Assembly of Peptidoglycan Fragments—A Synthetic Challenge


1
LAQV@REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Campus de Caparica, 2829-516 Caparica, Portugal
2
UCIBIO@REQUIMTE, Departamento de Ciências da Vida, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Campus de Caparica, 2829-516 Caparica, Portugal
3
Laboratory of Bacterial Cell Surfaces and Pathogenesis, Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, 2780-157 Oeiras, Portugal
*
Authors to whom correspondence should be addressed.
Pharmaceuticals 2020, 13(11), 392; https://0-doi-org.brum.beds.ac.uk/10.3390/ph13110392
Submission received: 6 October 2020
/
Revised: 9 November 2020
/
Accepted: 12 November 2020
/
Published: 15 November 2020
(This article belongs to the Special Issue Glycomimetics and Glycoconjugates in Drug Discovery)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Peptidoglycan (PGN) is a major constituent of most bacterial cell walls that is recognized as a primary target of the innate immune system. The availability of pure PGN molecules has become key to different biological studies. This review aims to (1) provide an overview of PGN biosynthesis, focusing on the main biosynthetic intermediates; (2) focus on the challenges for chemical synthesis posed by the unique and complex structure of PGN; and (3) cover the synthetic routes of PGN fragments developed to date. The key difficulties in the synthesis of PGN molecules mainly involve stereoselective glycosylation involving NAG derivatives. The complex synthesis of the carbohydrate backbone commonly involves multistep sequences of chemical reactions to install the lactyl moiety at the O-3 position of NAG derivatives and to control enantioselective glycosylation. Recent advances are presented and synthetic routes are described according to the main strategy used: (i) based on the availability of starting materials such as glucosamine derivatives; (ii) based on a particular orthogonal synthesis; and (iii) based on the use of other natural biopolymers as raw materials.
1. Introduction
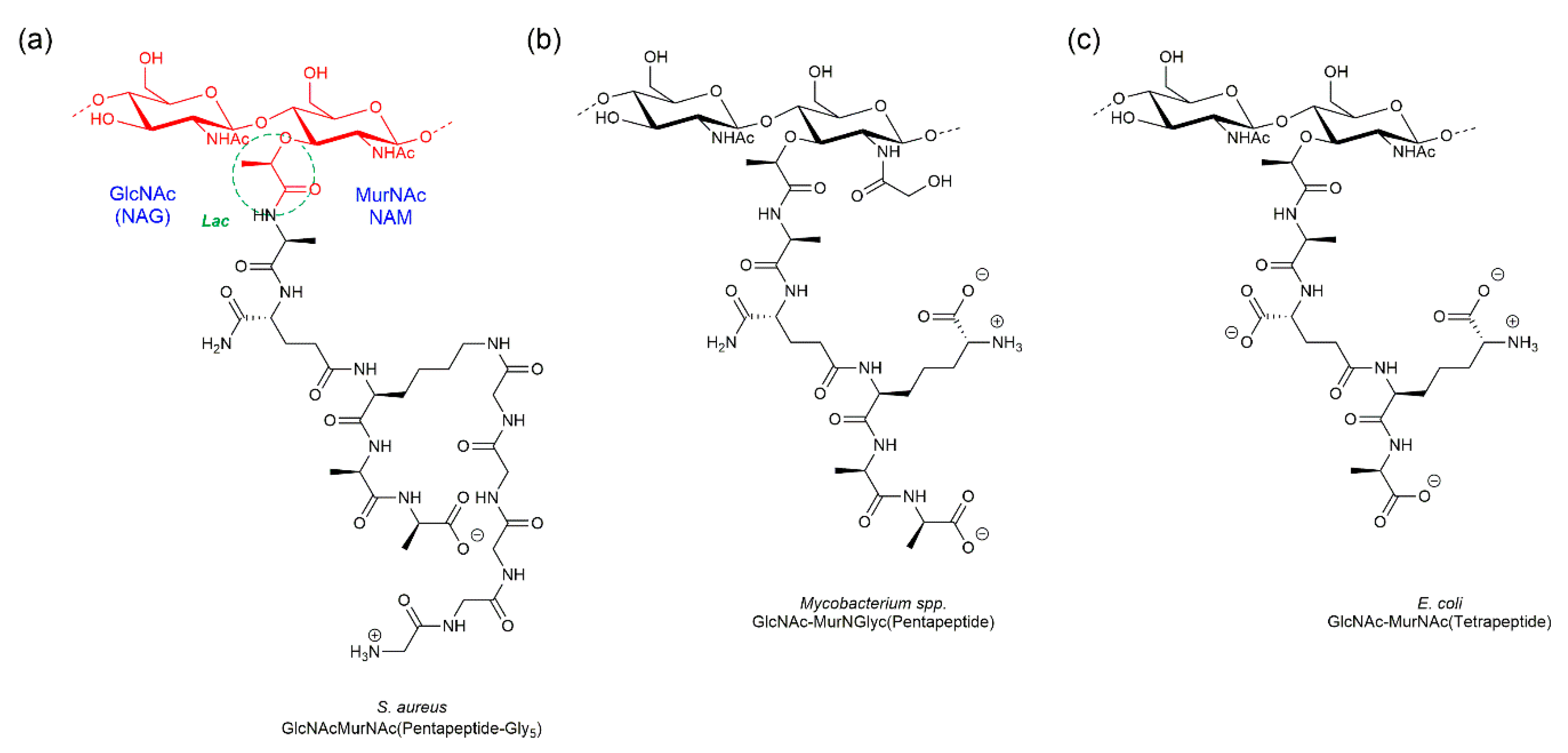
Peptidoglycan (PGN) is a major component of the cell wall that surrounds most bacteria. It is a macromolecule made of glycan chains, which consist of linear alternating copolymers of β-1,4-linked N-acetyl glucosamine (GlcNAc or NAG) and N-acetyl muramic acid (MurNAc or NAM) residues. Importantly, these glycan strands carry a lactyl group in the MurNAc moiety that is covalently bound to peptide stems via an amide bond. These stems are used to link glycan strands that are nearby, creating a net-like structure. The composition of the peptide stems is specific for each bacterial species (Figure 1) [1]. The unbranched peptide stems are typically pentapeptides of alternating L- and D-amino acids, usually L-alanine, D-glutamic acid, a diamino acid and two terminal D-alanines. The diamino acid in the third position is usually meso-diaminopimelic acid (DAP) in Gram-negative bacteria and l-Lysine is Gram-positive [2]. The diamino acids in the peptide stems allow PGN to “branch out” and connect to neighboring peptide stems, cross-linking adjacent glycan chains [2].
Bacterial PGN, a telltale molecule recognized by the innate immune system from different organisms, has important functions for the survival of bacteria. The structure of the PGN macromolecule determines the shape of these microorganisms; anchors different proteins, which are required for bacteria to interact with the surrounding environment and for the maintenance of envelope integrity; and provides the required mechanical strength to resist osmotic pressure [2]. Importantly, modification of the bacterial process of PGN synthesis, or of PGN composition, can be associated with an altered expression of resistance to different antibiotics. Examples of this effect include resistance to beta-lactams in methicillin-resistant Staphylococcus aureus (MRSA) strains, which is dependent on expression of different PGN synthetic enzymes [3], as well as resistance to vancomycin in isolates of Enterococcus species, which requires modification of the PGN precursors to reduce the affinity of the antibiotic to its target [4].
PGN metabolism requires (i) the synthesis of PGN building blocks inside bacteria (also known as muropeptide precursors), (ii) their transport across the bacterial membrane, (iii) the polymerization of the PGN scaffold, and (iv) the trimming or degradation of the PGN molecule. These processes take place during the bacterial cell cycle and ensure that PGN, which is a dynamic structure, is remodeled during bacterial growth.
Antibiotics that block PGN synthesis or that lead to changes in the composition of the bacterial cell surface may weaken the cell wall (for example by decreasing the number of crosslinking nodes between different glycan chains and reducing the robustness of the macromolecule to environment insults), lead to alterations of the bacterial shape, and, ultimately, cause bacterial death. Loss of viability may also result from cell lysis, due to the promoted activity of PGN hydrolases [5,6] or due to futile cycling of PGN synthesis and degradation, which depletes cellular resources [7]. PGN hydrolases, or ‘‘lysins’’, are capable of degrading the bacterial cell wall [8]. Lysins can (i) act on the sugar moiety, releasing either glycan fragments with NAG at the reducing end (endo-β-N-acetylglucosaminidases) or glycan fragments with NAM at the reducing end (N-acetylmuramidases, also known as lysozymes); (ii) act on the peptide moiety (endopeptidases or carboxypeptidases); (iii) hydrolyze the amide bond linking the lactyl group of NAM residues to the peptide stem (N-acetylmuramoyl-l-alanine amidases) [6,7,8].
Despite advances in the understanding of PGN biosynthesis and its inhibition by antibiotics, the study of the relevance of PGN structure in bacterial metabolism and of its role in host disease has been hampered by the lack of pure PGN fragments [9].
Chemical synthesis of NAG containing oligosaccharides [10,11,12,13], such as the NAG-NAM disaccharide, is a challenging task due to well-known limitations associated with the enantioselective glycosylation of NAG moieties: (i) the presence of a β-1,4 glycosidic bond requires a multistep synthetic sequence to obtain a regio- and stereoselective assembly of glycosidic bonds, crucial for biological activity; (ii) the use of 2-acetamido-2-deoxyglycosyl donors, due to the formation of 1,2-O,N-oxazoline intermediates [14,15,16,17], and the use of corresponding acceptors that are poor nucleophiles [18].
As mentioned above, the most demanding aspect in the synthesis of fragments of peptidoglycan is the control of the stereoselectivity of the glycosidic bond. The pursuit of straightforward and stereoselective methods for the assembly of PGN fragments remains an emerging research area. In this review, after an overview of the biosynthesis of bacterial PGN, we highlight recent advances in the synthesis of PGN fragments, focusing on the different strategies, including the selection of donors and acceptors, reaction conditions, stereoselectivity of the glycosylation reaction, and peptide coupling. Recently, chemoenzymatic approaches have been reported to solve problems associated with the orthogonal and multistep processes usually required for PGN synthesis, which will also be covered in this review.
2. Biosynthesis of the Bacterial Peptidoglycan
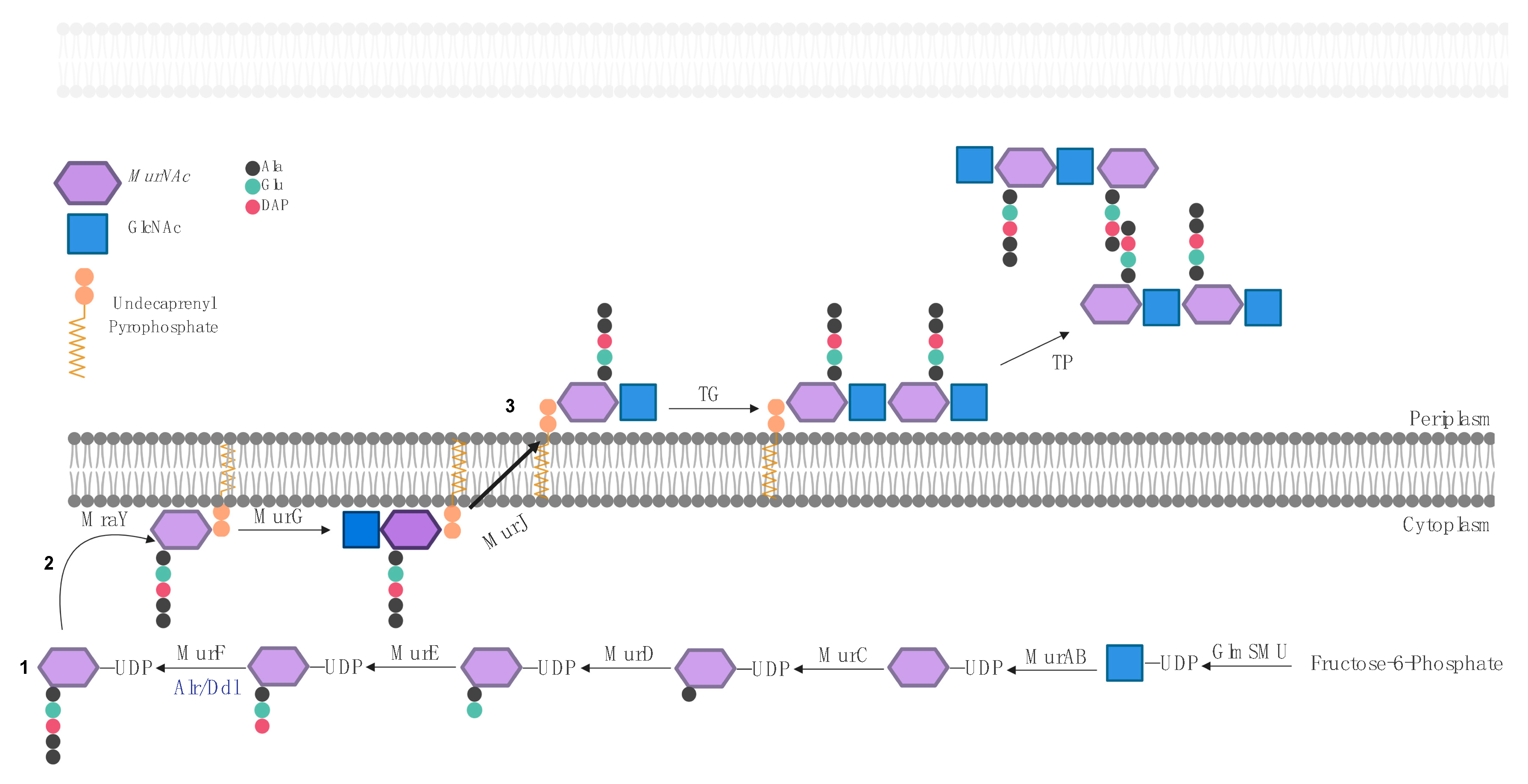
PGN biosynthesis is conserved among Gram-positive and Gram-negative bacteria and occurs in three different locations: (i) synthesis of uridine diphosphate (UDP)-activated PGN precursors in the cytoplasm; (ii) synthesis of lipid-linked precursors at the inner leaflet of the cytoplasmatic membrane; (iii) flipping to the outer leaflet of the cytoplasmatic membrane and incorporation of the precursor into nascent PGN (Figure 2).
PGN biosynthesis starts with the conversion of fructose-6-phosphate into uridine diphosphate-GlcNAc (UDP-GlcNAc) by GlmSMU enzymes [19,20,21,22,23,24]. UDP-GlcNAc is converted to uridine diphosphate-MurNAc (UDP-MurNAc) by MurAB enzymes [25,26,27]. The d-amino acids present in the PG are produced by the MurI and Alr racemases, which convert l-Glu to d-Glu and L-Ala to d-Ala, respectively [28,29,30]. The amino acids L-Ala, D-Glu, and L-Lys or DAP are added sequentially by the MurCDE enzymes, respectively, to the lactyl group of UDP-MurNAc [31,32,33,34]. The d-Ala-d-Ala dipeptide present at the carboxyl end of the PGN stem peptide is first formed by Ddl ligases [35] and then incorporated into the UDP-MurNAc-tripeptide by MurF [36,37], forming a UDP-MurNAc-pentapeptide known as the Park nucleotide.
The Park nucleotide is attached to undecaprenyl phosphate in the inner leaflet of the cytoplasmatic membrane by MraY with the release of UDP. The resulting MurNAc-(pentapeptide)-pyrophosphoryl-undecaprenol is known as lipid I [38,39,40]. MurG converts lipid I into lipid II (GlcNAc-β-(1,4)-MurNAc-(pentapeptide)-pyrophosphoryl-undecaprenol) by adding the GlcNAc of a UDP-GlcNAc to lipid I with the release of UDP [41,42,43].
The resulting product, lipid II, is flipped from the inner leaflet to the outer leaflet of the cytoplasmatic membrane. There is a debate regarding which enzyme is responsible for this step. Although FtsW and MurJ are the two strongest candidates, the literature favors MurJ as the bona fide flippase [44,45,46,47,48]. More recently, it has been shown that certain enzymes in particular organisms may also exhibit lipid II flippase activity [49].
Precursor incorporation into nascent PG is achieved by reactions of transglycosylation (TG), responsible for extending glycan chains, and reactions of transpeptidation (TP), responsible for connecting adjacent glycan chains through their stem peptides. TG connects the lipid II GlcNac C4 to the MurNAc C1 of the nascent PGN with the release of undecaprenyl pyrophosphate. After the incorporation of the PGN precursor into the PGN macromolecule, undecaprenyl pyrophosphate is subsequentially dephosphorylated, prior to its use in another cycle of glycan synthesis, leading to the removal of one phosphate group, and is recycled for another cycle of Lipid precursor synthesis [50,51].
3. Synthetic Approaches towards PGN Fragments from Glucosamine I
Currently, there is a pressing need for new approaches toward the chemical synthesis of PGN fragments. This will permit in vitro studies of the biochemical activity of PBP enzymes [52], which are important targets for the development of new antibiotics. It will also ensure that samples of PGN, a telltale molecule that flags an infected host to the presence of bacteria, are free of contaminants of bacterial origin [53,54].
However, attempts to fully synthesize the PGN molecule in the laboratory in a way that is independent of the use of components (substrates or enzymes) of bacterial origin have faced several restrictions.
The synthesis of NAG-containing oligosaccharides, such as the NAG-NAM disaccharide present in the PGN molecule, poses considerable limitations associated with the enantioselective glycosylation of NAG moieties with respect to both donor and acceptor units. Indeed, the glycosylation of complex aglycones with glycosyl donors bearing 2-acetamido-2-deoxy functionality is usually impractical due to the formation of a 1,2-O,N-oxazoline intermediate during glycosylation. This is a stable intermediate, which drastically decreases the rate and yield of the glycosylation step, especially when the chosen glycosyl acceptors have poor nucleophilicity or are sterically hindered. Furthermore, the use of glycosyl acceptors bearing 2-acetamido-2-deoxy functionality is considered not to be a good strategy as the amide group in the acceptor molecule can establish a hydrogen bond with the hydroxy group O-4, which decreases nucleophilicity at this position. In order to avoid the formation of the stable 1,2-O,N oxazoline intermediate, and improve the formation of 1,2-trans-glycoside, different authors have used various N-protecting groups in their attempt to synthesize PGN fragments. The N-protecting groups that have been developed differ in their ability to direct the stereochemical outcome of the glycosylation reactions and in their compatibility with diverse carbohydrate synthetic methods.
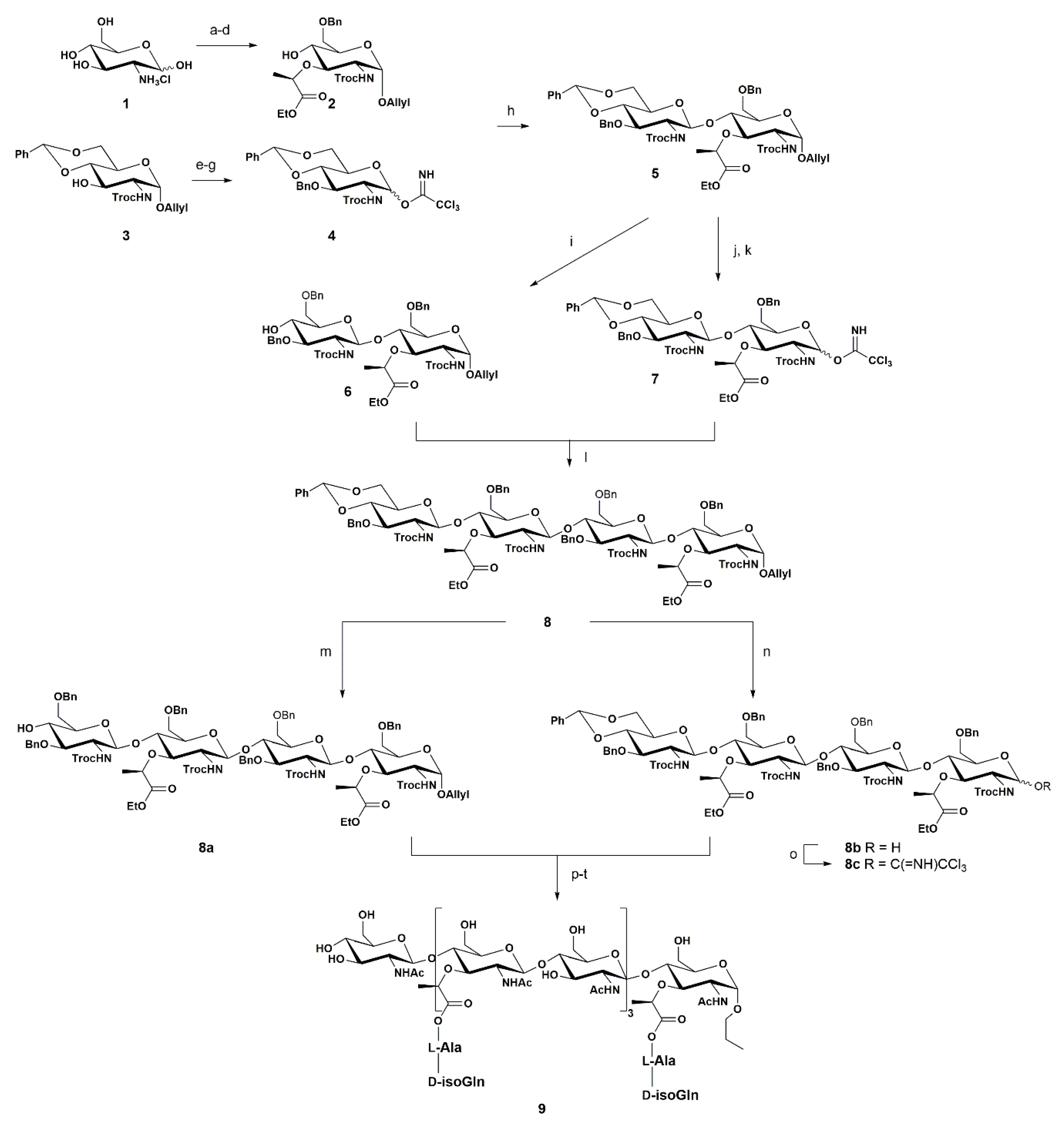
In 2001, Fukase and co-workers reported the synthesis of PGN fragments [55], in a follow-up of their previous work [56]. The authors developed an orthogonal synthesis that was able to create the building blocks required to achieve a PGN fragment—the complex octasaccharide (NAG-NAM)4 with two amino acid strands linked to every NAM unit 9 (Scheme 1).
The first step in the preparation of acceptor 2 consisted in the protection of the amine group of d-glucosamine with the allyloxycarbonyl (Alloc) group by treatment of glucosamine with AllocCl in the presence of NaHCO3 as a base. Next, the anomeric position was protected with the O-Allyl group, by reaction with allyl alcohol under acidic conditions using DOWEX as a proton exchange resin at high temperature. The O-4 and O-6 positions were regioselectively protected via acetylidene formation by treatment with benzaldehyde dimethyl acetal catalyzed by p-TsOH in 57% yield. This strategy is a well-known procedure for the regioselective protection of both the O-6 and O-4 positions.
For the introduction of the lactyl moiety at the O-3 position, the authors used trifluoromethanesulfonyl-l-(S)-2-propionic acid benzyl ester after hydroxy group deprotonation with NaH, and the product was isolated in 98% yield. The N-protection achieved with the introduction of the Alloc group is crucial to install the lactyl moiety at the O-3 position. However, the presence of this group will not permit the correct β-1,4 selectivity during the glycosylation reaction [14]. In order to obtain β-1,4 linkage, the authors replaced the Alloc group with the trichloroethoxycarbonyl (Troc) group. The Alloc group was first removed via treatment with a complex of Pd(PPh3)4 assisted by AcOH. The palladium complex coordinates with the double bond from the Alloc group, followed by hydrolysis with AcOH. Then TrocCl was added to provide the N-Troc product in 58% yield. The use of Troc as an N-protecting group at the acceptor moiety allowed for the simplified removal of the protecting groups in a later stage of the synthesis. Consequently, selective ring opening was performed to make the O-4 position free in preparation for the glycosylation reaction, using Me3N·BH3 to reduce the benzylic position, leaving the O-4 position free and providing the acceptor 2 in 87% yield.
The donor 4 was prepared via intermediate 3, which was obtained from d-glucosamine using a similar approach as that described for the acceptor, consisting of: N-protection with TrocCl, protection of the anomeric position with the O-Allyl group, and formation of the acetylidene. To prepare the donor unit, the authors protected the O-3 position with the Bn group, using BnBr, through a SN2 reaction catalyzed by AgO and obtained the product in 87% yield. The next step consisted in the conversion of the O-Allyl group into a trichloroacetimidate group. Thus, the authors removed the O-Allyl group with an Ir complex to isomerize the double bond, promoting the H2O nucleophilic attack at the anomeric position. The next step consisted in the treatment of the anomeric position of the donor with CCl3CN and Cs2CO3 as a base, leading to the product 4 in a quantitative yield.
The next step consisted in the glycosylation reaction using 2 as an acceptor and 4 as a donor. Since the trichloroacetimidate group is an excellent leaving group, TMSOTf was used in a catalytic amount, along with molecular sieves, to promote the glycosylation reaction, providing 5 in 88% yield.
Once the disaccharide 5 was obtained, the same rationale was used to produce the tetrasaccharide 8, including the selective ring opening to provide the acceptor 6, and replacement of the O-Allyl group with the trichloroacetimidate group to provide the donor 7. The synthesis of the octasaccharide followed a similar approach via preparation of both acceptor and donor tetrasaccharides 8a and 8c, respectively. After obtaining the octasaccharide, the authors attached the dipeptide via solid-phase peptide synthesis. The remaining benzyl groups were removed by a hydrogenation reaction.
Overall, the authors carried out 25 steps to achieve compound 9. This study [55] was a milestone in the synthesis of PGN fragments. To perform this synthetic plan, the authors had to carefully choose all the protecting groups, in particular the N-protecting group and the protection of the anomeric position. As the N-Alloc group, a well-known group that allows many chemical modifications, does not favor β-1,4 glycosidic bond formation in glycosylation reactions, the authors installed the Troc group which is a β-1,4 director.
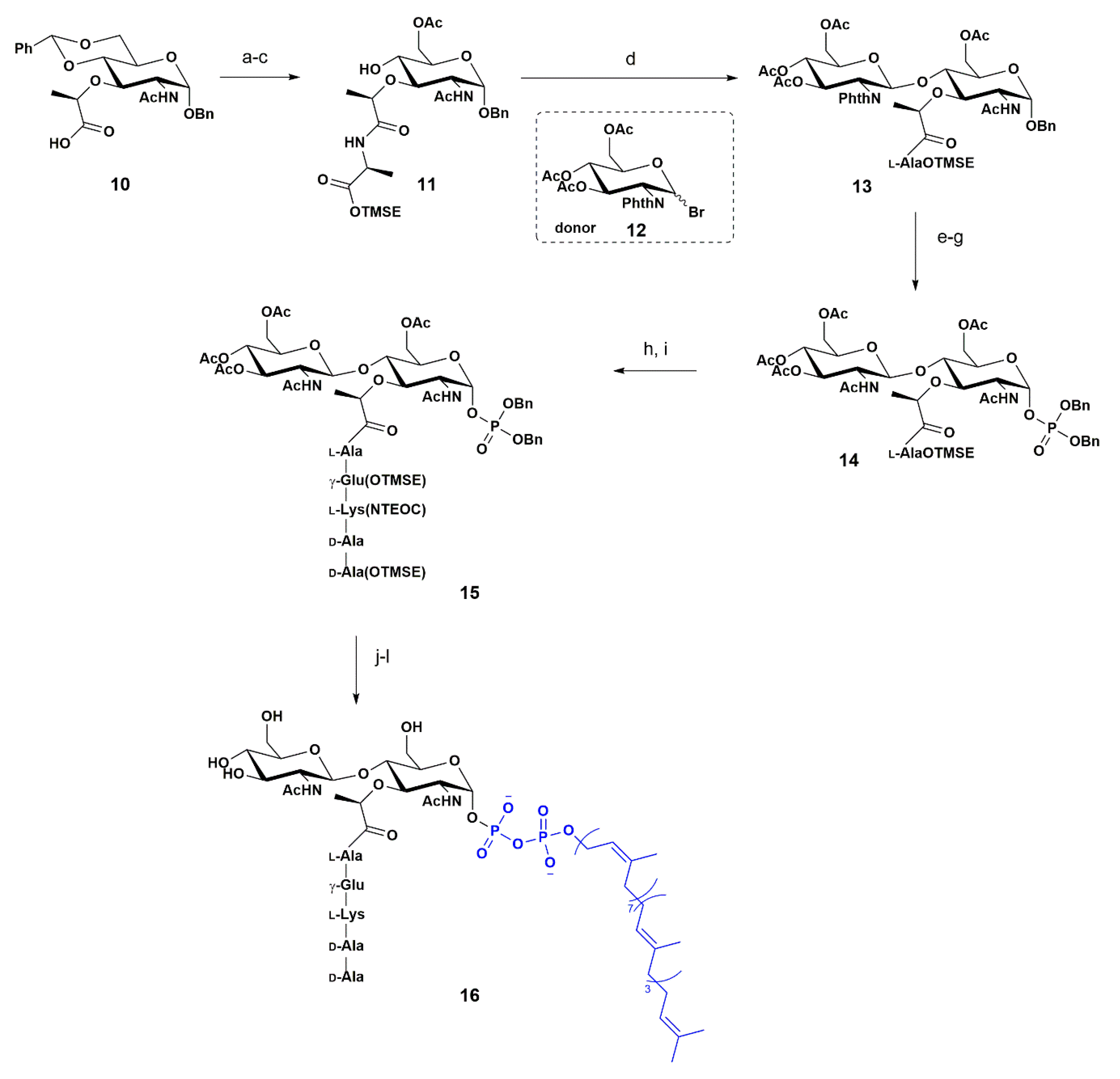
In 2001, Wang and co-workers reported the orthogonal synthesis of the Lipid II unit 16, Scheme 2 [57]. The authors aimed to develop an appropriate assay system for the observation of catalytic transformations involving PBP1b, a penicillin binding protein that in E. coli is involved in the transglycosylation and transeptidation of PGN precursors, since this area of study had been limited by the available amounts of Lipid II.
The synthesis started with the preparation of the acceptor unit 11 from the commercially available MurNAc derivative 10. The first step was to install the L-alanine unit to avoid the unwanted cyclization of compound 11, observed when the lactate was protected as an ester. This reaction was followed by removal of the arylidene group and acetylation of the free 6-hydroxy group, affording the acceptor 11 with a 57% yield after three steps.
As a donor, the authors used the commercially available donor 12, a glycosyl bromide possessing the N-phthalimide group. The glycosylation was carried in the presence of AgOTf and, with the participation of the N-phathalimido group in 13 that oriented the attack on the β face, the desired compound 13 was isolated in 60% yield. Next, the phtaloyl group in 13 was removed with concomitant N-acetylation using an amino-modified resin and acetic anhydride. Compound 13 was then converted to the desired phosphate 14 via consecutive anomeric deprotection and phosphorylation. Thus, hydrogenation of the benzyl group in 13, using the classical conditions 10% Pd/C afforded the free OH. Phosphorylation was performed in a one-pot, two-step procedure, consisting of the initial formation of the phosphite, followed by in situ oxidation with m-CPBA. The authors used (BnO)2PN(i-Pr)2, followed by m-CPBA oxidation, to provide 14 with 27% yield after three steps. Next, the silyl group from alanine was removed by means of treatment with TBAF, followed by pentapeptide coupling using the standard conditions reported by the authors. The final steps relied on the undecaprenyl unit coupling and removal of the protecting groups. The benzyl groups from 14 were removed by hydrogenation, followed by coupling with undecaprenyl phosphate using CDI as a coupling agent to obtain 15. After removal of the silyl group using TBAF and removal of the acetyl groups under Zemplén’s conditions the Lipid II unit 16 was obtained, consisting of the first synthesis with 12 steps.
Blaszczak and co-workers reported, in 2001, the synthesis of the compound 20 [58] and then in 2002 reported the final steps to afford the Lipid II unit 16 (Scheme 3) [59].
The synthesis of 16 started with the coupling of alanine to the commercially available monosaccharide 10 in 95% yield, followed by the reductive opening of arylidene using Et3SiH, giving the acceptor 17 in 61% yield. The glycosylation reaction with of 17 with the donor glycosyl bromide 18, containing the NHTroc group, was carried out in the presence of AgOTf as a catalyst, in 84% yield. Next, the O-6 benzyl group was removed, followed by acetylation of the positions O6 and N2 using ZnCl2 and Ac2O. The N-Troc group was removed using Zn dust in a multistep reaction, yielding 19 in 67% yield. The authors hydrogenated the OBn group, followed by insertion of the phophoryl group, affording 20 in 73% after two steps. The tetrapeptide was coupled using EDCI, followed by hydrogenation of the Bn groups, affording 21 in 41% yield (two steps). The final steps consisted in the coupling of the undecaprenyl moiety using undecaprenyl phosphate and CDI, followed by the removal of the acetyl groups under Zemplén’s conditions, to afford Lipid II unit 16. This synthesis was carried out in 10 steps.
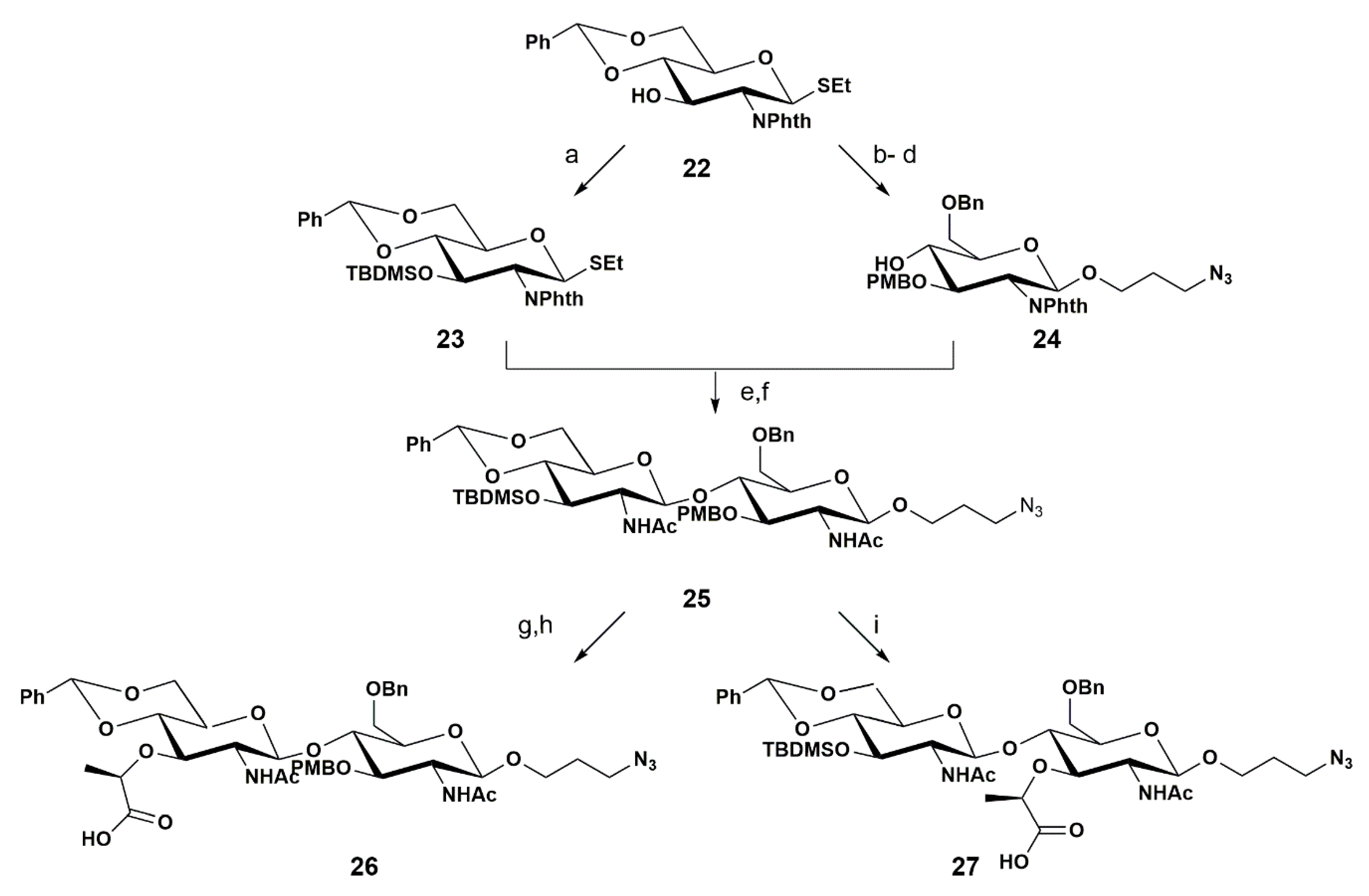
Other groups of researchers have reported alternative strategies to produce molecules that could be used in the synthesis of PGN. In 2002, the Boons group developed the synthesis of a high value disaccharide which can be converted into the intermediates 26 and 27, precursors of the NAM-NAG and NAG-NAM disaccharides, respectively (Scheme 4) [60].
In their report, the authors started the synthesis of PGN fragments with compound 22, which possesses the phthalimide (Phth) group as an N-protecting group, a 4,6-O arylidene ring, and an SEt group at the anomeric position. The O-3 position of 22 was protected with TBDMSOTf, affording 23 as the donor unit in 96% yield.
Concerning the preparation of acceptor 24, the synthesis started with the protection of the O-3 position of 22 with the PMB group, using PMBCl and NaH in 84% yield. The next step consisted in the opening of the arylidene ring to make the O-4 position free. This was achieved using Me3N·BH3 as a reducing agent, and the desired product was obtained in 72% yield. Next, the authors replaced the SEt group with 3-azido-propanol. Subsequent treatment with NIs oxidized SEt to S(O)Et, and this intermediate was attacked by 3-azido-propanol to afford 24 in 67% yield. Interestingly, the authors highlighted that due to the low reactivity of O-4, self-condensation was not observed.
Having both acceptor 24 and donor 23 in hand, the next step consisted in the glycosylation reaction. The activation of the anomeric group with NIS promoted the nucleophilic attack of the hydroxyl group of the acceptor 24 to the donor 23, affording the corresponding disaccharide in 62% yield. Once the synthesis of the disaccharide was obtained, the N-Phth group was removed by treatment with hydrazine at reflux, followed by an acetylation step, giving the N-acetylated product 25 in 70% yield.
The intermediate 25 was used to prepare both the NAM-NAG precursor 26 and the NAG-NAM precursor 27. To produce 26 the authors removed the TBDMS group using tetrabutylamoniun fluoride (TBAF) in 75% yield, followed by the introduction of the lactyl moiety. The authors used (S)-2-chloropropionic acid after deprotonation of O-3 with NaH, affording the corresponding product in 76% yield. To obtain 27 the authors removed the PMB group using DDQ in 79% yield, followed by the introduction of the lactyl moiety, using the same procedure used for 26, in 66% yield.
In their synthetic strategy, the authors selected the Phth group as the N-protecting group, since it blocks the nitrogen atom and is only removed by reduction with hydrazine. In addition, a thioglycoside was used as a glycosyl donor due to its versatility and stability and for being a good leaving group when activated with NIS.
Overall, the authors synthesized two disaccharides, which are similar to components of bacterial PGN, in eight steps. Their strategy consisted in first synthesizing the disaccharide unit and afterwards performing the insertion of the lactate moiety. The lactonization of muramic acid derivatives possessing a free O-4 position is a major obstacle. As mentioned above, Blaszczak and co-workers [58,59] addressed this problem by installing the l-alanine in the glycosyl acceptor for the synthesis of a disaccharide. The route developed by the Boons group addressed this challenge by synthesizing a disaccharide and thereafter incorporating the lactate unit; consequently, one disaccharide can be used for the synthesis of both PGN units (MurNAc-GlcNAc and GlcNAc-MurNAc).
The N-Troc group of the glycosyl donor is known to be very reactive [18], directing glycosylation exclusively to produce 1,2-trans linked glycosides via neighboring group participation. The authors found that glycosyl donors that were protected with phthalimido groups led to high-yielding glycosylations. This N-protecting group is compatible with strong basic conditions used for the incorporation of the lactate moiety, whereas the Troc group would not survive these conditions.
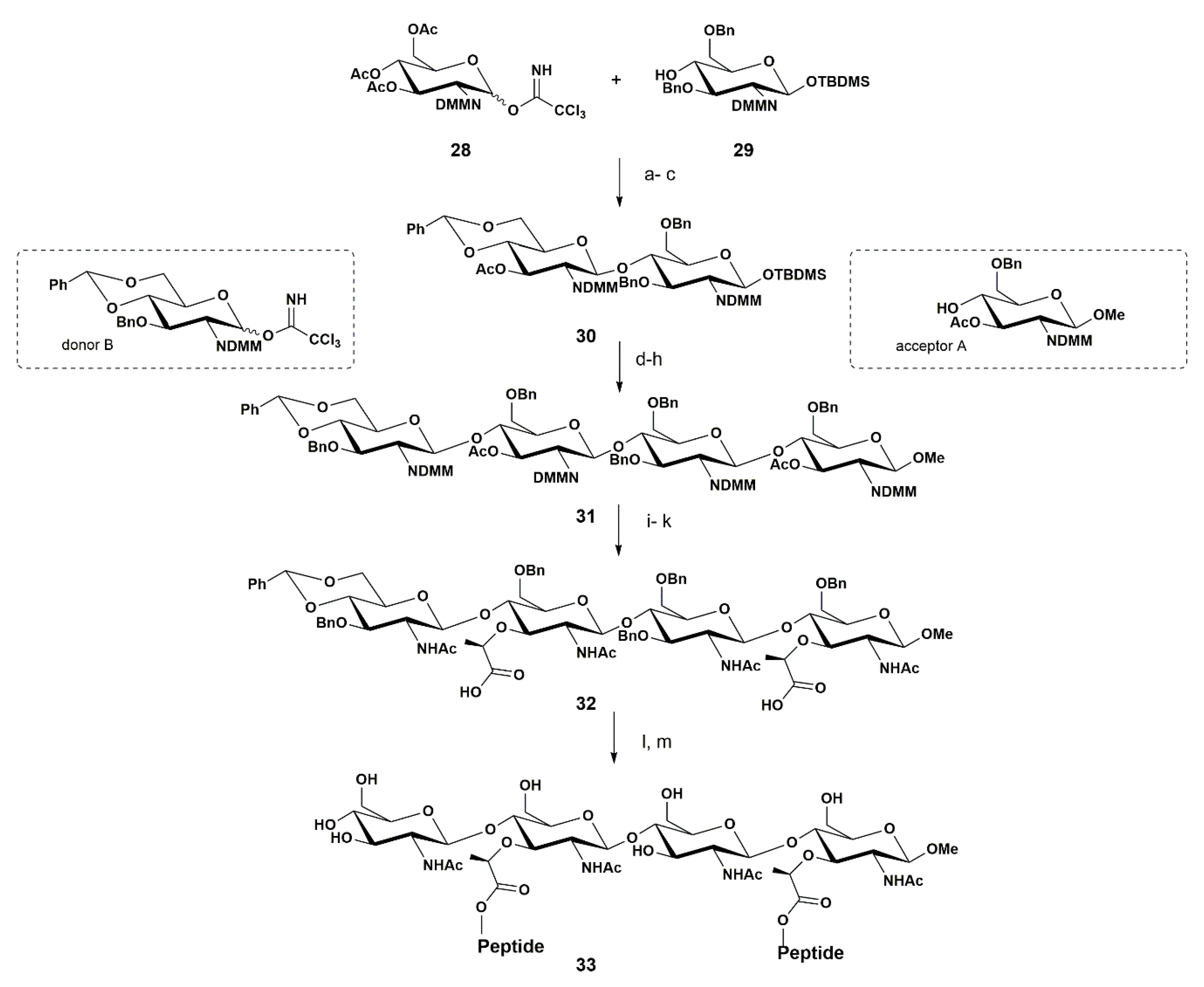
Soon after, in 2004, Mobashery and co-workers reported the synthesis of another PGN fragment—a tetrasaccharide, with two pentapeptides attached to the NAM units, 33 (Scheme 5) [61].
These authors started their synthesis by preparing both 28 and 29 from glucosamine via orthogonal synthesis. This began with protecting the amino group of glucosamine with the dimethyl maleimide (DMM) group, using 2,3-dimethylmaleic anhydride followed by acetylation of the hydroxy groups, affording the corresponding product in 55% yield after two steps. This N-DMM glucosamine intermediate was the precursor for both the donor 28 and the acceptor 29.
To prepare 28 the authors selectively deprotected the anomeric acetyl group using hydrazine acetate to give a free OH group at the anomeric position in 68% yield. Then, at this position, the trichloroacetimidate group was introduced by treatment with CCl3CN and DBU as a base, providing the donor 28 in 85% yield.
Concerning the preparation of acceptor 29, the authors selectively removed the anomeric acetyl group from peracetylated N-DMM glucosamine, using hydrazine acetate (68% yield), followed by silylation of the anomeric hydroxyl group using TBDMSCl and imidazole to afford the silylated product in 78% yield. Next, the remaining acetyl groups were removed using NaOMe in methanol and the product was obtained in 73% yield. The benzylation of the O-3 and O-6 was performed using Bu2SnO and BnBr, and the acceptor 29 was isolated in 73% yield.
Having both donor 28 and acceptor 29 in hand, the glycosylation reaction was performed. This step was carried using TfOH as a promotor, producing the desired disaccharide in 75% yield. Three additional steps were needed to produce disaccharide 30. In the first step there was the removal of the acetyl group using NaOMe that was quenched by the use of a proton exchange resin (Amberlite IR-120). In the second step, the authors executed the subsequent reaction with benzaldehyde dimethylacetal, leading to the formation of 4,6-O arylidene, and then O-6 and O-3 benzylation, (60%, in the two steps). The last step consisted in the acetylation of the remaining free O-3 position with acetic anhydride, affording 30 in 85% yield.
Glycan chain elongation using 30 as a substrate was then required. In order to achieve this, the TBDMS group was removed with TBAF in 75% yield. The authors observed that for long reaction times, the yield decreased due to hydrolysis of the acetyl group. Then, the trichloroacetimidate group was introduced, followed by a glycosylation reaction with an acceptor A (Scheme 5) possessing both O-6 benzylated and O-3 acetylated and an OMe group at the anomeric position, affording the corresponding trisaccharide in 55%. Next, the selective opening of the 4,6-O-arylidene ring with Me3N·BH3 gave the corresponding trisaccharide with the free O-4 position in 62% yield.
The next glycosylation reaction was performed using this trisaccharide as an acceptor and the monosaccharide B as a donor (Scheme 5), possessing a 4,6-O-arylidene ring, O-3 protected with a benzyl group, and the trichloroacetimidate group at the anomeric position. This glycosylation reaction was once again catalyzed by TfOH, affording 31 in 68%
At this stage the DMM group was no longer needed, and it was removed by treatment of 31 with NaOH in dioxane/water (4:1), quenched with HCl until reaching pH 3, followed by a reaction with acetic anhydride. The corresponding N-acetylated product was isolated in 48% yield. To introduce the lactyl moieties, the authors removed the O-acetyl groups using NaOMe with 74% yield, followed by treatment with (S)-2-chloropropionic acid (after deprotonation with NaH), and 32 was obtained in 46% yield.
The final steps involved the coupling of the tetrasaccharide with a synthesized pentapeptide (prepared as a CF3CO2H salt) in a 41% yield, and removal of the benzyl groups by hydrogenation, affording 33 in 76% yield.
The authors reported the synthesis of the tetrasaccharide 33 in 18 steps. Their strategy relied on the preparation of a disaccharide moiety possessing two O-3 positions that were differently protected (OAc and OBn), which once glycosylated with a donor and acceptor with the same O-3 protective groups led to a tetrasaccharide, which allowed alternated installations of the lactyl moiety. This was the first report on the synthesis of a tetrasaccharide with the pentapeptide linked to the two O-3 lactyl groups.
Several PGN fragments, mono-, di-, tetra-, and octasaccharides with the pentapeptide assembled were synthesized by the Fukase group in order to understand how the innate immune response was triggered [62]. The synthesis of these PGN fragments relied on a previous report from the same authors [55], using Troc as the protecting group for the amine moiety and the trichloroacetimidate group at the anomeric position. With these studies, the authors determined that the minimal recognition pattern for hPGRP-L, a human protein capable of binding bacterial PGN fragments, was the NAM unit linked to three amino acids. Furthermore, these authors also used the assembled synthetic PGN fragment to discover that the NAM unit linked to two amino acids (MDP) was recognized by NOD2, a protein that is involved in the activation of an innate immune response in murine cells.
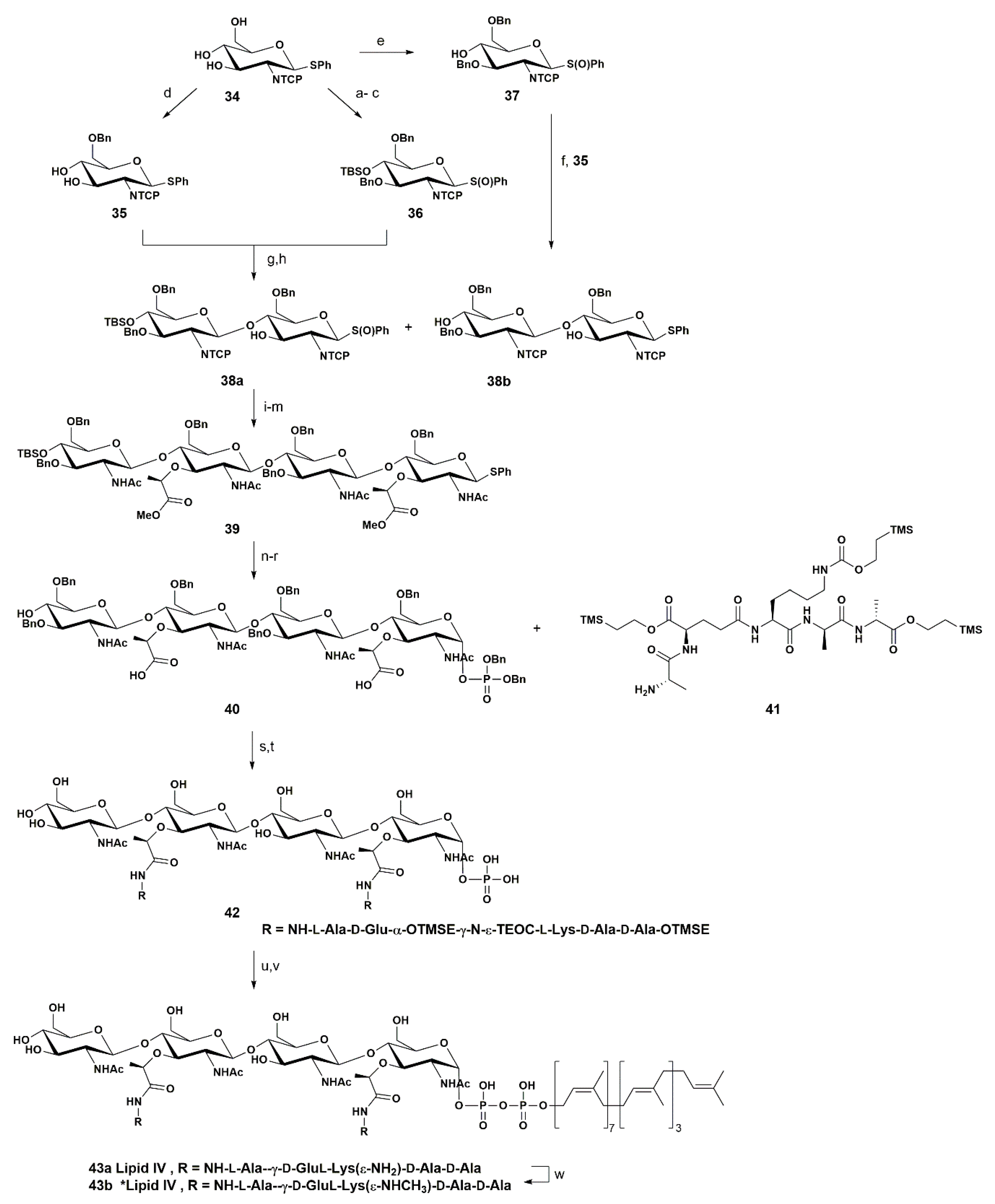
In 2007 Walker and co-workers reported the synthesis of a PGN precursor that may be used by bacterial enzymes that are involved in the assembly of the PGN macromolecule—the heptaprenyl-lipid IV 43b, which consists of a tetrasaccharide (NAG-NAM)4 linked to a diphospholipid at the anomeric position (Scheme 6) [63].
The strategy proposed by these authors relied on the preparation of a tetrasaccharide possessing alternated free hydroxyl groups at the O-3 position that could then be used to install the lactyl unit and the posterior attachment of the peptide chain, while the lipid moiety was introduced using a thioglycoside as donor.
The authors started the synthesis with the common starting material 34 that was converted into monosaccharides 35, 36, and 37—building blocks required to assemble the desired tetrasaccharide. Glucosamine derivative 34 was converted into 35 in three steps. In the first step, they used an O-6 and O-3 benzylation using Bu2SnO and BnBr in 51% yield. The second step involved O-4 silylation by means of treatment with TBSOTf and lutidine in 91% yield. In the third and final step, the authors performed the oxidation of the thioglycoside, with mCPBA affording 35 in 93% yield.
The production of 36 and 37 did not require so many steps. Treatment of 34 with (Bu3Sn)2O, followed by the addition of BnBr/TBAI (O-6 benzylation) led to 36 in 75% yield. The donor 37 was obtained by oxidation of 24 with mCPBA.
Glycosylation of 35 with 36 mediated by Tf2O followed by oxidation with mCPBA led to formation of the disaccharide 38a in 86% yield. N-protection with the tetrachlorophthalimide (TCP) group hindered the O-3 glycosylation reaction, which was expected due to the presence of the free hydroxyl group at O-3, and permitted the formation of a β-1,4 glyosidic bond. The disaccharide 38b was obtained in 58% yield via the inverted addition of 37 to the acceptor 35.
The glycosylation of the two disaccharides (38a and 38b) afforded the desired tetrasaccharide in 77% yield. Next the N-TCP group was replaced by the N-acetyl group via treatment of the tetrasaccharide with hydrazine and subsequent N-acetylation with acetic anhydride in 75% yield (two steps). The generated tetrasaccharide possessed alternated free O-3 positions, which were ready to receive the lactyl moiety. The authors introduced the lactyl moiety using S-(–)-2-bromo-propionic acid, followed by an O-alkylation step, affording 39 in 70% yield.
In order to install the phosphonate moiety at the anomeric position, which yielded compound 40, the compound 39 was subjected to a sequence of five synthetic steps, involving the oxidation of SPh with NIS in 75% yield, treatment with (i-Pr)2NP(OBn)2 and subsequent oxidation with mCPBA in 84% yield (two steps), TBS group removal, and hydrolysis of the ester groups.
The coupling of the silyl-protected pentapeptide 41, which had been previously synthesized using Fmoc chemistry, with tetrasaccharide 40 was performed using standard peptide coupling conditions. This approach led to tetrasaccharide 42, which was obtained after global hydrogenolysis in 44% yield.
Next, the coupling of the heptaprenyl phosphate [64] to the anomeric phosphate present in compound 42 was performed with 1,1′-carbonyl diimidazole in 50% yield. The final steps consisted in the removal of the silyl groups, which resulted in the heptaprenyl—Lipid IV 43a. In order to perform certain enzymatic assays, the authors further converted 43a into 43b by treatment with [14] C-acetic anhydride.
In this report the authors established an orthogonal synthetic plan based on the use of a building block possessing the amino group protected with tetrachlorophthalimide (TCP) and the thiophenol group at the anomeric position. The authors then used the synthesized PGN building blocks, together with a bacterial enzyme, PBP1a, a peptidoglycan glycosyltransferase from E. coli, to elongate the PGN strand. They successfully mimicked the natural process and were able to convert the synthetic tetrasaccharide into octasaccharide in 51% yield.
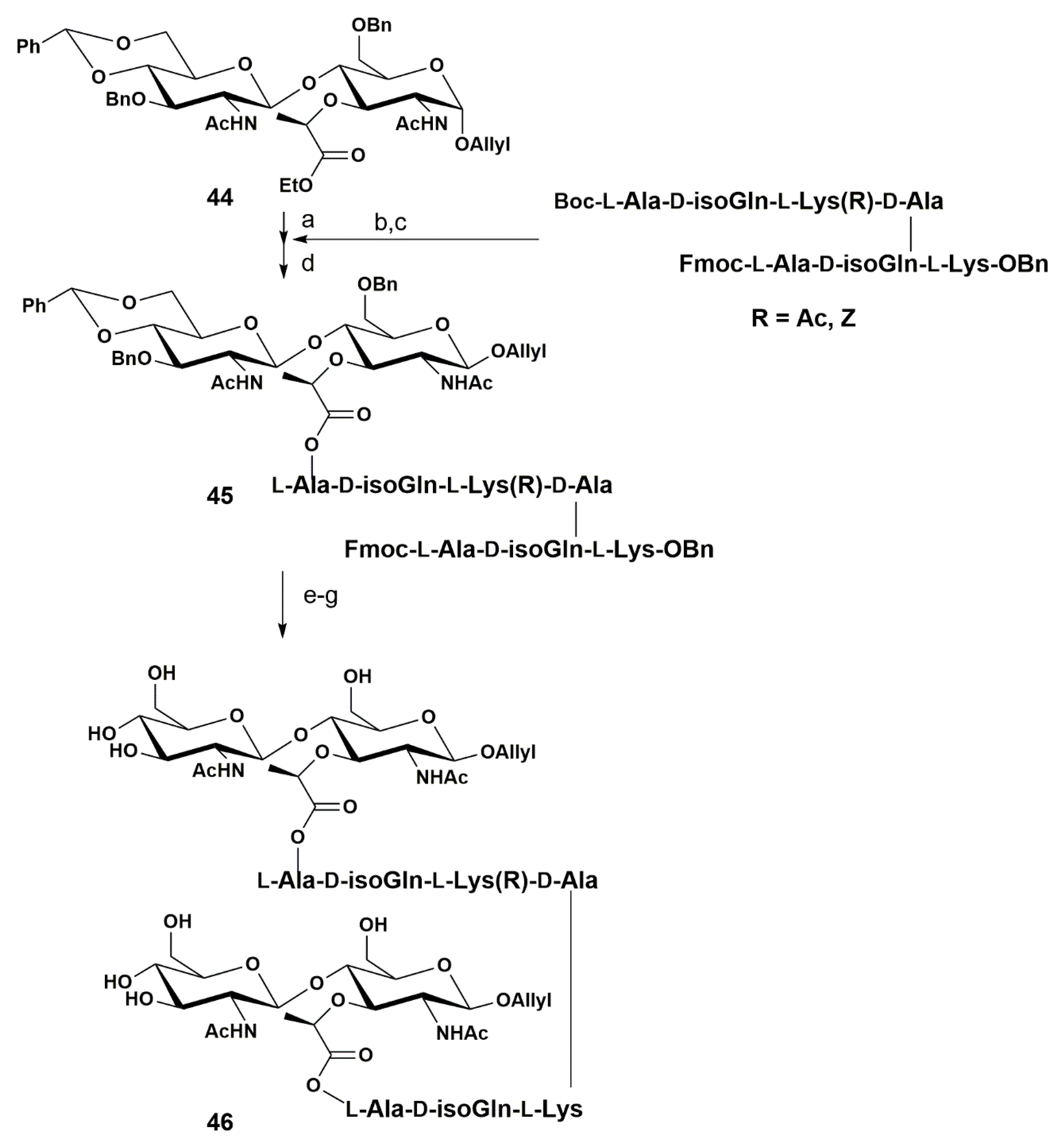
More recently, Fukase and co-workers reported the first synthesis of cross-linked PGN fragments that are frequently found at the cell wall of Streptococcus pneumoniae bacteria [65]. The authors were able to use the disaccharide 44 from previous reports [55,66] as a key intermediate in order to cross-link the peptide chains (Scheme 7).
The ester group from disaccharide 44 was hydrolyzed to the carboxylic acid and the Boc group from the peptide substrate was removed. The peptide was then coupled to the disaccharide using the standard procedure with HATU, affording 45 in 75% yield. The authors further attempted to introduce two glycan chains to the two l-Ala residues of the linked structure. However, the desired compounds were not obtained due to insufficient reactivity of the amino groups.
Next, the benzylidene acetal was removed after treatment with 10% TFA. The authors reported difficulties associated with the removal of the Fmoc group from the peptide chain due to low solubility, but it was accomplished after treatment with 20% piperidine.
The coupling of the second disaccharide moiety was carried out using the same conditions as before, affording the corresponding product in 26–42% yield, depending on the lysine protecting group (Z or Ac). Final hydrogenation removed all the benzyl groups and 46 was afforded in quantitative yield.
The authors investigated the biological activity of all the synthetic PGN fragments and found that the cross-linked PGN fragment is not the major ligand of human NOD2.
This synthetic route developed by Fukase and coworkers established a plan that allows access to longer PGN structures, which might be useful for the investigation in innate immunostimulation of PGN. Indeed, on the basis of this study and PGN fragments, other biological activities can be investigated, as well as the determination of the structural requirements for other types of PGN recognition molecules including peptidoglycan recognition proteins (PGRPs) and PGN-recognizing lectins.
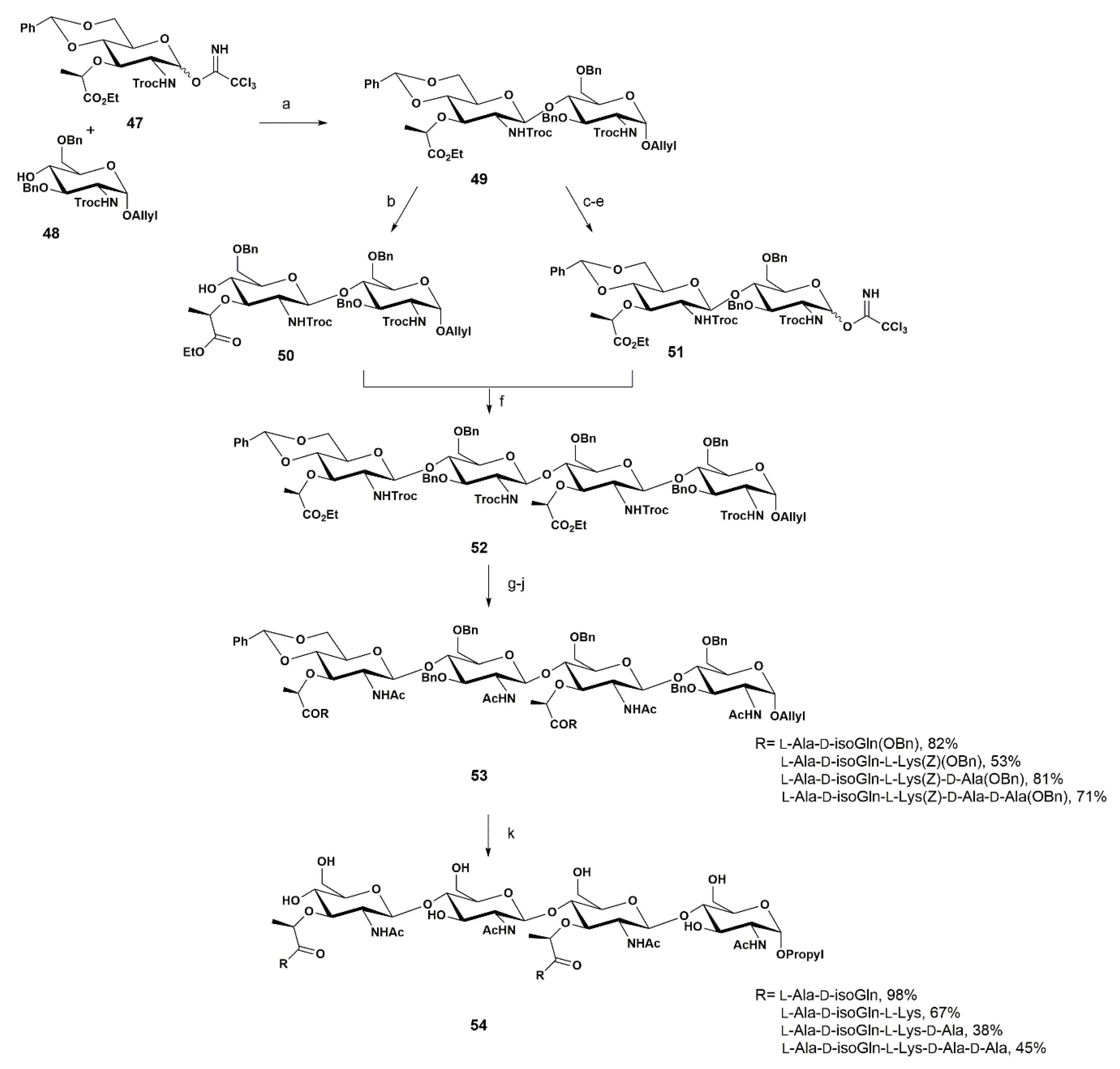
Some years later, the Fukase group reported the synthesis of several disaccharide and tetrasaccharide fragments, which were used to investigate their ability to stimulate human Nod2 (hNod2)-dependent responses [67]. Nod2 can recognize specific fragments of the bacterial cell-wall PGN (the minimum ligand is the muramyl dipeptide (MDP)). Enzymatic digestion of PGN appears to be important for Nod2 recognition. PGN is degraded by muramidase or glucosamidase, producing two types of glycan sequence—glycans containing NAGβ(1-4)NAM or NAMβ(1-4)NAG. Thus, the authors prepared fragments of diverse composition to understand whether the sequence of these PGN fragments could affect an immune response that was dependent on the innate immune receptor Nod2 (Scheme 8).
In their work, the authors applied previously reported synthetic studies [68,69]. Compound 49 was synthesized in 84% yield via the glycosylation reaction of 47 with 48 mediated by TMSOTf as a catalytic promotor. Disaccharide 49 was converted into the acceptor 50 and donor 51, to assemble the desired tetrasaccharide 52. Preparation of acceptor 50 involved the regioselective opening of the arylidene ring to free the O-4 position under standard reductive conditions using Me3N·BH3 in ACN for 1 h, affording 50 in 83% yield. The donor 51 was prepared in three steps from 49, involving different reactions: (1) the isomerization of the allyl double bond, using an Ir complex; (2) the subsequent hydrolysis assisted by iodine, in 81% yield for these two steps; and (3) the final installation of the trichloroacetimidate group at the anomeric position in a quantitative yield.
Tetrasaccharide 52 was obtained after glycosylation using TMSOTf as a promotor in 16% yield. In their work, the authors also described the use of glycosyl N-phenyltrifluoroacetoamidate as a donor, due to its high reactivity and better stability when compared to the corresponding trichloroacetimidates. The formation of byproducts such as the N-glycosyl trifluoroacetamides is suppressed, since the N-phenyl group of the eliminated N-phenyltrifluoroacetamide prevents the undesirable attack of the amide on the cationic intermediates. The authors also performed the glycosylation using a N-phenyltrifluoroacetimidate donor by increasing the equivalent amount of acceptor 50 (donor/acceptor ratio 1:1.5), with an improved yield of 61%, and preserving the selectivity.
Next, the Troc group from tetrasaccharide 52 was removed using a Zn/Cu in AcOH for 3 h, followed by an N-acetylation reaction (acetic anhydride in pyridine for 2 h). The saponification of the ethyl esters with LiOH afforded the corresponding product in a quantitative yield.
The following steps involved the condensation of the tetrasaccharide with a library of peptides with a composition found in the bacterial PGN (i.e., di-, tri-, tetra-, and pentapeptides). Thus, different protocols were used depending on the peptide chain required. After the final hydrogenation, a library of PGN fragments 54 was obtained in yields ranging from 38% to 98%.
Fukase and co-workers demonstrated that hNod2 recognition is dependent on the glycan sequence. The authors compared the activities of glycans with the same peptide moieties, and showed that (MurNAcβ(1-4)GlcNAc)2-containing structures exhibited stronger activity than those containing (GlcNAcβ(1-4)MurNAc)2. These results indicate that differences in the enzymatic degradation process may affect the host’s immunomodulation process.
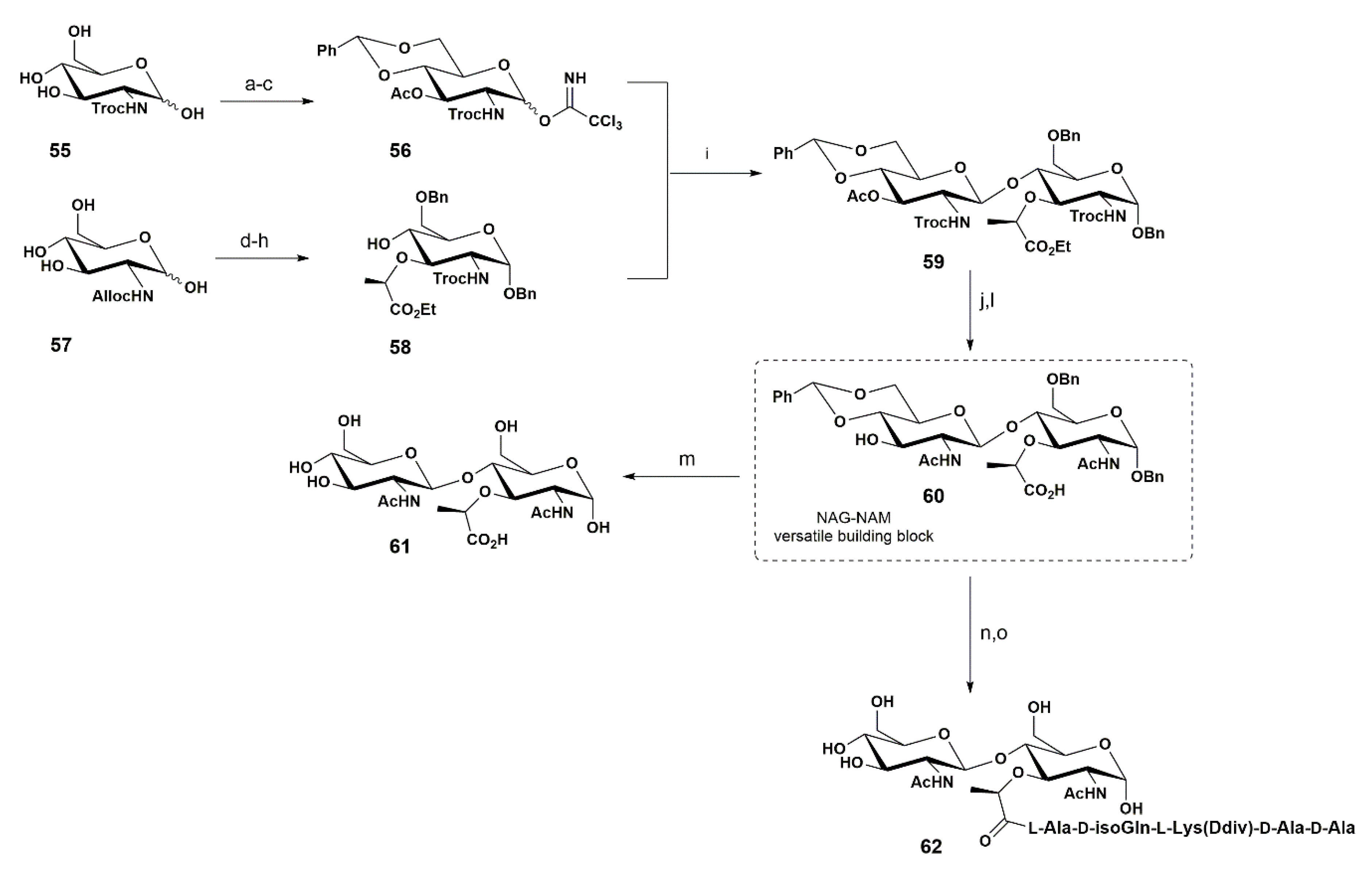
More recently, our group reported the synthesis of a NAGβ-(1-4)-NAM moiety 61, which is crucial for the preparation of synthetic components of a bacterial peptidoglycan (Scheme 9) [70].
In 2016, Fukase and co-workers reported a chemoenzymatic approach to the large-scale synthesis of a meso-DAP type PGN from Mycobacterium [71]. Since meso-DAP has been reported to be the most potent activator of Nod2, there was a need to develop a large-scale synthesis protocol. The use of l-aminocyclase and d-aminocyclase was a key factor in the production of meso-DAP on a large scale in order to proceed with the biological studies.
The proposed route involved the preparation of both donor 56 and acceptor 58 from N-Troc glucosamine 55 and N-Alloc glucosamine 57, respectively. The synthetic sequence relied on standard orthogonal protection of the hydroxyl groups, involving four steps for the preparation of 56: (i) the formation of the O-4,6 arylidene; (ii) an O-3 and O-1 acetylation, via treatment with benzaldehyde dimethyl acetal and ZnCl2, the crude dissolved in pyridine and acetic anhydride, in 78% yield (two steps); (iii) the selective removal of the anomeric acetyl group using morpholine, in 87% yield; and (iv) the final establishment of the desired glycosyl trichloroacetamidate using CCl3CN/Cs2CO3, affording 56 in 74% yield, which was used in the glycosylation reaction without further purification.
The acceptor 58 was prepared in five steps, involving the following reactions: (i) the benzylation of the anomeric position using BnOH and AcCl in 61% yield; (ii) the formation of the 4,6-O arylidene, in 72% yield; (iii) the introduction of the lactyl moiety at O-3 using trifluoromethanesulfonyl-l-(S)-2-propionic acid ethyl ester, in 68% yield; (iv) the removal of the Alloc group by treatment with Pd(PPh3)4. followed by in situ N-protection with TrocCl, affording the corresponding product in 82% yield; and (v) a final selective arylidene ring opening using the classical procedure with Me3N·BH3, affording 58 in 72% yield.
The glycosylation reaction was carried using TMSOTf as a promoter, giving the product 59 with the desired β-1,4 selectivity, in 52% yield. The exchange of both trichloroethyl carbamate groups by the natural N-acetyl group was performed by addition of Zn–Cu in AcOH/Ac2O/THF, followed by N-acetylation, in 63% yield. The key intermediate 60 was obtained after hydrolysis with LiOH, which simultaneously led to the removal of the O-3 acetyl group and lactic acid formation, in 69% yield.
Disaccharide 60 constitutes a versatile intermediate, allowing further regioselective manipulation at O-3 and O-4 (after arylidene ring opening) positions, as well as the insertion of a peptide chain at the lactate moiety.
Accordingly, NAG-NAM disaccharide 61 was obtained in quantitative yield by hydrogenation using Pd(OH)2 on charcoal (removal of the benzyl groups at the O-1 and O-6 positions of the muramyl residue and the benzylidene ring at the O-6 and O-4 positions of the NAG unit).
Disaccharide 60 was also used to prepare the glycopeptide 62 by coupling with a pentapeptide, previously attained via solid-phase synthesis using HMPB-AM resin, followed by cleavage from the resin upon treatment with trifluoroacetic acid solution.
This work was a follow-up to previous approaches reported by us, such as one-pot regioselective protection and orthogonal approaches towards key intermediates to achieve NAG-NAM moieties [14,72]. In this approach, the conjugation of the different protecting groups allowed for a shorter synthetic sequence and a complete removal of protecting groups in a late stage of the synthesis. In particular, the choice of the anomeric benzyl group facilitated the removal of protecting groups in one step. This work represents a useful route to obtain the NAG-NAM disaccharide moiety, as well as non-natural derivatives.
4. Synthetic Approaches towards the Carbohydrate Backbone of PGN from Biopolymers
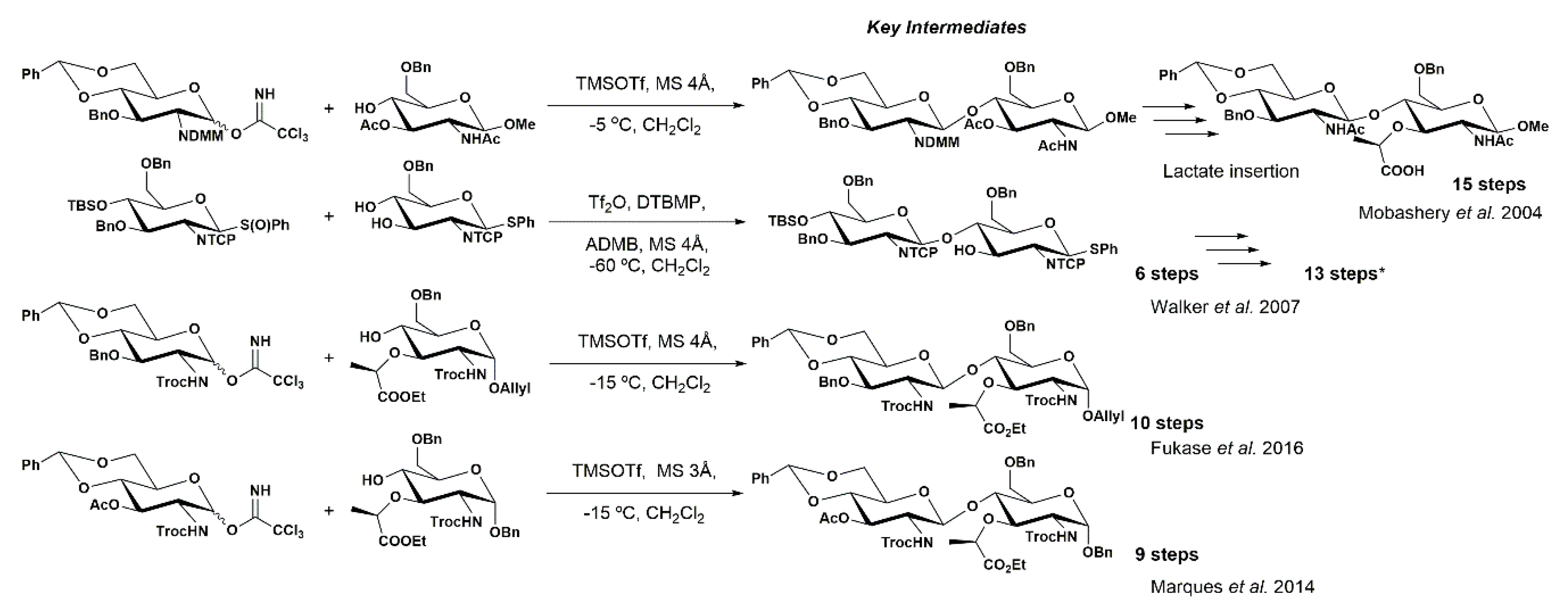
In the previous section, we described the different approaches developed to address the synthesis of the complex PGN structure from a scaffold of residues of NAG derivatives. The main difficulty in the synthesis of PGN fragments involves stereoselective glycosylation involving NAG derivatives [10,73,74]. Approaches to assembling the complex PGN fragments have been described, and include one-pot regioselective protection and orthogonal approaches to achieve key intermediates as precursors of NAG-NAM moieties [12,14,15,16,17,61,63,65,70,71,75,76].
We and others have been focused on establishing straightforward routes to achieve PGN fragments and also on protection–deprotection methodologies of NAG-containing saccharides (Scheme 10) [12,61,63,65,70,71,72,75,76]. All these synthetic methods involve a high number of steps required to achieve a key intermediate that possesses the lactyl moiety installed at the O-3 position of the NAG moiety. These long multistep sequences are a puzzling game of compatible protecting groups, due to the difficulties associated with the establishment of the β-1,4 glyosidic bond of NAG-NAM. The success of a particular synthetic sequence relies on the proper choice of protecting groups, in particular the N-protecting group (e.g., Troc, TCP, MDM) [14,77,78], which enable enantioselective control over glycosylation, as well as regioselective insertion of the lactyl moiety at the O-3 position of a modified and alternated (in the case of oligosaccharides) NAG unit.
During recent years, our group embraced the challenge of synthetizing NAG-NAM containing oligosaccharides and its precursors, using as starting material biopolymers, with the β-1,4 glycosidic bond already installed.
In order to explore unprecedented synthetic studies, in order to develop alternatives to the synthesis of PGN fragments, we were inspired by the particular sugar arrangement of PGN (Figure 1) and its resemblance to other biopolymers found in nature, such as chitin and chitosan. Chitin is one of the most abundant biopolymers in the world and consists of β-(1,4)-N-acetylglucosamine (NAG), sharing the same basic carbohydrate backbone as that of PGN.
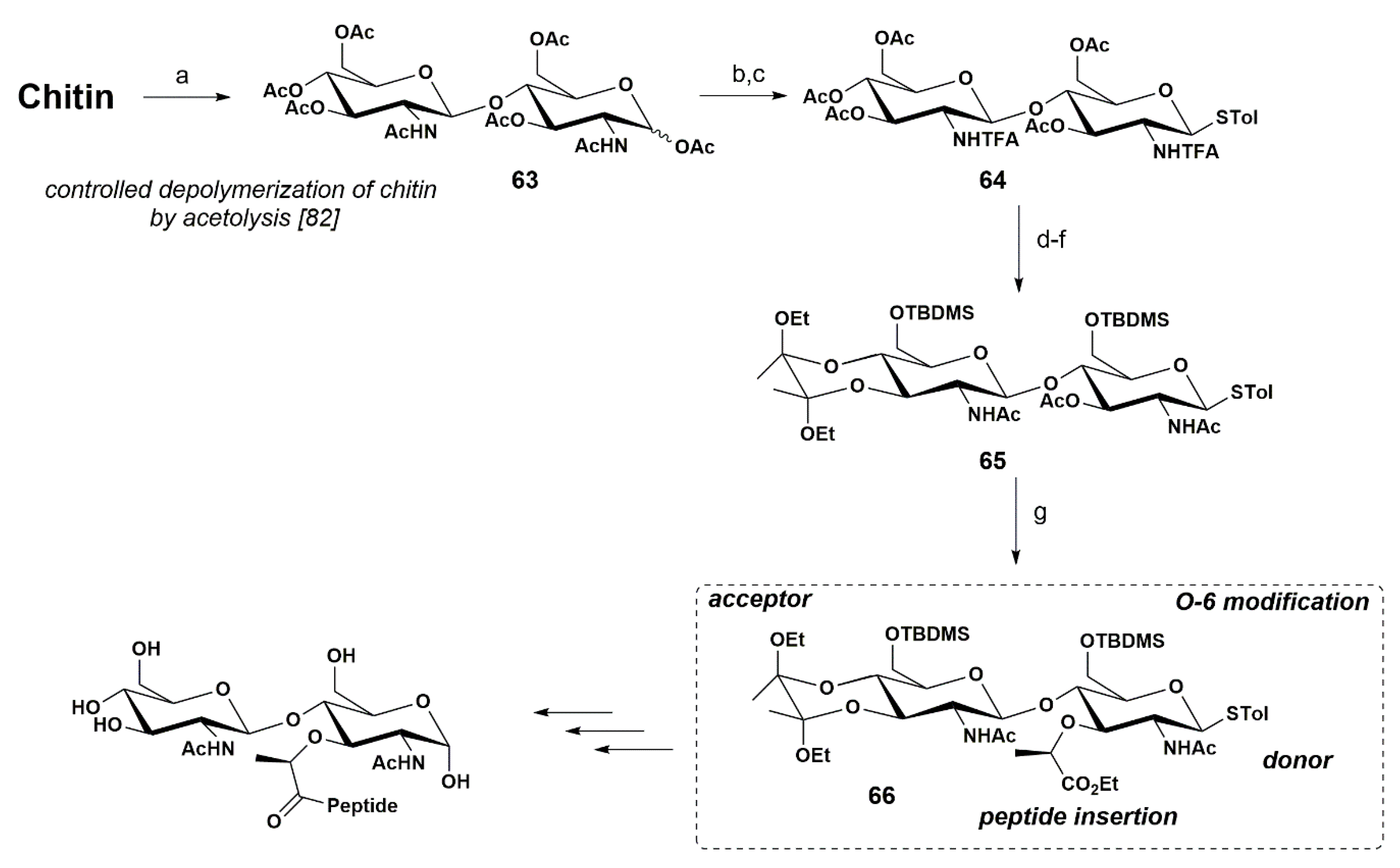
In 2018 we established a procedure to achieve an advanced precursor of the NAG-NAM unit via orthogonal synthesis, from peracetylated chitobiose, obtained after chitin chemical hydrolysis, as shown in Scheme 11 [79].
Peracetylated chitobiose 63 was obtained on a gram scale from the chemical and controlled depolimerization of chitin via acetolysis [80,81], using an improved procedure from that originally described by Beau at al. [82].
We performed chemoselective N-trans-acylation using a reported protocol [82], and converted 63 into the N-trifluoroacetyl derivative (NHTFA). This modification relied on direct and chemoselective N-trans-acylation by the reaction of 63 with trifluoroacetyl anhydride, allowing an increased solubility, while facilitating the following steps. Next, we produced disaccharide 64 by installing the STolyl (STol) group at the anomeric position, using p-methylthiophenol under BF3·OEt2 conditions, in 80% yield. Methanolysis of 64 and successive protection of 3,4-O positions with the butane-2,3-diacetal moiety and protection of O-6 with TBDMS, using TBDMSCl, TEA, and DMAP in DMF for 16 h afforded the corresponding product. This procedure was executed in 56% yield.
In our first attempts, we were not successful when introducing the lactyl moiety directly into the precursor possessing the NTFA group. Thus, to avoid the random introduction of the lactate moiety, the NTFA group was replaced by the NAc group, by treatment with NaOH in THF at 80 °C, followed by N-acetylation with acetic anhydride in pyridine, affording 65 in 56% yield. The lactyl unit was then introduced by treatment of 65 with propionic ester triflate, and 66 was attained in 38% yield. Intermediate 66 possesses all functional groups (O-lactyl and NAc) required to prepare the NAG-NAM disaccharide, constituting an excellent precursor of this moiety.
In our strategy, we took advantage of the already established β-1,4 glycosidic bond present in the easily-obtained peracetylated chitobiose 63, avoiding one of the most challenging steps in the synthesis of PGN fragments. This strategy resulted in a considerable reduction of the number of steps required to obtain a NAG-NAM-containing disaccharide, constituting an atom-economy procedure.
In the follow-up of our synthesis of an advanced NAG-NAM precursor from chitobiose, we next reported the synthesis of NAG-NAM-containing oligosaccharides from chitosan via a top-down approach involving both chemical and enzymatic reactions (Scheme 12) [83]. This new route was able to overcome the difficulties in obtaining sufficient long fragments to replace samples of biological origin, which also constituted a limitation for further biological investigations [9].
Given the natural resemblance between the carbohydrate backbone of the PGN and chitosan, we envisaged that a properly functionalized chitosan derivative would provide a suitable platform for an alternate attachment of the Lac unit. This new polymer, after removal of the protecting groups, could then be recognized as a PGN substrate by enzymes involved in the degradation of bacterial PGN [84,85,86,87], which would afford the production of smaller oligosaccharides containing NAG-NAM units. PGN hydrolases, such as lysozyme or mutanolysin, can efficiently hydrolyze the β-1,4 glycosidic bond in the natural PGN substrate, between NAM and NAG residues [84,85,86,87,88] and in certain conditions and to some extent in chitin and derivatives [89,90].
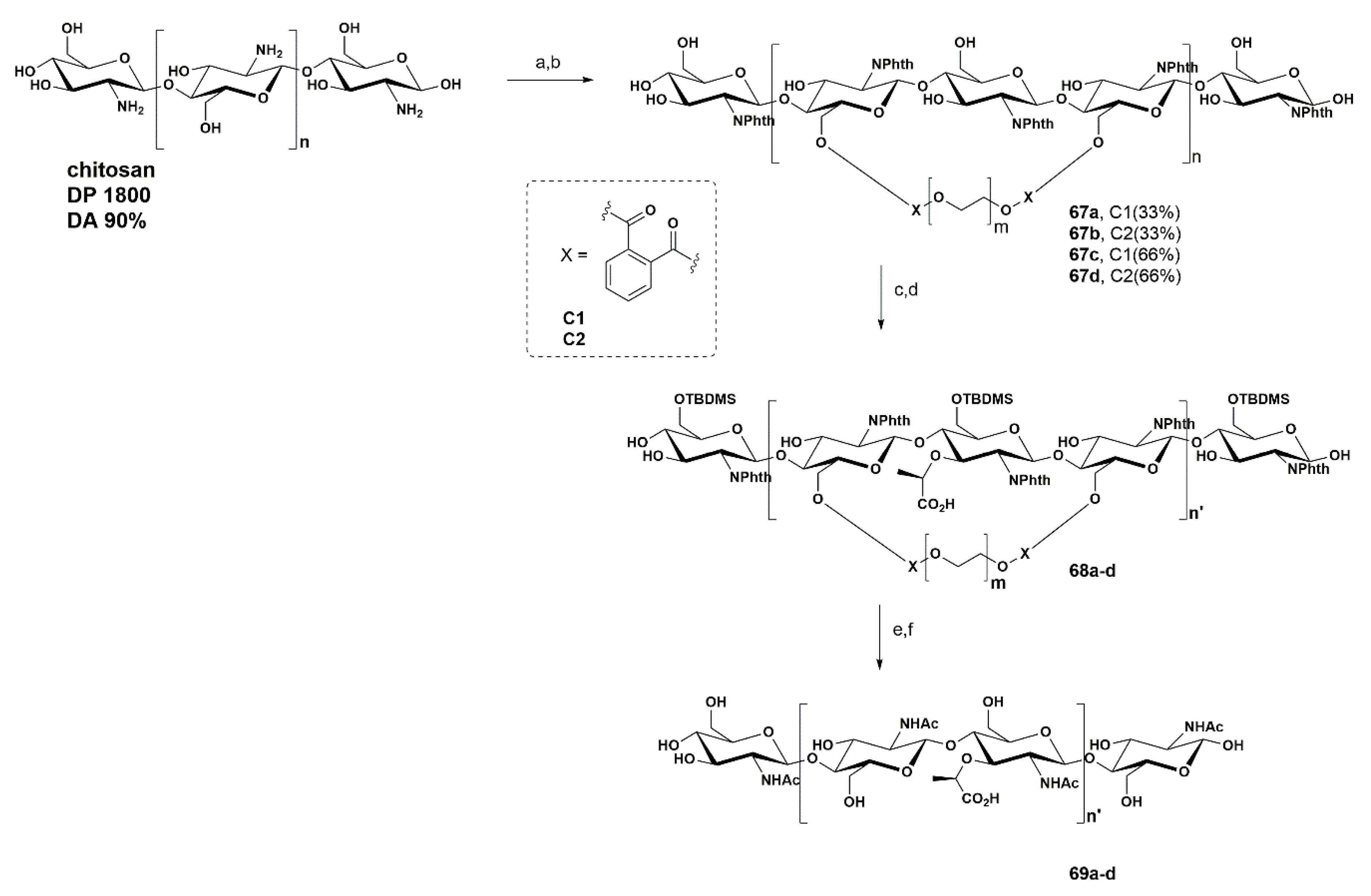
We initiated the synthesis by using the phthaloyl group as the N-protecting group of the commercial chitosan, preventing the problems associated with the poor nucleophilicity of NAG derivatives. In order to promote the attachment of the Lac unit in alternate glucosamine units, we explored the use of a molecular clamp strategy, consisting of the formation of a stable bridge between two 6-OH positions of alternate glucosamine units via an ester linkage. Thus, two clamps were synthesized possessing different lengths C1 (m = 1) and C2 (m = 2). Different amounts of C1 and C2 were tested to maximize the attachment of the Lac moiety in alternate positions. Thus, treatment of the N-phthaloyl chitosan with pre-activated acids C1 or C2 with CDI in DMF led to the formation of compounds 67. In order to prevent the introduction of Lac moieties in NAG residues that had not reacted with clamps C1 and C2, compounds 67 were further silylated at the remaining free 6-OH positions by reaction with TBDMSCl in the presence of imidazole, in DMF. Next, we performed the introduction of the Lac moiety, by treatment with NaH in DMF, using (S)-(−)2-chloropropionic acid under different conditions and equivalents, affording compounds 68a–d. The sequential removal of the phthalimide group by treatment with hydrazine solution and simultaneous removal of the ester clamp, followed by N-acetylation with acetic anhydride and finally silyl group removal, by treatment with a TBAF solution, afforded compounds 69a–d. Due to the polymeric character of the substrates, the structural characterization was performed by FT-IR and 13C CP/MAS NMR (cross polarization magic angle spinning nuclear magnetic resonance).
The ratio of NAM/NAG in samples 69a–d was determined by high performance anion exchange chromatography coupled with pulsed amperometric detection (HPAEC-PAD) and compared with values determined in samples of bacterial PGN purified from Staphylococcus aureus and Escherichia coli (values of 1.2 and 1, respectively, used as positive controls). We found that the NAM/NAG ratio determined in samples 69a–d was dependent on the amounts of C1/C2 and loadings of the (S)-(−)2-chloropropionic acid used in our synthetic procedure. The best results were obtained when a higher loading of C1, together with 2 equiv. of (S)-(−)2-chloropropionic acid, were used. The sample 69 produced in these conditions had a NAM/NAG ratio of 1.21, similar to that found in bacterial PGN purified from Staphylococcus aureus bacteria.
Having confirmed that our synthetic procedures were able to result in a significant incorporation of NAM residues on the chitosan skeleton, indicated by the ratio of NAM/NAG, we next investigated whether alternated NAM units had been installed. This was performed by using PGN lytic enzymes, such as mutanolysin and lysozyme, and by analysis of the fragments released by LC-MS. We found that 55% of the digested 69 generated the disaccharide NAG-NAM in 7% yield, as well as the trisaccharide NAG-NAG-NAM in 33% yield.
This work highlights that the combination of chemical and chemical/enzymatic approaches is an extremely attractive option, compared to traditional orthogonal synthesis, in the synthesis of complex and biological important molecules. Of note, several enzymatic strategies, which use particular purified bacterial enzymes for the production of PGN precursors/fragments, have also been recently developed [91,92,93].
5. Conclusions
PGN is a major constituent of the bacterial cell wall. Although its biosynthesis is assisted by several enzymes that sequentially transform specific sugar precursors and assemble them into the complex structure of the natural PGN, its chemical synthesis poses several problems. Despite all efforts developed by us and others, the synthesis of PGN fragments remains a challenge. The key difficulty is based on stereoselective glycosylation involving NAG derivatives. Indeed, the complex synthesis of the carbohydrate backbone commonly involves multistep sequences to install the lactyl moiety at the O-3 position and to control enantioselective glycosylation.
This review presents the biosynthetic pathways, as well as covering several approaches used to assemble the complex PGN fragments, including one-pot regioselective protection, orthogonal approaches, and the recently reported chemoenzymatic route towards key intermediates to achieve NAG-NAM moieties.
The synthesis of bacterial PGN fragments is crucial in order to better understand the role of this macromolecule in the metabolism of bacteria and their recognition by different hosts. This review collects the approaches, the difficulties imposed by the natural PGN structure, and the discoveries made by the community to address the synthesis of PGN molecules.
Author Contributions
All authors have equally contributed to this review, G.C. and S.R.F. are responsible for the subchapter of the biosynthesis of PGN; and F.Q. and M.M.B.M. contributed for the sections dedicated to the synthetic approaches to fragments of PGN. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors acknowledge the Fundação para a Ciência e Tecnologia (FC&T) for the fellowships SFRH/BD/89518/2012, SFRH/BD/52207/2013 (to FQ and GC) and for the projects (PTDC/QEQ-QOR/2132/2012, PTDC/BIA-MIC/30746/2017 and UID/DTP/04138/2013). This work was supported by the Associate Laboratory for Green Chemistry, LAQV, which is financed by national funds from FCT/MCTES (UID/QUI/50006/2019) and co-financed by the ERDF under the PT2020 Partnership Agreement (POCI-01-0145-FEDER—007265). We also acknowledge the support from Applied Molecular Biosciences Unit-UCIBIO, which is financed by national funds from FCT/MCTES (UID/Multi/04378/2019).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schleifer, K.H.; Kandler, O. Peptidoglycan types of bacterial cell-walls and their taxonomic implications. Bacteriol. Rev. 1972, 36, 407–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtje, J.V. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol. Mol. Biol. Rev. 1998, 62, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roemer, T.; Schneider, T.; Pinho, M.G. Auxiliary factors: A chink in the armor of MRSA resistance to beta-lactam antibiotics. Curr. Opin. Microbiol. 2013, 16, 538–548. [Google Scholar] [CrossRef] [PubMed]
- Courvalin, P. Vancomycin resistance in gram-positive cocci. Clin. Infect. Dis. 2006, 42, S25–S34. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef] [Green Version]
- Vollmer, W.; Joris, B.; Charlier, P.; Foster, S. Bacterial peptidoglycan (murein) hydrolases. Fems Microbiol. Rev. 2008, 32, 259–286. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Uehara, T.; Bernhardt, T.G. Beta-Lactam Antibiotics Induce a Lethal Malfunctioning of the Bacterial Cell Wall Synthesis Machinery. Cell 2014, 159, 1300–1311. [Google Scholar] [CrossRef] [Green Version]
- Catalao, M.J.; Gil, F.; Moniz-Pereira, J.; Sao-Jose, C.; Pimentel, M. Diversity in bacterial lysis systems: Bacteriophages show the way. Fems Microbiol. Rev. 2013, 37, 554–571. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.H.; Weber, A.N.; Atilano, M.L.; Filipe, S.R.; Gay, N.J.; Ligoxygakis, P. Sensing of Gram-positive bacteria in Drosophila: GNBP1 is needed to process and present peptidoglycan to PGRP-SA. Embo J. 2006, 25, 5005–5014. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Yu, B. Recent advances in the synthesis of chitooligosaccharides and congeners. Tetrahedron 2014, 70, 1023–1046. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, T.A.; Yang, Y.L.; Wu, Q.Y.; Yang, Q.; Yu, B.A. Synthesis, Evaluation, and Mechanism of N,N,N-Trimethyl-D-glucosamine-(1 -> 4)-chitooligosaccharides as Selective Inhibitors of Glycosyl Hydrolase Family 20 beta-N-Acetyl-D-hexosaminidases. Chembiochem 2011, 12, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.W.; Chen, K.T.; Cheng, T.J.R.; Wong, C.H.; Cheng, W.C. A New Synthetic Approach toward Bacterial Transglycosylase Substrates, Lipid II and Lipid IV. Org. Lett. 2011, 13, 4600–4603. [Google Scholar] [CrossRef] [PubMed]
- Berthelot, N.; Brossay, A.; Gasciolli, V.; Bono, J.J.; Baron, A.; Beau, J.M.; Urban, D.; Boyer, F.D.; Vauzeilles, B. Synthesis of lipo-chitooligosaccharide analogues and their interaction with LYR3, a high affinity binding protein for Nod factors and Myc-LCOs. Org. Biomol. Chem. 2017, 15, 7802–7812. [Google Scholar] [CrossRef] [PubMed]
- Enugala, R.; Carvalho, L.C.R.; Pires, M.J.D.; Marques, M.M.B. Stereoselective Glycosylation of Glucosamine: The Role of the N-Protecting Group. Chem. Asian J. 2012, 7, 2482–2501. [Google Scholar] [CrossRef] [PubMed]
- Bongat, A.F.G.; Demchenko, A.V. Recent trends in the synthesis of O-glycosides of 2-amino-2-deoxysugars. Carbohydr. Res. 2007, 342, 374–406. [Google Scholar] [CrossRef] [PubMed]
- Arihara, R.; Nakamura, S.; Hashimoto, S. Direct and stereoselective synthesis of 2-acetamido-2-deoxy-beta-D-glycopyranosides by using the phosphite method. Angew. Chem. Int. Ed. 2005, 44, 2245–2249. [Google Scholar] [CrossRef] [PubMed]
- Stevenin, A.; Boyer, F.D.; Beau, J.M. Highly Selective Formation of beta-Glycosides of N-Acetylglucosamine Using Catalytic Iron(III) Triflate. Eur. J. Org. Chem. 2012, 2012, 1699–1702. [Google Scholar] [CrossRef]
- Crich, D.; Dudkin, V. Why are the hydroxy groups of partially protected N-acetylglucosamine derivatives such poor glycosyl accepters, and what can be done about it? A comparative study of the reactivity of N-acetyl-, N-phthalimido-, and 2-azido-2-deoxy-glucosamine derivatives in glycosylation. 2-picolinyl ethers as reactivity-enhancing replacements for benzyl ethers. J. Am. Chem. Soc. 2001, 123, 6819–6825. [Google Scholar] [CrossRef]
- Badet, B.; Vermoote, P.; Legoffic, F. Glucosamine synthetase from escherichia-coli kinetic mechanism and inhibition by n3-fumaroyl-l-2,3-diaminopropionic derivatives. Biochemistry 1988, 27, 2282–2287. [Google Scholar] [CrossRef]
- Badetdenisot, M.A.; Badet, B. Chemical modification of glucosamine-6-phosphate synthase by diethyl pyrocarbonate—Evidence of histidine requirement for enzymatic-activity. Arch. Biochem. Biophys. 1992, 292, 475–478. [Google Scholar] [CrossRef]
- Gehring, A.M.; Lees, W.J.; Mindiola, D.J.; Walsh, C.T.; Brown, E.D. Acetyltransfer precedes uridylyltransfer in the formation of UDP-N-acetylglucosamine in separable active sites of the bifunctional GlmU protein of Escherichia coli. Biochemistry 1996, 35, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Jolly, L.; Ferrari, P.; Blanot, D.; van Heijenoort, J.; Fassy, F.; Mengin-Lecreulx, D. Reaction mechanism of phosphoglucosamine mutase from Escherichia coli. Eur. J. Biochem. 1999, 262, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Menginlecreulx, D.; Vanheijenoort, J. Copurification of glucosamine-1-phosphate acetyltransferase and n-acetylglucosamine-1-phosphate uridyltransferase activities of escherichia-coli-characterization of the glmu gene-product as a bifunctional enzyme catalyzing 2 subsequent steps in the pathway for udp-n-acetylglucosamine synthesis. J. Bacteriol. 1994, 176, 5788–5795. [Google Scholar] [CrossRef] [Green Version]
- MenginLecreulx, D.; vanHeijenoort, J. Characterization of the essential gene glmM encoding phosphoglucosamine mutase in Escherichia coli. J. Biol. Chem. 1996, 271, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Benson, T.E.; Marquardt, J.L.; Marquardt, A.C.; Etzkorn, F.A.; Walsh, C.T. Overexpression, purification, and mechanistic study of udp-n-acetylenolpyruvylglucosamine reductase. Biochemistry 1993, 32, 2024–2030. [Google Scholar] [CrossRef] [PubMed]
- Benson, T.E.; Walsh, C.T.; Massey, V. Kinetic characterization of wild-type and S229A mutant MurB: Evidence for the role of ser 229 as a general acid. Biochemistry 1997, 36, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Krekel, F.; Samland, A.K.; Macheroux, P.; Amrhein, N.; Evans, J.N.S. Determination of the pK(a) value of C115 in MurA (UDP-N-acetylglucosamine enolpyruvyltransferase) from Enterobacter cloacae. Biochemistry 2000, 39, 12671–12677. [Google Scholar] [CrossRef]
- Doublet, P.; Vanheijenoort, J.; Bohin, J.P.; Menginlecreulx, D. The muri gene of escherichia-coli is an essential gene that encodes a glutamate racemase activity. J. Bacteriol. 1993, 175, 2970–2979. [Google Scholar] [CrossRef] [Green Version]
- Doublet, P.; Vanheijenoort, J.; Menginlecreulx, D. The glutamate racemase activity from escherichia-coli is regulated by peptidoglycan precursor udp-n-acetylmuramoyl-l-alanine. Biochemistry 1994, 33, 5285–5290. [Google Scholar] [CrossRef]
- Scaletti, E.R.; Luckner, S.R.; Krause, K.L. Structural features and kinetic characterization of alanine racemase from Staphylococcus aureus (Mu50). Acta Crystallogr. Sect. D Struct. Biol. 2012, 68, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Kouidmi, I.; Levesque, R.C.; Paradis-Bleau, C. The biology of Mur ligases as an antibacterial target. Mol. Microbiol. 2014, 94, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Ruane, K.M.; Lloyd, A.J.; Fulop, V.; Dowson, C.G.; Barreteau, H.; Boniface, A.; Dementin, S.; Blanot, D.; Mengin-Lecreulx, D.; Gobec, S.; et al. Specificity Determinants for Lysine Incorporation in Staphylococcus aureus Peptidoglycan as Revealed by the Structure of a MurE Enzyme Ternary Complex. J. Biol. Chem. 2013, 288, 33439–33448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertrand, J.A.; Auger, G.; Martin, L.; Fanchon, E.; Blanot, D.; La Beller, D.; van Heijenoort, J.; Dideberg, O. Determination of the MurD mechanism through crystallographic analysis of enzyme complexes. J. Mol. Biol. 1999, 289, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Emanuele, J.J.; Jin, H.Y.; Jacobson, B.L.; Chang, C.Y.Y.; Einspahr, H.M.; Villafranca, J.J. Kinetic and crystallographic studies of Escherichia coli UDP-N-acetylmuramate: L-alanine ligase. Protein Sci. 1996, 5, 2566–2574. [Google Scholar] [CrossRef] [Green Version]
- Zawadzke, L.E.; Bugg, T.D.H.; Walsh, C.T. Existence of 2 d-alanine-d-alanine ligases in escherichia-coli-cloning and sequencing of the ddla gene and purification and characterization of the ddla and ddlb enzymes. Biochemistry 1991, 30, 1673–1682. [Google Scholar] [CrossRef]
- Anderson, M.S.; Eveland, S.S.; Onishi, H.R.; Pompliano, D.L. Kinetic mechanism of the Escherichia coli UDPMurNAc-tripeptide D-alanyl-D-alanine-adding enzyme: Use of a glutathione S-transferase fusion. Biochemistry 1996, 35, 16264–16269. [Google Scholar] [CrossRef]
- Albar, O.A.M.; Oconnor, C.D.; Giles, I.G.; Akhtar, M. D-alanine-d-alanine ligase of escherichia-coli-expression, purification and inhibitory studies on the cloned enzyme. Biochem. J. 1992, 282, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Al-Dabbagh, B.; Henry, X.; El Ghachi, M.; Auger, G.; Blanot, D.; Parquet, C.; Mengin-Lecreulx, D.; Bouhss, A. Active site mapping of MraY, a member of the polyprenyl-phosphate N-acetylhexosamine 1-phosphate transferase superfamily, catalyzing the first membrane step of peptidoglycan biosynthesis. Biochemistry 2008, 47, 8919–8928. [Google Scholar] [CrossRef]
- Chung, B.C.; Zhao, J.S.; Gillespie, R.A.; Kwon, D.Y.; Guan, Z.Q.; Hong, J.Y.; Zhou, P.; Lee, S.Y. Crystal Structure of MraY, an Essential Membrane Enzyme for Bacterial Cell Wall Synthesis. Science 2013, 341, 1012–1016. [Google Scholar] [CrossRef] [Green Version]
- Bouhss, A.; Crouvoisier, M.; Blanot, D.; Mengin-Lecreulx, D. Purification and characterization of the bacterial MraY translocase catalyzing the first membrane step of peptidoglycan biosynthesis. J. Biol. Chem. 2004, 279, 29974–29980. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.S.; Matsuhashi, M.; Haskin, M.A.; Strominger, J.L. Lipid-phosphoacetylmuramyl-pentapeptide and lipid-phosphodisaccharide-pentapeptide–presumed membrane transport intermediates in cell wall synthesis. Proc. Natl. Acad. Sci. USA 1965, 53, 881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Heijenoort, J. Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol. Mol. Biol. Rev. 2007, 71, 620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menginlecreulx, D.; Texier, L.; Rousseau, M.; Vanheijenoort, J. The murg gene of escherichia-coli codes for the udp-n-acetylglucosamine-n-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol n-acetylglucosamine transferase involved in the membrane steps of peptidoglycan synthesis. J. Bacteriol. 1991, 173, 4625–4636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadi, T.; van Dam, V.; Sijbrandi, R.; Vernet, T.; Zapun, A.; Bouhss, A.; Diepeveen-de Bruin, M.; Nguyen-Disteche, M.; de Kruijff, B.; Breukink, E. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. Embo J. 2011, 30, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, N. Bioinformatics identification of MurJ (MviN) as the peptidoglycan lipid II flippase in Escherichia coli. Proc. Natl. Acad. Sci. USA 2008, 105, 15553–15557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sham, L.T.; Butler, E.K.; Lebar, M.D.; Kahne, D.; Bernhardt, T.G.; Ruiz, N. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science 2014, 345, 220–222. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, N. Lipid Flippases for Bacterial Peptidoglycan Biosynthesis. Lipid Insights 2015, 8, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Teo, A.C.K.; Roper, D.I. Core Steps of Membrane-Bound Peptidoglycan Biosynthesis: Recent Advances, Insight and Opportunities. Antibiot. Basel 2015, 4, 495–520. [Google Scholar] [CrossRef]
- Meeske, A.J.; Sham, L.T.; Kimsey, H.; Koo, B.M.; Gross, C.A.; Bernhardt, T.G.; Rudner, D.Z. MurJ and a novel lipid II flippase are required for cell wall biogenesis in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2015, 112, 6437–6442. [Google Scholar] [CrossRef] [Green Version]
- Egan, A.J.F.; Errington, J.; Vollmer, W. Regulation of peptidoglycan synthesis and remodelling. Nat. Rev. Microbiol. 2020, 18, 446–460. [Google Scholar] [CrossRef]
- Manat, G.; Roure, S.; Auger, R.; Bouhss, A.; Barreteau, H.; Mengin-Lecreulx, D.; Touzé, T. Deciphering the metabolism of undecaprenyl-phosphate: The bacterial cell-wall unit carrier at the membrane frontier. Microb. Drug Resist. 2014, 20, 199–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebar, M.D.; May, J.M.; Meeske, A.J.; Leiman, S.A.; Lupoli, T.J.; Tsukamoto, H.; Losick, R.; Rudner, D.Z.; Walker, S.; Kahne, D. Reconstitution of Peptidoglycan Cross-Linking Leads to Improved Fluorescent Probes of Cell Wall Synthesis. J. Am. Chem. Soc. 2014, 136, 10874–10877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travassos, L.H.; Girardin, S.E.; Philpott, D.J.; Blanot, D.; Nahori, M.A.; Werts, C.; Boneca, I.G. Toll-like receptor 2-dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep. 2004, 5, 1000–1006. [Google Scholar] [CrossRef] [Green Version]
- Wolf, A.J.; Underhill, D.M. Peptidoglycan recognition by the innate immune system. Nat. Rev. Immunol. 2018, 18, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Inamura, S.; Fukase, K.; Kusumoto, S. Synthetic study of peptidoglycan partial structures. Synthesis of tetrasaccharide and octasaccharide fragments. Tetrahedron Lett. 2001, 42, 7613–7616. [Google Scholar] [CrossRef]
- Kusumoto, S.; Yamamoto, K.; Imoto, M.; Inage, M.; Tsujimoto, M.; Kotani, S.; Shiba, T. Chemical synthesis and biological-activities of 2 disaccharide dipeptides corresponding to the repeating units of bacterial peptidoglycan. Bull. Chem. Soc. Jpn. 1986, 59, 1411–1417. [Google Scholar] [CrossRef]
- Schwartz, B.; Markwalder, J.A.; Wang, Y. Lipid II: Total synthesis of the bacterial cell wall precursor and utilization as a substrate for glycosyltransfer and transpeptidation by penicillin binding protein (PBP) 1b of Eschericia coli. J. Am. Chem. Soc. 2001, 123, 11638–11643. [Google Scholar] [CrossRef]
- Saha, S.L.; Van Nieuwenhze, M.S.; Hornback, W.J.; Aikins, J.A.; Blaszczak, L.C. Synthesis of an orthogonally protected precursor to the glycan repeating unit of the bacterial cell wall. Org. Lett. 2001, 3, 3575–3577. [Google Scholar] [CrossRef]
- VanNieuwenhze, M.S.; Mauldin, S.C.; Zia-Ebrahimi, M.; Winger, B.E.; Hornback, W.J.; Saha, S.L.; Aikins, J.A.; Blaszczak, L.C. The first total synthesis of lipid II: The final monomeric intermediate in bacterial cell wall biosynthesis. J. Am. Chem. Soc. 2002, 124, 3656–3660. [Google Scholar] [CrossRef]
- Chowdhury, A.R.; Siriwardena, A.; Boons, G.J. A highly convergent approach for the synthesis of disaccharide repeating units of peptidoglycan. Tetrahedron Lett. 2002, 43, 7805–7807. [Google Scholar] [CrossRef]
- Hesek, D.; Lee, M.J.; Morio, K.I.; Mobashery, S. Synthesis of a fragment of bacterial cell wall. J. Org. Chem. 2004, 69, 2137–2146. [Google Scholar] [CrossRef] [PubMed]
- Inamura, S.; Fujimoto, Y.; Kawasaki, A.; Shiokawa, Z.; Woelk, E.; Heine, H.; Lindner, B.; Inohara, N.; Kusumoto, S.; Fukase, K. Synthesis of peptidoglycan fragments and evaluation of their biological activity. Org. Biomol. Chem. 2006, 4, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fechter, E.J.; Wang, T.S.A.; Barrett, D.; Walker, S.; Kahne, D.E. Synthesis of heptaprenyl-lipid IV to analyze peptidoglycan glycosyltransferases. J. Am. Chem. Soc. 2007, 129, 3080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.Y.; Lo, M.C.; Brunner, L.; Walker, D.; Kahne, D.; Walker, S. Better substrates for bacterial transglycosylases. J. Am. Chem. Soc. 2001, 123, 3155–3156. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Konishi, Y.; Kubo, O.; Hasegawa, M.; Inohara, N.; Fukase, K. Synthesis of crosslinked peptidoglycan fragments for investigation of their immunobiological functions. Tetrahedron Lett. 2009, 50, 3631–3634. [Google Scholar] [CrossRef]
- Uehara, A.; Yang, S.; Fujimoto, Y.; Fukase, K.; Kusumoto, S.; Shibata, K.; Sugawara, S.; Takada, H. Muramyldipeptide and diaminopimelic acid-containing desmuramylpeptides in combination with chemically synthesized Toll-like receptor agonists synergistically induced production of interleukin-8 in a NOD2- and NOD1-dependent manner, respectively, in human monocytic cells in culture. Cell. Microbiol. 2005, 7, 53–61. [Google Scholar] [CrossRef]
- Wang, N.; Huang, C.; Hasegawa, M.; Inohara, N.; Fujimoto, Y.; Fukase, K. Glycan Sequence-Dependent Nod2 Activation Investigated by Using a Chemically Synthesized Bacterial Peptidoglycan Fragment Library. Chembiochem 2013, 14, 482–488. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Pradipta, A.R.; Inohara, N.; Fukase, K. Peptidoglycan as Nod1 ligand; fragment structures in the environment, chemical synthesis, and their innate immunostimulation. Nat. Prod. Rep. 2012, 29, 568–579. [Google Scholar] [CrossRef]
- Fujimoto, Y.; Inamura, S.; Kawasaki, A.; Shiokawa, Z.; Shimoyama, A.; Hashimoto, T.; Kusumoto, S.; Fukase, K. Chemical synthesis of peptidoglycan fragments for elucidation of the immunostimulating mechanism. J. Endotoxin Res. 2007, 13, 189–196. [Google Scholar] [CrossRef]
- Enugala, R.; Pires, M.J.D.; Marques, M.M.B. Synthesis of the NAG-NAM disaccharide via a versatile intermediate. Carbohydr. Res. 2014, 384, 112–118. [Google Scholar] [CrossRef]
- Wang, Q.Q.; Matsuo, Y.; Pradipta, A.R.; Inohara, N.; Fujimoto, Y.; Fukase, K. Synthesis of characteristic Mycobacterium peptidoglycan (PGN) fragments utilizing with chemoenzymatic preparation of meso-diaminopimelic acid (DAP), and their modulation of innate immune responses. Org. Biomol. Chem. 2016, 14, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Enugala, R.; Marques, M.M.B. Synthesis of a 3-hydroxyl-free N-acetyl glucosamine disaccharide. Arkivoc 2012, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Marqvorsen, M.H.S.; Pedersen, M.J.; Rasmussen, M.R.; Kristensen, S.K.; Dahl-Lassen, R.; Jensen, H.H. Why Is Direct Glycosylation with N-Acetylglucosamine Donors Such a Poor Reaction and What Can Be Done about It? J. Org. Chem. 2017, 82, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Xolin, A.; Stevenin, A.; Pucheault, M.; Norsikian, S.; Boyer, F.D.; Beau, J.M. Glycosylation with N-acetyl glycosamine donors using catalytic iron(III) triflate: From microwave batch chemistry to a scalable continuous-flow process. Org. Chem. Front. 2014, 1, 992–1000. [Google Scholar] [CrossRef]
- Enugala, R.; Carvalho, L.C.R.; Marques, M.M.B. Towards Glucosamine Building Blocks: Regioselective One-Pot Protection and Deallylation Procedures. Synlett 2010, 2711–2716. [Google Scholar] [CrossRef]
- Carvalho, L.R.; Corvo, M.C.; Enugala, R.; Marques, M.M.B.; Cabrita, E.J. Application of HR-MAS NMR in the solid-phase synthesis of a glycopeptide using Sieber amide resin. Magn. Reson. Chem. 2010, 48, 323–330. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Wang, C.C.; Sabbavarapu, N.M.; Podilapu, A.R.; Liao, P.H.; Hung, S.C. “One-Pot” Protection, Glycosylation, and Protection-Glycosylation Strategies of Carbohydrates. Chem. Rev. 2018, 118, 8025–8104. [Google Scholar] [CrossRef]
- Ito, Y.; Kanie, O.; Ogawa, T. Orthogonal glycosylation strategy for rapid assembly of oligosaccharides on a polymer support. Angew. Chem. Int. Ed. Engl. 1996, 35, 2510–2512. [Google Scholar] [CrossRef]
- Carvalho, L.C.R.; Queda, F.; Almeida, C.V.; Filipe, S.R.; Marques, M.M.B. From a Natural Polymer to Relevant NAG-NAM Precursors. Asian J. Org. Chem. 2018, 7, 2544–2551. [Google Scholar] [CrossRef]
- Nishimura, S.I.; Kuzuhara, H.; Takiguchi, Y.; Shimahara, K. Peracetylated chitobiose-preparation by specific degradations of chitin, and chemical manipulations. Carbohydr. Res. 1989, 194, 223–231. [Google Scholar] [CrossRef]
- Chang, R.; Yeager, A.R.; Finney, N.S. Probing the mechanism of a fungal glycosyltransferase essential for cell wall biosynthesis. UDP-chitobiose is not a substrate for chitin synthase. Org. Biomol. Chem. 2003, 1, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Despras, G.; Alix, A.; Urban, D.; Vauzeilles, B.; Beau, J.M. From Chitin to Bioactive Chitooligosaccharides and Conjugates: Access to Lipochitooligosaccharides and the TMG-chitotriomycin. Angew. Chem. Int. Ed. 2014, 53, 11912–11916. [Google Scholar] [CrossRef] [PubMed]
- Queda, F.; Covas, G.; Silva, T.; Santos, C.A.; Bronze, M.R.; Canada, F.J.; Corvo, M.C.; Filipe, S.R.; Marques, M.M.B. A top-down chemo-enzymatic approach towards N-acetylglucosamine-N-acetylmuramic oligosaccharides: Chitosan as a reliable template. Carbohydr. Polym. 2019, 224. [Google Scholar] [CrossRef] [PubMed]
- Goodman, H.; Pollock, J.J.; Iacono, V.J.; Wong, W.; Shockman, G.D. Peptidoglycan loss during hen egg-white lysozyme-inorganic salt lysis of streptococcus-mutans. J. Bacteriol. 1981, 146, 755–763. [Google Scholar] [CrossRef] [Green Version]
- Kurita, K.; Ikeda, H.; Yoshida, Y.; Shimojoh, M.; Harata, M. Chemoselective protection of the amino groups of chitosan by controlled phthaloylation: Facile preparation of a precursor useful for chemical modifications. Biomacromolecules 2002, 3, 1–4. [Google Scholar] [CrossRef]
- Binette, A.; Gagnon, J. Regioselective silylation of N-phthaloylchitosan with TBDMS and TBDPS groups. Biomacromolecules 2007, 8, 1812–1815. [Google Scholar] [CrossRef]
- Lee, M.; Artola-Recolons, C.; Carrasco-Lopez, C.; Martinez-Caballero, S.; Hesek, D.; Spink, E.; Lastochkin, E.; Zhang, W.L.; Hellman, L.M.; Boggess, B.; et al. Cell-Wall Remodeling by the Zinc-Protease AmpDh3 from Pseudomonas aeruginosa. J. Am. Chem. Soc. 2013, 135, 12604–12607. [Google Scholar] [CrossRef] [Green Version]
- Vocadlo, D.J.; Davies, G.J.; Laine, R.; Withers, S.G. Catalysis by hen egg-white lysozyme proceeds via a covalent intermediate. Nature 2001, 412, 835–838. [Google Scholar] [CrossRef]
- Maeda, R.; Matsumoto, M.; Kondo, K. Enzymatic hydrolysis reaction of water-soluble chitin derivatives with egg white lysozyme. J. Ferment. Bioeng. 1997, 84, 478–479. [Google Scholar] [CrossRef]
- Amano, K.I.; Ito, E. Action of lysozyme on partially deacetylated chitin. Eur. J. Biochem. 1978, 85, 97–104. [Google Scholar] [CrossRef]
- Raymond, J.B.; Price, N.P.; Pavelka, M.S. A method for the enzymatic synthesis and HPLC purification of the peptidoglycan precursor UDP-N-acetylmuramic acid. Fems Microbiol. Lett. 2003, 229, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.Y.; Huang, S.H.; Chang, Y.C.; Cheng, W.C.; Cheng, T.J.R.; Wong, C.H. Enzymatic Synthesis of Lipid II and Analogues. Angew. Chem. Int. Ed. 2014, 53, 8060–8065. [Google Scholar] [CrossRef] [PubMed]
- Unsleber, S.; Borisova, M.; Mayer, C. Enzymatic synthesis and semi-preparative isolation of N-acetylmuramic acid 6-phosphate. Carbohydr. Res. 2017, 445, 98–103. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Representation of the monomeric muropeptides frequently found in the peptidoglycan (PGN) of different bacteria. (a) the structure of the Staphylococcus aureus PGN (a Lys-type PGN) unit is presented. N-acetylglucosamine (NAG)–N-acetylmuramic (NAM) disaccharide units (red) are linked to each other via NAG-(β-1,4)–NAM linkage. Stem peptides (black), of which the composition is specific for each bacteria species, are attached to the d-lactyl (Lac – highlighted in the green circle) moiety of each NAM. The sequence of the peptide stem attached to the lactyl carboxylate of NAM is l-Ala-d-iGln-l-Lys-d-Ala-d-Ala. (b) the structure of a muropeptide of Gram-positive Mycobacterium spp., with an L-Ala-D-iGln-meso-DAP-d-Ala-d-Ala stem peptide sequence. (c) a representative muropeptide of the Gram-negative bacterium Escherichia coli.
Figure 1.
Representation of the monomeric muropeptides frequently found in the peptidoglycan (PGN) of different bacteria. (a) the structure of the Staphylococcus aureus PGN (a Lys-type PGN) unit is presented. N-acetylglucosamine (NAG)–N-acetylmuramic (NAM) disaccharide units (red) are linked to each other via NAG-(β-1,4)–NAM linkage. Stem peptides (black), of which the composition is specific for each bacteria species, are attached to the d-lactyl (Lac – highlighted in the green circle) moiety of each NAM. The sequence of the peptide stem attached to the lactyl carboxylate of NAM is l-Ala-d-iGln-l-Lys-d-Ala-d-Ala. (b) the structure of a muropeptide of Gram-positive Mycobacterium spp., with an L-Ala-D-iGln-meso-DAP-d-Ala-d-Ala stem peptide sequence. (c) a representative muropeptide of the Gram-negative bacterium Escherichia coli.

Figure 2.
Schematic representation of PGN biosynthesis in Gram-negative bacteria: (1) synthesis of a PG precursor in the cytoplasm; (2) synthesis of lipid-linked precursors at the inner leaflet of the cytoplasmatic membrane; (3) flipping to the outer leaflet of the cytoplasmatic membrane and incorporation of the precursor into nascent PGN by transglycosylation (TG) and transpeptidation (TP) reactions. Enzymes shown in blue synthesize substrates added on the depicted reaction: Alr racemases convert l-Ala to d-Ala and Ddl ligases form the d-Ala-d-Ala dipeptide.
Figure 2.
Schematic representation of PGN biosynthesis in Gram-negative bacteria: (1) synthesis of a PG precursor in the cytoplasm; (2) synthesis of lipid-linked precursors at the inner leaflet of the cytoplasmatic membrane; (3) flipping to the outer leaflet of the cytoplasmatic membrane and incorporation of the precursor into nascent PGN by transglycosylation (TG) and transpeptidation (TP) reactions. Enzymes shown in blue synthesize substrates added on the depicted reaction: Alr racemases convert l-Ala to d-Ala and Ddl ligases form the d-Ala-d-Ala dipeptide.

Scheme 1.
Synthesis of PGN fragment 9 reported by Fukase [55]. (a) (i) AllocCl, NaHCO3, H2O; (ii) AllylOH, DOWEX 50W X8 200–400 mesh, 80 °C; (iii) PhCH(OMe)2, p-TsOH, 57%; (b) NaH then trifluoromethanesulfonyl-l-(S)-2-propionic acid benzyl ester, 98%; (c) Pd(PPh3)4, AcOH, DCM, then TrocCl, 58%; (d) Me3N·BH3, BF3·Et2O, ACN, 87%; (e) BnBr, AgO, DCM, 87%; (f) Ir complex, H2, THF, then I2, H2O, 99%; (g) CCl3CN, Cs2CO3, DCM, quantitative; (h) TMSOTf (0.1 equiv.), MS 4 Å, DCM, −15 °C, 88%; (i) Me3N·BH3, BF3·Et2O, ACN, 52%; (j) Ir complex, H2, THF, then I2, H2O, 91%; (k) CCl3CN, Cs2CO3, DCM, 85%; (l) TMSOTf (0.1 equiv.), MS 4 Å, DCM, −15 °C, 79%; (m) Me3N·BH3, BF3·Et2O, ACN, 60%; (n) Ir complex, H2, THF, then I2, H2O, 81%; (o) CCl3CN, Cs2CO3, DCM, 83%; (p) TMSOTf (0.1 equiv.), MS 4 Å, DCM, −15 °C, 70%; (o) Zn–Cu, AcOH, then Ac2O, Py, 82%; (r) 1 M NaOMe, THF, H2O: LiOH, dioxane, THF, H2O, 97%; (s) HCl·H-l-Ala-d-Glu(OBn)-NH2, WSCI·HCl, HOBt, TEA, DCM, 31%; (t) Pd(OH)2, AcOH, H2 (10 atm), 48%.
Scheme 1.
Synthesis of PGN fragment 9 reported by Fukase [55]. (a) (i) AllocCl, NaHCO3, H2O; (ii) AllylOH, DOWEX 50W X8 200–400 mesh, 80 °C; (iii) PhCH(OMe)2, p-TsOH, 57%; (b) NaH then trifluoromethanesulfonyl-l-(S)-2-propionic acid benzyl ester, 98%; (c) Pd(PPh3)4, AcOH, DCM, then TrocCl, 58%; (d) Me3N·BH3, BF3·Et2O, ACN, 87%; (e) BnBr, AgO, DCM, 87%; (f) Ir complex, H2, THF, then I2, H2O, 99%; (g) CCl3CN, Cs2CO3, DCM, quantitative; (h) TMSOTf (0.1 equiv.), MS 4 Å, DCM, −15 °C, 88%; (i) Me3N·BH3, BF3·Et2O, ACN, 52%; (j) Ir complex, H2, THF, then I2, H2O, 91%; (k) CCl3CN, Cs2CO3, DCM, 85%; (l) TMSOTf (0.1 equiv.), MS 4 Å, DCM, −15 °C, 79%; (m) Me3N·BH3, BF3·Et2O, ACN, 60%; (n) Ir complex, H2, THF, then I2, H2O, 81%; (o) CCl3CN, Cs2CO3, DCM, 83%; (p) TMSOTf (0.1 equiv.), MS 4 Å, DCM, −15 °C, 70%; (o) Zn–Cu, AcOH, then Ac2O, Py, 82%; (r) 1 M NaOMe, THF, H2O: LiOH, dioxane, THF, H2O, 97%; (s) HCl·H-l-Ala-d-Glu(OBn)-NH2, WSCI·HCl, HOBt, TEA, DCM, 31%; (t) Pd(OH)2, AcOH, H2 (10 atm), 48%.

Scheme 2.
Synthesis of the Lipid II unit 16 (undecaprenyl phosphate, highlighted in blue) reported by Wang [57]. (a) HAlaOTMSE, DIEA, HOBt, PyBOP, 1:1 THF:DCM, r.t. (room temperature), 2 h, 81%. (b) Catalyst TsOH, MeOH, 75 °C, 30 min, 89%. (c) Pyridine, 1 equiv. of AcCl, −30 °C, 15 min, then warm to r.t., 80%. (d) AgOTf (3 equiv.), donor (2 equiv.), MS 4 Å, DCM, −40 °C, drybox, 3 h, then overnight, 60%. (e) Amino-modified resin, MS 4 Å, butanol, 85 °C, 24 h, then pyridine, Ac2O, r.t., overnight, 53%. (f) 10%Pd/C, H2, MeOH, 1 h, 93%. (g) Tetrazole, (BnO)2PN(i-Pr)2, DCM, −30 °C to r.t., 1 h, then m-CPBA, −40 °C to r.t., 1 h, 55%. (h) TBAF, THF, r.t., 45 min, 87%. (i) H-γ-d-Glu(OTMSE)-Lys(TEOC)-d-Ala-d-AlaOTMSE, DIEA, HOBt, PyBOP, 1:1 THF:DCM, r.t., 2 h, 61%. (j) 10%Pd/C, H2, MeOH, 1 h, 100%. (k) CDI, dry DMF, r.t., 4 h, then MeOH, then undecaprenyl phosphate, r.t., 48 h, 39%. (l) TBAF, DMF, r.t., 24 h, then 3% NaOMe, MeOH, 0 °C, 1 h, 35%.
Scheme 2.
Synthesis of the Lipid II unit 16 (undecaprenyl phosphate, highlighted in blue) reported by Wang [57]. (a) HAlaOTMSE, DIEA, HOBt, PyBOP, 1:1 THF:DCM, r.t. (room temperature), 2 h, 81%. (b) Catalyst TsOH, MeOH, 75 °C, 30 min, 89%. (c) Pyridine, 1 equiv. of AcCl, −30 °C, 15 min, then warm to r.t., 80%. (d) AgOTf (3 equiv.), donor (2 equiv.), MS 4 Å, DCM, −40 °C, drybox, 3 h, then overnight, 60%. (e) Amino-modified resin, MS 4 Å, butanol, 85 °C, 24 h, then pyridine, Ac2O, r.t., overnight, 53%. (f) 10%Pd/C, H2, MeOH, 1 h, 93%. (g) Tetrazole, (BnO)2PN(i-Pr)2, DCM, −30 °C to r.t., 1 h, then m-CPBA, −40 °C to r.t., 1 h, 55%. (h) TBAF, THF, r.t., 45 min, 87%. (i) H-γ-d-Glu(OTMSE)-Lys(TEOC)-d-Ala-d-AlaOTMSE, DIEA, HOBt, PyBOP, 1:1 THF:DCM, r.t., 2 h, 61%. (j) 10%Pd/C, H2, MeOH, 1 h, 100%. (k) CDI, dry DMF, r.t., 4 h, then MeOH, then undecaprenyl phosphate, r.t., 48 h, 39%. (l) TBAF, DMF, r.t., 24 h, then 3% NaOMe, MeOH, 0 °C, 1 h, 35%.

Scheme 3.
Synthesis of the Lipid II unit 16 reported by Blaszczak [58,59]. (a) N-methylmorpholine, 2-chloro-4,6-dimethoxy-1,3,5-triazine, DCM, 2 h, 95%; (b) Et3SiH (3 equiv.), TFA (6 equiv.), DCM, 0 °C, 5 h, 61%; (c) AgOTf (3 equiv.), donor (3 equiv.), MS 4 Å (5.5 wt equiv.), DCM, 25 °C, 20 h 84%; (d) (1) anhydrous ZnCl2,Ac2O/AcOH (2:1), 25 °C, 20 h; (2) Zn dust (20 reducing equiv.), 25 °C, 4 h, 67%. (e) H2, Pd/C, MeOH/THF, 94% yield; (f) (1) dibenzyl-N,N-diethylphosphoramidite, 1H-tetrazole, DCM; (2) 30% H2O2, THF, −78 °C to room temperature, 78% yield; (g) (1) DBU, DCM; (2) EDCI, NHS, DMF, then peptide and iPr2NEt, 46% yield; (h) H2, Pd/C, MeOH, then pyridine, 91% yield; (i) (1) CDI/DMF/THF; (2) undecaprenyl mono-phosphate (bis-amonium salt); (j) NaOH/H2O/1,4-dioxane, 24% overall yield from 10.
Scheme 3.
Synthesis of the Lipid II unit 16 reported by Blaszczak [58,59]. (a) N-methylmorpholine, 2-chloro-4,6-dimethoxy-1,3,5-triazine, DCM, 2 h, 95%; (b) Et3SiH (3 equiv.), TFA (6 equiv.), DCM, 0 °C, 5 h, 61%; (c) AgOTf (3 equiv.), donor (3 equiv.), MS 4 Å (5.5 wt equiv.), DCM, 25 °C, 20 h 84%; (d) (1) anhydrous ZnCl2,Ac2O/AcOH (2:1), 25 °C, 20 h; (2) Zn dust (20 reducing equiv.), 25 °C, 4 h, 67%. (e) H2, Pd/C, MeOH/THF, 94% yield; (f) (1) dibenzyl-N,N-diethylphosphoramidite, 1H-tetrazole, DCM; (2) 30% H2O2, THF, −78 °C to room temperature, 78% yield; (g) (1) DBU, DCM; (2) EDCI, NHS, DMF, then peptide and iPr2NEt, 46% yield; (h) H2, Pd/C, MeOH, then pyridine, 91% yield; (i) (1) CDI/DMF/THF; (2) undecaprenyl mono-phosphate (bis-amonium salt); (j) NaOH/H2O/1,4-dioxane, 24% overall yield from 10.

Scheme 4.
Synthesis of the disaccharides 26 and 27—precursors of NAM-NAG and NAG-NAM reported by Boons [60]. (a) TBDMSOTf, 2,6-lutidine, DCM, 96%; (b) PMBCl, NaH, TBAI, THF, reflux, 84%; (c) Me3N·BH3, AlCl3, THF, 72%; (d) HO(CH2)3N3, NIS, TMSOTf, 67%; (e) NIS, TMSOTf, 62%; (f) H2N(CH2)2NH2, EtOH then Ac2O, MeOH, 70%; (g) Bu4NF, THF, 75%; (h) NaH, (S)-2-chloropropionic acid, dioxane, 14 (76%) and 15 (66%); (i) DDQ, DCM, H2O, 79%.
Scheme 4.
Synthesis of the disaccharides 26 and 27—precursors of NAM-NAG and NAG-NAM reported by Boons [60]. (a) TBDMSOTf, 2,6-lutidine, DCM, 96%; (b) PMBCl, NaH, TBAI, THF, reflux, 84%; (c) Me3N·BH3, AlCl3, THF, 72%; (d) HO(CH2)3N3, NIS, TMSOTf, 67%; (e) NIS, TMSOTf, 62%; (f) H2N(CH2)2NH2, EtOH then Ac2O, MeOH, 70%; (g) Bu4NF, THF, 75%; (h) NaH, (S)-2-chloropropionic acid, dioxane, 14 (76%) and 15 (66%); (i) DDQ, DCM, H2O, 79%.

Scheme 5.
Synthesis of the tetrasaccharide with pentapeptides attached to NAM units, 33, reported by Mobashery [61]. (a) TfOH, 71%. (b) (1) NaOMe, Amberlite IR-120 (H+), (2) benzaldehyde dimethylacetal, p-TsOH, 60% in two steps; (c) Ac2O, 85%; (d) n-Bu4NF, 75%; (e) Cl3CCN, DBU; (f) acceptor A, TfOH, 55% in two steps; (g) BH3.NMe3, BF3.OEt2, 62%; (h) donor B, TfOH, 68%; (i) (1) NaOH, dioxane/water (4:1); HCl, pH 3.0; (2) Ac2O, 48% in two steps; (j) NaOMe, 74%; (k) NaH, (S)-2-chloropropionic acid, 46%; (m) 4-nitrophenyl trifluoroacetate, pyridine; peptide (prepared as CF3CO2H salt), 41%; (l) 60% AcOH; H2, Pd/C, 76%.
Scheme 5.
Synthesis of the tetrasaccharide with pentapeptides attached to NAM units, 33, reported by Mobashery [61]. (a) TfOH, 71%. (b) (1) NaOMe, Amberlite IR-120 (H+), (2) benzaldehyde dimethylacetal, p-TsOH, 60% in two steps; (c) Ac2O, 85%; (d) n-Bu4NF, 75%; (e) Cl3CCN, DBU; (f) acceptor A, TfOH, 55% in two steps; (g) BH3.NMe3, BF3.OEt2, 62%; (h) donor B, TfOH, 68%; (i) (1) NaOH, dioxane/water (4:1); HCl, pH 3.0; (2) Ac2O, 48% in two steps; (j) NaOMe, 74%; (k) NaH, (S)-2-chloropropionic acid, 46%; (m) 4-nitrophenyl trifluoroacetate, pyridine; peptide (prepared as CF3CO2H salt), 41%; (l) 60% AcOH; H2, Pd/C, 76%.

Scheme 6.
Synthesis of the heptaprenyl—Lipid IV 43b reported by Walker [63]. (a) Bu2SnO, toluene, reflux, then Bu4NI, BnBr, reflux, 51%; (b) TBSOTf, 2,6-lutidine, CH2Cl2, 91%; (c) mCPBA, CH2Cl2, −78 °C to −60 °C, 93%; (d) (1) (Bu3Sn)2O, MeOH, reflux; (2) Bu4NI, BnBr, toluene, 91 °C, 75%; (e) mCPBA, CH2Cl2, −78 °C to −60 °C, 91%; (f) Tf2O, DTBMP, ADMB, MS 4Å, CH2Cl2, −60 °C to −40 °C, 58%; (g) Tf2O, DTBMP, ADMB, MS 4Å, CH2Cl2, −60 °C to −30 °C, 75%; (h) mCPBA, CH2Cl2, −78 °C to −60 °C, 86%; (i) Tf2O, DTBMP, ADMB, MS 4Å, CH2Cl2, −40 °C, 77%; (j) (NH2CH2)2, THF/CH3CN/EtOH (1:2:1), 60 °C; (k) Ac2O, MeOH/H2O (5:1), r.t., 75% two steps; (l) NaH, S-(−)-2-bromo-propionic acid, THF, 0 °C to r.t.; (m) TMSCHN2, benzene/MeOH (3:1), 0 °C, 70% two steps; (n) NIS, CH3CN/H2O (5:1), r.t., 75%; o) 1H-tetrazole, (i-Pr)2NP(OBn)2, CH2Cl2, −40 to −20 °C; (p) mCPBA, CH2Cl2, −40 °C to r.t., 84% two steps; (q) TBAF, THF, 0 °C to r.t.; (r) 1.3 M KOH, THF/H2O (10:1), r.t., 64% two steps; (s) HATU, DIEA, DMF, r.t., 60%; (t) Pd(OH)2/C, H2, 44%; (u) ammonium heptaprenyl phosphate, 1,1′-carbonyl diimidazole, THF, r.t., then 30, SnCl2, DMF, r.t., 50%; (v) TBAF, DMF, r.t., 69%; (w) (CH3[14]CO)2O, toluene/16 mM NaOH in MeOH (1:1), sonication, 37 °C, 50%.
Scheme 6.
Synthesis of the heptaprenyl—Lipid IV 43b reported by Walker [63]. (a) Bu2SnO, toluene, reflux, then Bu4NI, BnBr, reflux, 51%; (b) TBSOTf, 2,6-lutidine, CH2Cl2, 91%; (c) mCPBA, CH2Cl2, −78 °C to −60 °C, 93%; (d) (1) (Bu3Sn)2O, MeOH, reflux; (2) Bu4NI, BnBr, toluene, 91 °C, 75%; (e) mCPBA, CH2Cl2, −78 °C to −60 °C, 91%; (f) Tf2O, DTBMP, ADMB, MS 4Å, CH2Cl2, −60 °C to −40 °C, 58%; (g) Tf2O, DTBMP, ADMB, MS 4Å, CH2Cl2, −60 °C to −30 °C, 75%; (h) mCPBA, CH2Cl2, −78 °C to −60 °C, 86%; (i) Tf2O, DTBMP, ADMB, MS 4Å, CH2Cl2, −40 °C, 77%; (j) (NH2CH2)2, THF/CH3CN/EtOH (1:2:1), 60 °C; (k) Ac2O, MeOH/H2O (5:1), r.t., 75% two steps; (l) NaH, S-(−)-2-bromo-propionic acid, THF, 0 °C to r.t.; (m) TMSCHN2, benzene/MeOH (3:1), 0 °C, 70% two steps; (n) NIS, CH3CN/H2O (5:1), r.t., 75%; o) 1H-tetrazole, (i-Pr)2NP(OBn)2, CH2Cl2, −40 to −20 °C; (p) mCPBA, CH2Cl2, −40 °C to r.t., 84% two steps; (q) TBAF, THF, 0 °C to r.t.; (r) 1.3 M KOH, THF/H2O (10:1), r.t., 64% two steps; (s) HATU, DIEA, DMF, r.t., 60%; (t) Pd(OH)2/C, H2, 44%; (u) ammonium heptaprenyl phosphate, 1,1′-carbonyl diimidazole, THF, r.t., then 30, SnCl2, DMF, r.t., 50%; (v) TBAF, DMF, r.t., 69%; (w) (CH3[14]CO)2O, toluene/16 mM NaOH in MeOH (1:1), sonication, 37 °C, 50%.

Scheme 7.
Synthesis of cross-linked PGN fragment 46 reported by Fukase [65]. (a) LiOH, THF/1,4-dioxane/H2O = 4/2/1, quantitative; (b) TFA; (c) HCl.Et2O; (d) Peptide, HATU, Et3N, DMF/DMSO (1:1), 75%, three steps; (e) 10% TFA/DCM; (f) 20% piperidine/DMSO; (g) H2, Pd(OH)2, AcOH.
Scheme 7.
Synthesis of cross-linked PGN fragment 46 reported by Fukase [65]. (a) LiOH, THF/1,4-dioxane/H2O = 4/2/1, quantitative; (b) TFA; (c) HCl.Et2O; (d) Peptide, HATU, Et3N, DMF/DMSO (1:1), 75%, three steps; (e) 10% TFA/DCM; (f) 20% piperidine/DMSO; (g) H2, Pd(OH)2, AcOH.

Scheme 8.
Synthesis of the PGN fragments 54 reported by Fukase [67]. (a) TMSOTf, CH2Cl2, −15 °C, MS 4Å, 20 min, 84%; (b) Me3N BH3/BF3·Et2O, ACN, 1 h, 83%; (c) [Ir(cod)H-(MePh2P)2]PF6, THF, 1.5 h; (d) I2/H2O, 30 min, 81%; (e) CCl3CN/Cs2CO3, DCM, 30 min, quantitative; (f) TMSOTf, DCM, MS 4Å, 40 min, 16%; (g) Zn/Cu, AcOH,3 h, Ac2O/Py, 2 h, 62%; (h) LiOH, quantitative. For the condensation: (i) WSCD/HOBt/DMF/TEA, HCl·l-Ala-d-isoGln(OBn) or HCl·L-Ala-d-isoGln-l-Lys(Z)(OBn); (j) HATU/DMF/TEA, HCl·l-Ala-d-isoGln-l-Lys(Z)-d-Ala(OBn) or HCl·l-Ala-d-isoGln-l-Lys(Z)-d-Ala-d-Ala(OBn); (k) H2/Pd(OH)2, AcOH.
Scheme 8.
Synthesis of the PGN fragments 54 reported by Fukase [67]. (a) TMSOTf, CH2Cl2, −15 °C, MS 4Å, 20 min, 84%; (b) Me3N BH3/BF3·Et2O, ACN, 1 h, 83%; (c) [Ir(cod)H-(MePh2P)2]PF6, THF, 1.5 h; (d) I2/H2O, 30 min, 81%; (e) CCl3CN/Cs2CO3, DCM, 30 min, quantitative; (f) TMSOTf, DCM, MS 4Å, 40 min, 16%; (g) Zn/Cu, AcOH,3 h, Ac2O/Py, 2 h, 62%; (h) LiOH, quantitative. For the condensation: (i) WSCD/HOBt/DMF/TEA, HCl·l-Ala-d-isoGln(OBn) or HCl·L-Ala-d-isoGln-l-Lys(Z)(OBn); (j) HATU/DMF/TEA, HCl·l-Ala-d-isoGln-l-Lys(Z)-d-Ala(OBn) or HCl·l-Ala-d-isoGln-l-Lys(Z)-d-Ala-d-Ala(OBn); (k) H2/Pd(OH)2, AcOH.

Scheme 9.
Synthesis of the PGN fragment 62 reported by Marques [70]. (a) (1) PhCHO, ZnCl2, MS 3Å, r.t., overnight; (2) Ac2O, pyridine, r.t., overnight 78%; (b) morpholine, EtOAc, r.t., overnight, 87%; (c) CCl3CN, Cs2CO3, CH2Cl2, r.t., 2 h, 74%; (d) BnOH, AcCl, 80 °C, 3 h, 61%; (e) PhCHO, ZnCl2, overnight, 72%; (f) ethyl l-(S)-2- trifluoromethanesulphonyloxy-propionate, NaH, CH2Cl2, r.t., 3 h, 68%; (g) Pd(PPh3)4, AcOH, TrocCl, r.t., 82%; (h) BH3Me2N, BF3OEt2, r.t., 3 h, 72%; (i) TMSOTf, DCM, MS 3 Å, -15 °C, 52%; (j) Zn-Cu, THF: AcOH: Ac2O (1:1:1), then Ac2O/pyridine, 63%; (l) LiOH, THF:1,4-dioxane:H2O, 69%; (m) Pd(OH)2, H2, AcOH, r.t., quantitative; (n) peptide coupling via Fmoc-solid-phase synthesis; (o) TFA (1% CH2Cl2), r.t.
Scheme 9.
Synthesis of the PGN fragment 62 reported by Marques [70]. (a) (1) PhCHO, ZnCl2, MS 3Å, r.t., overnight; (2) Ac2O, pyridine, r.t., overnight 78%; (b) morpholine, EtOAc, r.t., overnight, 87%; (c) CCl3CN, Cs2CO3, CH2Cl2, r.t., 2 h, 74%; (d) BnOH, AcCl, 80 °C, 3 h, 61%; (e) PhCHO, ZnCl2, overnight, 72%; (f) ethyl l-(S)-2- trifluoromethanesulphonyloxy-propionate, NaH, CH2Cl2, r.t., 3 h, 68%; (g) Pd(PPh3)4, AcOH, TrocCl, r.t., 82%; (h) BH3Me2N, BF3OEt2, r.t., 3 h, 72%; (i) TMSOTf, DCM, MS 3 Å, -15 °C, 52%; (j) Zn-Cu, THF: AcOH: Ac2O (1:1:1), then Ac2O/pyridine, 63%; (l) LiOH, THF:1,4-dioxane:H2O, 69%; (m) Pd(OH)2, H2, AcOH, r.t., quantitative; (n) peptide coupling via Fmoc-solid-phase synthesis; (o) TFA (1% CH2Cl2), r.t.

Scheme 10.
Reported multistep synthesis of NAG-NAM intermediates via orthogonal routes from glucosamine.
Scheme 10.
Reported multistep synthesis of NAG-NAM intermediates via orthogonal routes from glucosamine.

Scheme 11.
Synthesis of the NAG-NAM precursor 66 from peracetylated chitobiose 63 reported by Marques group [79]. (a) Ac2O, H2SO4, 55 °C, 3 h, then r.t., 16 h, and 55 °C, 1 h, 13%; (b) TFAA, Pyr, 120 °C, 15 min, 76%; (c) TolSH, BF3OEt2, DCM, r.t., overnight, 80%; (d) (1) MeONa, MeOH, r.t., overnight. (2) butane-2,3-dione, p-TsOH, triethylorthoformate, EtOH, 60 °C, 16 h, 58%; (e) TBDMSCl, DMAP, TEA, CH2Cl2, 56%; (f) (1) NaOH 2 M, THF, 80 °C, 3 h, (2) Ac2O, pyridine, CH2Cl2, r.t., overnight, 56%; (g) (1) NaH, DCM, 30 min; (2) Ethyl (S)-2-(trifluoromethylsulfonyloxy)propionate, r.t., 16 h, 38%.
Scheme 11.
Synthesis of the NAG-NAM precursor 66 from peracetylated chitobiose 63 reported by Marques group [79]. (a) Ac2O, H2SO4, 55 °C, 3 h, then r.t., 16 h, and 55 °C, 1 h, 13%; (b) TFAA, Pyr, 120 °C, 15 min, 76%; (c) TolSH, BF3OEt2, DCM, r.t., overnight, 80%; (d) (1) MeONa, MeOH, r.t., overnight. (2) butane-2,3-dione, p-TsOH, triethylorthoformate, EtOH, 60 °C, 16 h, 58%; (e) TBDMSCl, DMAP, TEA, CH2Cl2, 56%; (f) (1) NaOH 2 M, THF, 80 °C, 3 h, (2) Ac2O, pyridine, CH2Cl2, r.t., overnight, 56%; (g) (1) NaH, DCM, 30 min; (2) Ethyl (S)-2-(trifluoromethylsulfonyloxy)propionate, r.t., 16 h, 38%.

Scheme 12.
Synthesis of NAG-NAM oligosaccharides (69) by Marques group [83]. (a) Phthalic anhydride, DMF 5% H2O, 120 °C, overnight; (b) C1 (m = 1) or C2 (m = 2), CDI, DMF, r.t., 24 h; (c) TBDMSCl, imidazole, DMF, N2, r.t., 72 h; (d) (S)-2-chloropropionic acid, NaH, DMF, r.t., 48 h; (e) (1) NH2NH2, H2O, reflux, 24 h; (2) Ac2O, pyridine, r.t., overnight; (f) TBAF, pyridine, r.t., overnight.
Scheme 12.
Synthesis of NAG-NAM oligosaccharides (69) by Marques group [83]. (a) Phthalic anhydride, DMF 5% H2O, 120 °C, overnight; (b) C1 (m = 1) or C2 (m = 2), CDI, DMF, r.t., 24 h; (c) TBDMSCl, imidazole, DMF, N2, r.t., 72 h; (d) (S)-2-chloropropionic acid, NaH, DMF, r.t., 48 h; (e) (1) NH2NH2, H2O, reflux, 24 h; (2) Ac2O, pyridine, r.t., overnight; (f) TBAF, pyridine, r.t., overnight.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Queda, F.; Covas, G.; Filipe, S.R.; Marques, M.M.B. Assembly of Peptidoglycan Fragments—A Synthetic Challenge. Pharmaceuticals 2020, 13, 392. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13110392
AMA Style
Queda F, Covas G, Filipe SR, Marques MMB. Assembly of Peptidoglycan Fragments—A Synthetic Challenge. Pharmaceuticals. 2020; 13(11):392. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13110392
Chicago/Turabian StyleQueda, Fausto, Gonçalo Covas, Sérgio R. Filipe, and M. Manuel B. Marques. 2020. "Assembly of Peptidoglycan Fragments—A Synthetic Challenge" Pharmaceuticals 13, no. 11: 392. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13110392
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.