Funding
Conflicts of Interest
References
- Lessmann, V.; Gottmann, K.; Malcangio, M. Neurotrophin secretion: Current facts and future prospects. Progr. Neurobiol. 2003, 69, 341–374. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophin receptors: A window into neuronal differentiation. Neuron 1992, 9, 583–593. [Google Scholar] [CrossRef]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.J.; Reichardt, L.F. Trk Receptors: Roles in Neuronal Signal Transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef]
- Fenner, B.M. Truncated TrkB: Beyond a dominant negative receptor. Cytokine Growth Factor Rev. 2012, 23, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Lakkaniga, N.R.; Carlomagno, F.; Santoro, M.; McDonald, N.Q.; Lv, F.; Gunaganti, N.; Frett, B.; Li, H.-Y. Insights into Current Tropomyosin Receptor Kinase (TRK) Inhibitors: Development and Clinical Application. J. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef]
- Poo, M.-M. Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2001, 2, 24–32. [Google Scholar] [CrossRef]
- Zhang, F.; Kang, Z.; Li, W.; Xiao, Z.; Zhou, X. Roles of brain-derived neurotrophic factor/tropomyosin-related kinase B (BDNF/TrkB) signalling in Alzheimer’s disease. J. Clin. Neurosci. 2012, 19, 946–949. [Google Scholar] [CrossRef]
- Song, J.-H.; Yu, J.-T.; Tan, L. Brain-Derived Neurotrophic Factor in Alzheimer’s Disease: Risk, Mechanisms, and Therapy. Mol. Neurobiol. 2015, 52, 1477–1493. [Google Scholar] [CrossRef] [PubMed]
- Murer, M.G.; Yan, Q.; Raisman-Vozari, R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Progr. Neurobiol. 2001, 63, 71–124. [Google Scholar] [CrossRef]
- Reinhart, V.; Bove, S.E.; Volfson, D.; Lewis, D.A.; Kleiman, R.J.; Lanz, T.A. Evaluation of TrkB and BDNF transcripts in prefrontal cortex, hippocampus, and striatum from subjects with schizophrenia, bipolar disorder, and major depressive disorder. Neurobiol. Dis. 2015, 77, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Oyesiku, N.M.; Evans, C.-O.; Houston, S.; Darrell, R.S.; Smith, J.S.; Fulop, Z.L.; Dixon, C.E.; Stein, D.G. Regional changes in the expression of neurotrophic factors and their receptors following acute traumatic brain injury in the adult rat brain. Brain Res. 1999, 833, 161–172. [Google Scholar] [CrossRef]
- Deng, V.; Matagne, V.; Banine, F.; Frerking, M.; Ohliger, P.; Budden, S.; Pevsner, J.; Dissen, G.A.; Sherman, L.S.; Ojeda, S.R. FXYD1 is an MeCP2 target gene overexpressed in the brains of Rett syndrome patients and Mecp2-null mice. Hum. Mol. Genet. 2007, 16, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.K.; You, Y.; Gupta, V.B.; Klistorner, A.; Graham, S.L. TrkB Receptor Signalling: Implications in Neurodegenerative, Psychiatric and Proliferative Disorders. Int. J. Mol. Sci. 2013, 14, 10122–10142. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, I.; Marín, C.; Rey, M.J.; Ribalta, T.; Goutan, E.; Blanco, R.; Tolosa, E.; Martí, E. BDNF and Full-length and Truncated TrkB Expression in Alzheimer Disease. Implications in Therapeutic Strategies. J. Neuropathol. Exp. Neurol. 1999, 58, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Allen, S.J.; Wilcock, G.K.; Dawbarn, D. Profound and Selective Loss of Catalytic TrkB Immunoreactivity in Alzheimer′s Disease. Biochem. Biophys. Res. Commun. 1999, 264, 648–651. [Google Scholar] [CrossRef]
- Savaskan, E.; Müller-Spahn, F.; Olivieri, G.; Bruttel, S.; Otten, U.; Rosenberg, C.; Hulette, C.; Hock, C. Alterations in Trk A, Trk B and Trk C Receptor Immunoreactivities in Parietal Cortex and Cerebellum in Alzheimer’s Disease. Eur. Neurol. 2000, 44, 172–180. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Che, S.; Wuu, J.; Counts, S.E.; Mufson, E.J. Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer′s disease. J. Neurochem. 2006, 97, 475–487. [Google Scholar] [CrossRef]
- Castello, N.A.; Nguyen, M.H.; Tran, J.D.; Cheng, D.; Green, K.N.; LaFerla, F.M. 7,8-Dihydroxyflavone, a Small Molecule TrkB Agonist, Improves Spatial Memory and Increases Thin Spine Density in a Mouse Model of Alzheimer Disease-Like Neuronal Loss. PLoS ONE 2014, 9, e91453. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, G.-T. The novel squamosamide derivative FLZ enhances BDNF/TrkB/CREB signaling and inhibits neuronal apoptosis in APP/PS1 mice. Acta Pharmacol. Sin. 2010, 31, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Massa, S.M.; Yang, T.; Xie, Y.; Shi, J.; Bilgen, M.; Joyce, J.N.; Nehama, D.; Rajadas, J.; Longo, F.M. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J. Clin. Investig. 2010, 120, 1774–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahara, A.H.; Tuszynski, M.H. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat. Rev. Drug Discov. 2011, 10, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Ohno, M. TrkB reduction exacerbates Alzheimer’s disease-like signaling aberrations and memory deficits without affecting β-amyloidosis in 5XFAD mice. Transl. Psychiatry 2015, 5, e562. [Google Scholar] [CrossRef] [PubMed]
- Géral, C.; Angelova, A.; Lesieur, S. From Molecular to Nanotechnology Strategies for Delivery of Neurotrophins: Emphasis on Brain-Derived Neurotrophic Factor (BDNF). Pharmaceutics 2013, 5, 127–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xiao, Z.; Chen, G.; Han, Z.; Liu, Y.; Zhang, C.; Sun, Y.; Song, Y.; Wang, K.; Fang, F.; et al. A PET imaging approach for determining EGFR mutation status for improved lung cancer patient management. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Tejeda, G.S.; Díaz-Guerra, M. Integral Characterization of Defective BDNF/TrkB Signalling in Neurological and Psychiatric Disorders Leads the Way to New Therapies. Int. J. Mol. Sci. 2017, 18, 268. [Google Scholar] [CrossRef]
- Bernard-Gauthier, V.; Boudjemeline, M.; Rosa-Neto, P.; Thiel, A.; Schirrmacher, R. Towards tropomyosin-related kinase B (TrkB) receptor ligands for brain imaging with PET: Radiosynthesis and evaluation of 2-(4-[18F]fluorophenyl)-7,8-dihydroxy-4H-chromen-4-one and 2-(4-([N-methyl-11C]-dimethylamino)phenyl)-7,8-dihydroxy-4H-chromen-4-one. Bioorganic Med. Chem. 2013, 21, 7816–7829. [Google Scholar] [CrossRef]
- Jang, S.-W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chan, C.-B.; Jang, S.-W.; Pradoldej, S.; Huang, J.; He, K.; Phun, L.H.; France, S.; Xiao, G.; Jia, Y.; et al. A Synthetic 7,8-Dihydroxyflavone Derivative Promotes Neurogenesis and Exhibits Potent Antidepressant Effect. J. Med. Chem. 2010, 53, 8274–8286. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chan, C.-B.; Qi, Q.; Xiao, G.; Luo, H.R.; He, X.; Ye, K. Optimization of a Small Tropomyosin-Related Kinase B (TrkB) Agonist 7,8-Dihydroxyflavone Active in Mouse Models of Depression. J. Med. Chem. 2012, 55, 8524–8537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boltaev, U.; Meyer, Y.; Tolibzoda, F.; Jacques, T.; Gassaway, M.; Xu, Q.; Wagner, F.; Zhang, Y.-L.; Palmer, M.; Holson, E.; et al. Multiplex quantitative assays indicate a need for reevaluating reported small-molecule TrkB agonists. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Shia, C.-S.; Tsai, S.-Y.; Kuo, S.-C.; Hou, Y.-C.; Chao, P.-D.L. Metabolism and Pharmacokinetics of 3,3′,4′,7-Tetrahydroxyflavone (Fisetin), 5-Hydroxyflavone, and 7-Hydroxyflavone and Antihemolysis Effects of Fisetin and Its Serum Metabolites. J. Agric. Food Chem. 2009, 57, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Jäger, A.; Saaby, L. Flavonoids and the CNS. Molecules 2011, 16, 1471–1485. [Google Scholar] [CrossRef] [Green Version]
- Aldrich, C.; Bertozzi, C.; Georg, G.I.; Kiessling, L.; Lindsley, C.; Liotta, D.; Merz, K.M., Jr.; Schepartz, A.; Wang, S. The Ecstasy and Agony of Assay Interference Compounds. ACS Cent. Sci. 2017, 3, 143–147. [Google Scholar] [CrossRef]
- Bothwell, M. Recent advances in understanding neurotrophin signaling. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Altar, C.A.; Burton, L.E.; Bennett, G.L.; Dugich-Djordjevic, M. Recombinant human nerve growth factor is biologically active and labels novel high-affinity binding sites in rat brain. Proc. Nat. Acad. Sci. USA 1991, 88, 281–285. [Google Scholar] [CrossRef]
- Altar, C.; Dugich-Djordjevic, M.; Armanini, M.; Bakhit, C. Medial-to-lateral gradient of neostriatal NGF receptors: Relationship to cholinergic neurons and NGF-like immunoreactivity. J. Neurosci. 1991, 11, 828–836. [Google Scholar] [CrossRef]
- Merlio, J.P.; Ernfors, P.; Jaber, M.; Persson, H. Molecular cloning of rat trkC and distribution of cells expressing messenger RNAs for members of the trk family in the rat central nervous system. Neuroscience 1992, 51, 513–532. [Google Scholar] [CrossRef]
- Anderson, K.D.; Alderson, R.F.; Altar, C.A.; DiStefano, P.S.; Corcoran, T.L.; Lindsay, R.M.; Wiegand, S.J. Differential distribution of exogenous BDNF, NGF, and NT-3 in the brain corresponds to the relative abundance and distribution of high-affinity and low-affinity neurotrophin receptors. J. Comp. Neurol. 1995, 357, 296–317. [Google Scholar] [CrossRef] [PubMed]
- Altar, C.A.; Siuciak, J.A.; Wright, P.; Ip, N.Y.; Lindsay, R.M.; Wiegand, S.J. In Situ Hybridization of trkB and trkC Receptor mRNA in Rat Forebrain and Association with High-affinity Binding of [125I]BDNF, [125I]NT-4/5 and [125I]NT-3. Eur. J. Neurosci. 1994, 6, 1389–1405. [Google Scholar] [CrossRef]
- Zhang, L.; Villalobos, A.; Beck, E.M.; Bocan, T.; Chappie, T.A.; Chen, L.; Grimwood, S.; Heck, S.D.; Helal, C.J.; Hou, X.; et al. Design and Selection Parameters to Accelerate the Discovery of Novel Central Nervous System Positron Emission Tomography (PET) Ligands and Their Application in the Development of a Novel Phosphodiesterase 2A PET Ligand. J. Med. Chem. 2013, 56, 4568–4579. [Google Scholar] [CrossRef] [PubMed]
- Pike, V.W. PET radiotracers: Crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Rankovic, Z. CNS Drug Design: Balancing Physicochemical Properties for Optimal Brain Exposure. J. Med. Chem. 2015, 58, 2584–2608. [Google Scholar] [CrossRef] [PubMed]
- Wager, T.T.; Hou, X.; Verhoest, P.R.; Villalobos, A. Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach to Enable Alignment of Druglike Properties. ACS Chem. Neurosci. 2010, 1, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Wood, E.R.; Kuyper, L.; Petrov, K.G.; Hunter, R.N.; Harris, P.A.; Lackey, K. Discovery and in vitro evaluation of potent TrkA kinase inhibitors: Oxindole and aza-oxindoles. Bioorg. Med. Chem. Lett. 2004, 14, 953–957. [Google Scholar] [CrossRef]
- Bernard-Gauthier, V.; Aliaga, A.; Aliaga, A.; Boudjemeline, M.; Hopewell, R.; Kostikov, A.; Rosa-Neto, P.; Thiel, A.; Schirrmacher, R. Syntheses and Evaluation of Carbon-11- and Fluorine-18-Radiolabeled pan-Tropomyosin Receptor Kinase (Trk) Inhibitors: Exploration of the 4-Aza-2-oxindole Scaffold as Trk PET Imaging Agents. ACS Chem. Neurosci. 2015, 6, 260–276. [Google Scholar] [CrossRef]
- Prakash, Y.; Thompson, M.A.; Meuchel, L.; Pabelick, C.M.; Mantilla, C.B.; Zaidi, S.; Martin, R.J. Neurotrophins in lung health and disease. Expert Rev. Respir. Med. 2010, 4, 395–411. [Google Scholar] [CrossRef] [Green Version]
- Stachel, S.J.; Sanders, J.M.; Henze, D.A.; Rudd, M.T.; Su, H.-P.; Li, Y.; Nanda, K.K.; Egbertson, M.S.; Manley, P.J.; Jones, K.L.G.; et al. Maximizing Diversity from a Kinase Screen: Identification of Novel and Selective pan-Trk Inhibitors for Chronic Pain. J. Med. Chem. 2014, 57, 5800–5816. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Gauthier, V.; Schirrmacher, R. 5-(4-((4-[18F]fluorobenzyl)oxy)-3-methoxybenzyl)pyrimidine-2,4-diamine: A selective dual inhibitor for potential PET imaging of Trk/CSF-1R. Bioorg. Med. Chem. Lett. 2014, 24, 4784–4790. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Gauthier, V.; Mahringer, A.; Vesnaver, M.; Fricker, G.; Schirrmacher, R. Design and synthesis of a fluorinated quinazoline-based type-II Trk inhibitor as a scaffold for PET radiotracer development. Bioorg. Med. Chem. Lett. 2017, 27, 2771–2775. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.I.; Hunt, J.P.; Herrgard, S.; Ciceri, P.; Wodicka, L.M.; Pallares, G.; Hocker, M.; Treiber, D.K.; Zarrinkar, P.P. Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2011, 29, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, C.; Libert, L.; Plenevaux, A.; Aerts, J.; Franci, X.; Luxen, A. Fast and reliable method for the preparation of ortho- and para-[18F]fluorobenzyl halide derivatives: Key intermediates for the preparation of no-carrier-added PET aromatic radiopharmaceuticals. J. Fluor. Chem. 2012, 138, 48–55. [Google Scholar] [CrossRef]
- Baindur, N.; Gaul, M.D.; Kreutter, K.D.; Baumann, C.A.; Kim, A.J.; Xu, G.; Tuman, R.W.; Johnson, D.L. Alkylquinoline and Alkylquinazoline Kinase Modulators. U.S. Patent US2006281772, 14 December 2006. [Google Scholar]
- Bernard-Gauthier, V.; Bailey, J.J.; Aliaga, A.; Kostikov, A.; Rosa-Neto, P.; Wuest, M.; Brodeur, G.M.; Bedell, B.J.; Wuest, F.; Schirrmacher, R. Development of subnanomolar radiofluorinated (2-pyrrolidin-1-yl)imidazo[1,2-b]pyridazine pan-Trk inhibitors as candidate PET imaging probes. MedChemComm 2015, 6, 2184–2193. [Google Scholar] [CrossRef]
- Bernard-Gauthier, V.; Bailey, J.J.; Mossine, A.V.; Lindner, S.; Vomacka, L.; Aliaga, A.; Shao, X.; Quesada, C.A.; Sherman, P.; Mahringer, A.; et al. A Kinome-Wide Selective Radiolabeled TrkB/C Inhibitor for in Vitro and in Vivo Neuroimaging: Synthesis, Preclinical Evaluation, and First-in-Human. J. Med. Chem. 2017, 60, 6897–6910. [Google Scholar] [CrossRef]
- Preshlock, S.; Tredwell, M.; Gouverneur, V. 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev. 2016, 116, 719–766. [Google Scholar] [CrossRef]
- Brooks, A.F.; Topczewski, J.J.; Ichiishi, N.; Sanford, M.S.; Scott, P.J.H. Late-stage [(18)F]Fluorination: New Solutions to Old Problems. Chem. Sci. 2014, 5, 4545–4553. [Google Scholar]
- Zlatopolskiy, B.D.; Zischler, J.; Krapf, P.; Zarrad, F.; Urusova, E.A.; Kordys, E.; Endepols, H.; Neumaier, B. Copper-Mediated Aromatic Radiofluorination Revisited: Efficient Production of PET Tracers on a Preparative Scale. Chem. A Eur. J. 2015, 21, 5972–5979. [Google Scholar] [CrossRef] [PubMed]
- Tredwell, M.; Preshlock, S.M.; Taylor, N.J.; Gruber, S.; Huiban, M.; Passchier, J.; Mercier, J.; Génicot, C.; Gouverneur, V. A General Copper-Mediated Nucleophilic 18F Fluorination of Arenes. Angew. Chem. Int. Ed. 2014, 53, 7751–7755. [Google Scholar] [CrossRef] [PubMed]
- Ichiishi, N.; Brooks, A.F.; Topczewski, J.J.; Rodnick, M.E.; Sanford, M.S.; Scott, P.J.H. Copper-catalyzed [18F]fluorination of (mesityl)(aryl)iodonium salts. Org. Lett. 2014, 16, 3224–3227. [Google Scholar] [CrossRef] [PubMed]
- Bernard-Gauthier, V.; Mossine, A.V.; Mahringer, A.; Aliaga, A.; Bailey, J.J.; Shao, X.; Stauff, J.; Arteaga, J.; Sherman, P.; Grand’Maison, M.; et al. Identification of [18F]TRACK, a Fluorine-18-Labeled Tropomyosin Receptor Kinase (Trk) Inhibitor for PET Imaging. J. Med. Chem. 2018, 61, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Mossine, A.V.; Brooks, A.F.; Bernard-Gauthier, V.; Bailey, J.J.; Ichiishi, N.; Schirrmacher, R.; Sanford, M.S.; Scott, P.J.H. Automated Synthesis of PET Radiotracers by Copper-mediated 18F-Fluorination of Organoborons: Importance of the Order of Addition and Competing Protodeborylation. J. Label. Compd. Radiopharm. 2018, 63, 228–236. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schirrmacher, R.; Bailey, J.J.; Mossine, A.V.; Scott, P.J.H.; Kaiser, L.; Bartenstein, P.; Lindner, S.; Kaplan, D.R.; Kostikov, A.; Fricker, G.; et al. Radioligands for Tropomyosin Receptor Kinase (Trk) Positron Emission Tomography Imaging. Pharmaceuticals 2019, 12, 7. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12010007
Schirrmacher R, Bailey JJ, Mossine AV, Scott PJH, Kaiser L, Bartenstein P, Lindner S, Kaplan DR, Kostikov A, Fricker G, et al. Radioligands for Tropomyosin Receptor Kinase (Trk) Positron Emission Tomography Imaging. Pharmaceuticals. 2019; 12(1):7. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12010007
Chicago/Turabian StyleSchirrmacher, Ralf, Justin J. Bailey, Andrew V. Mossine, Peter J. H. Scott, Lena Kaiser, Peter Bartenstein, Simon Lindner, David R. Kaplan, Alexey Kostikov, Gert Fricker, and et al. 2019. "Radioligands for Tropomyosin Receptor Kinase (Trk) Positron Emission Tomography Imaging" Pharmaceuticals 12, no. 1: 7. https://0-doi-org.brum.beds.ac.uk/10.3390/ph12010007