Proteomics Reveals the Potential Protective Mechanism of Hydrogen Sulfide on Retinal Ganglion Cells in an Ischemia/Reperfusion Injury Animal Model



Abstract
:1. Introduction
2. Results
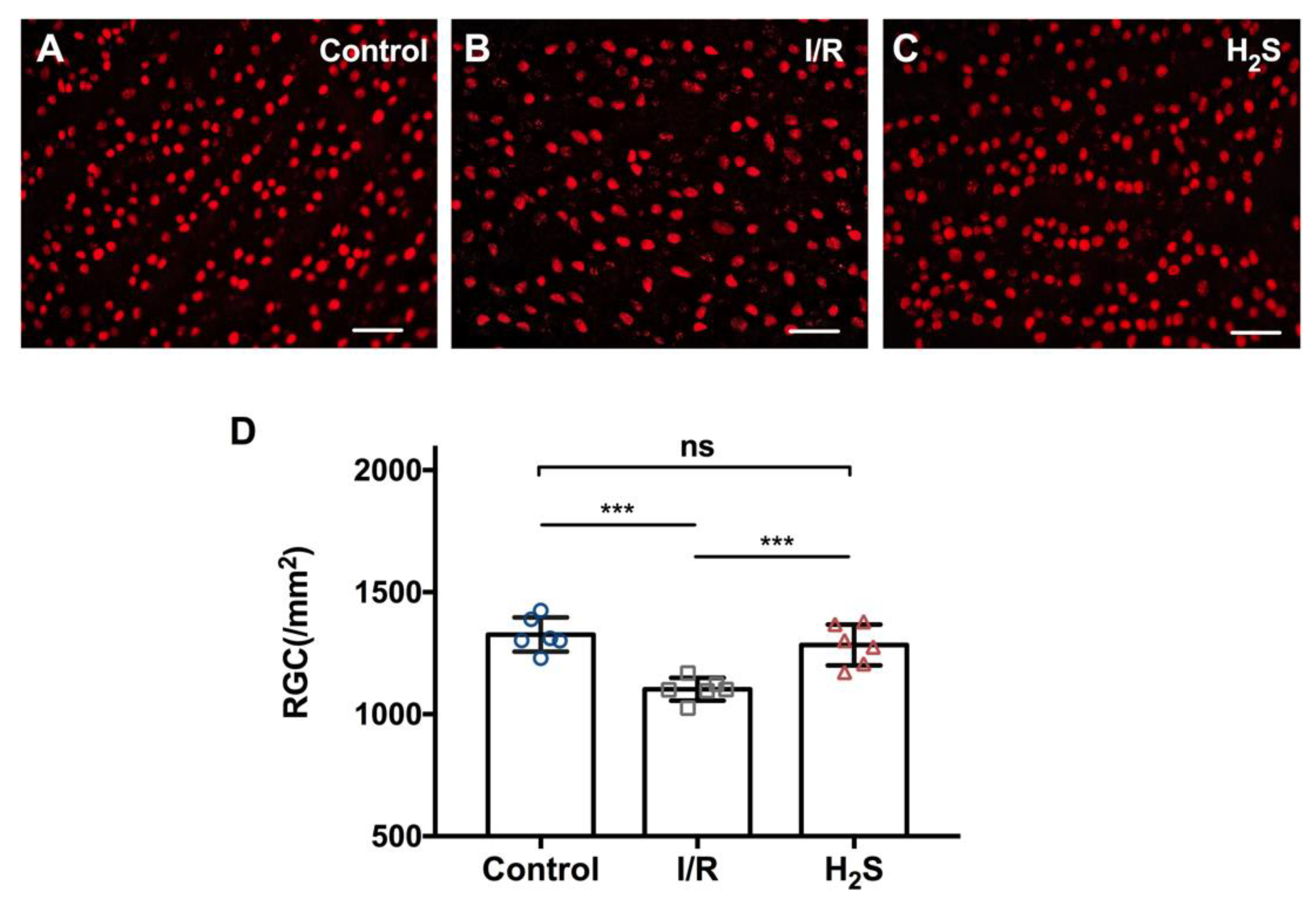
2.1. Pretreatment of H2S Improves RGC Survival against Ischemia-Reperfusion Injury
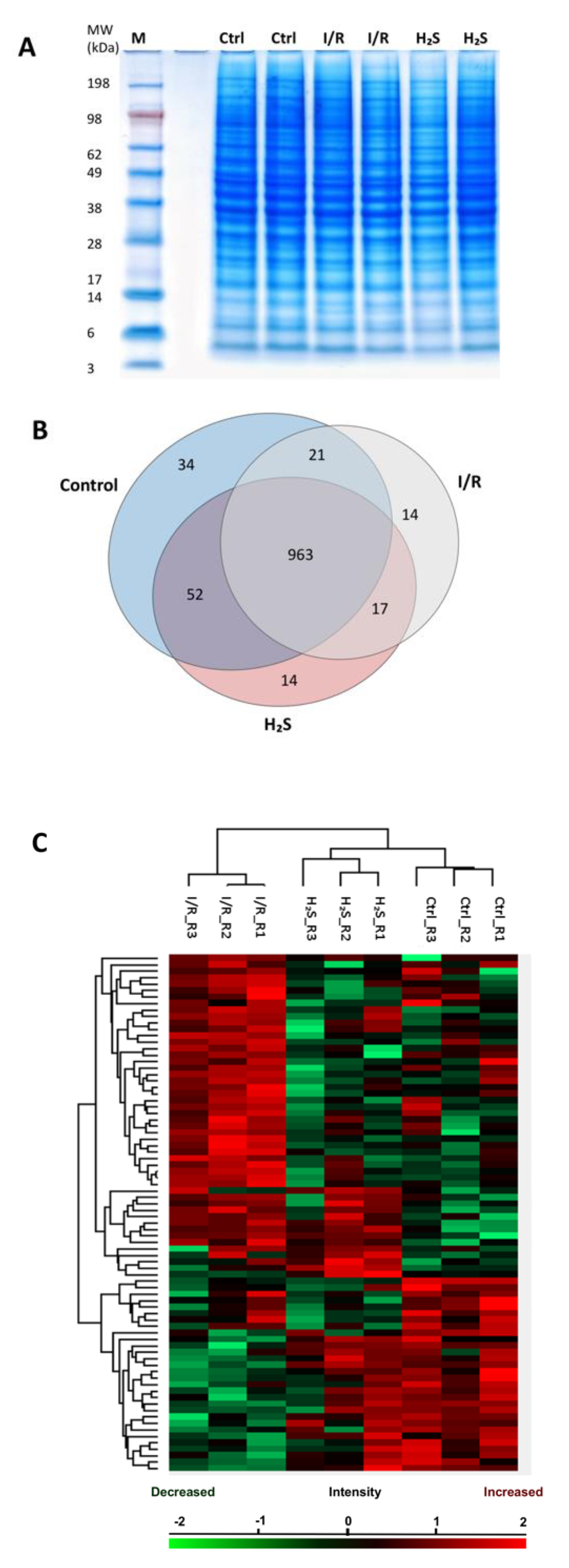
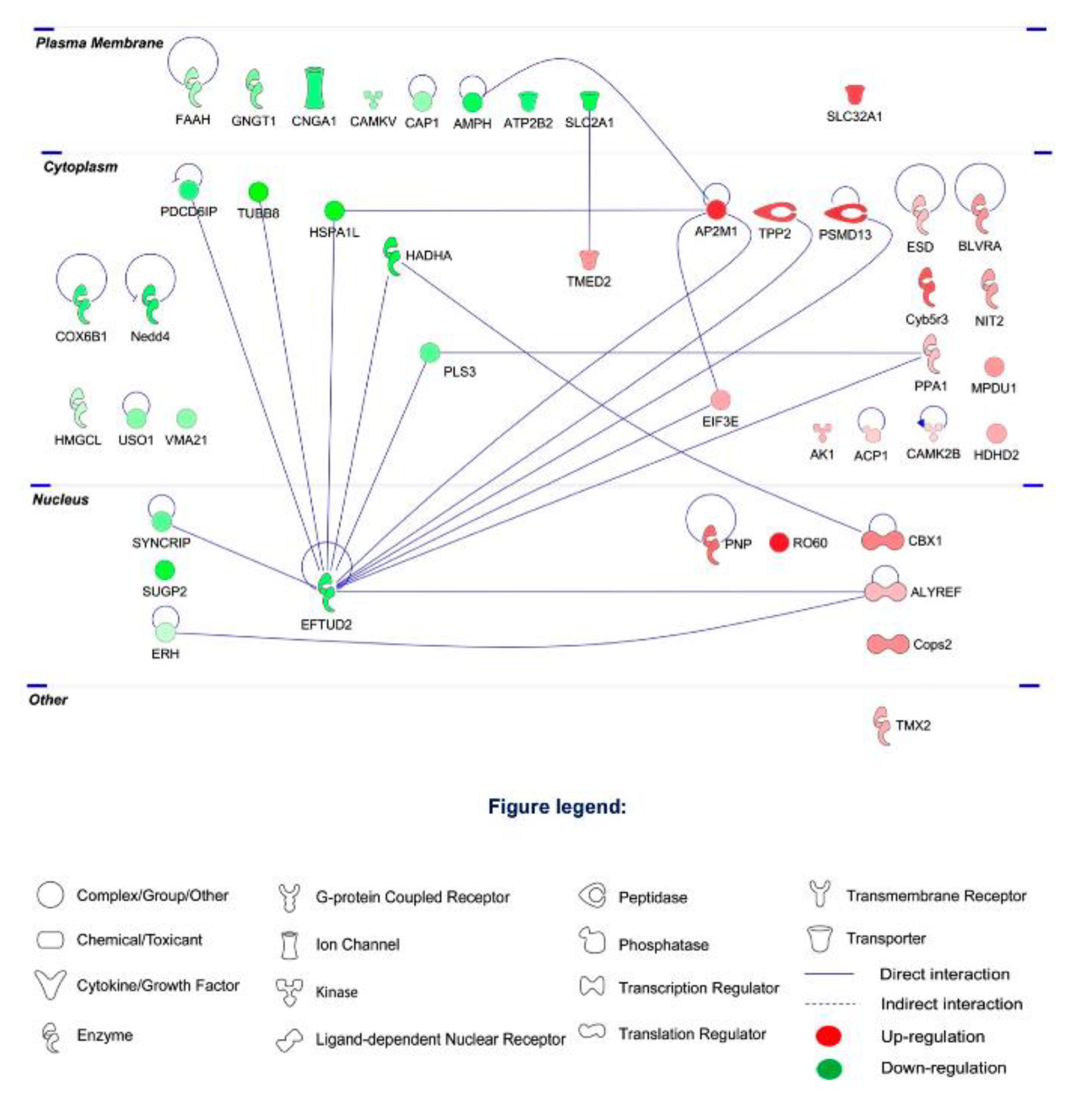
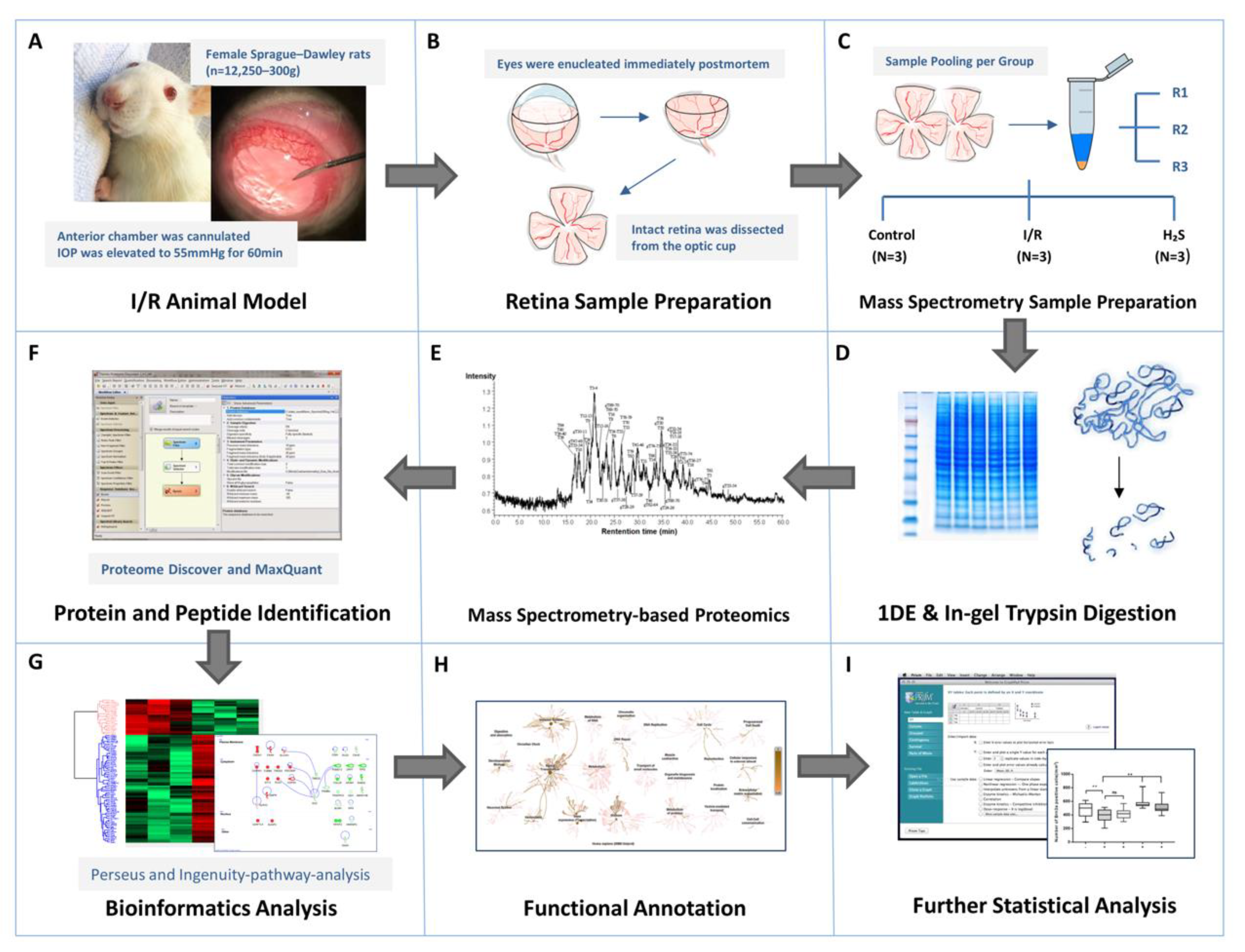
2.2. Proteomic Profiling
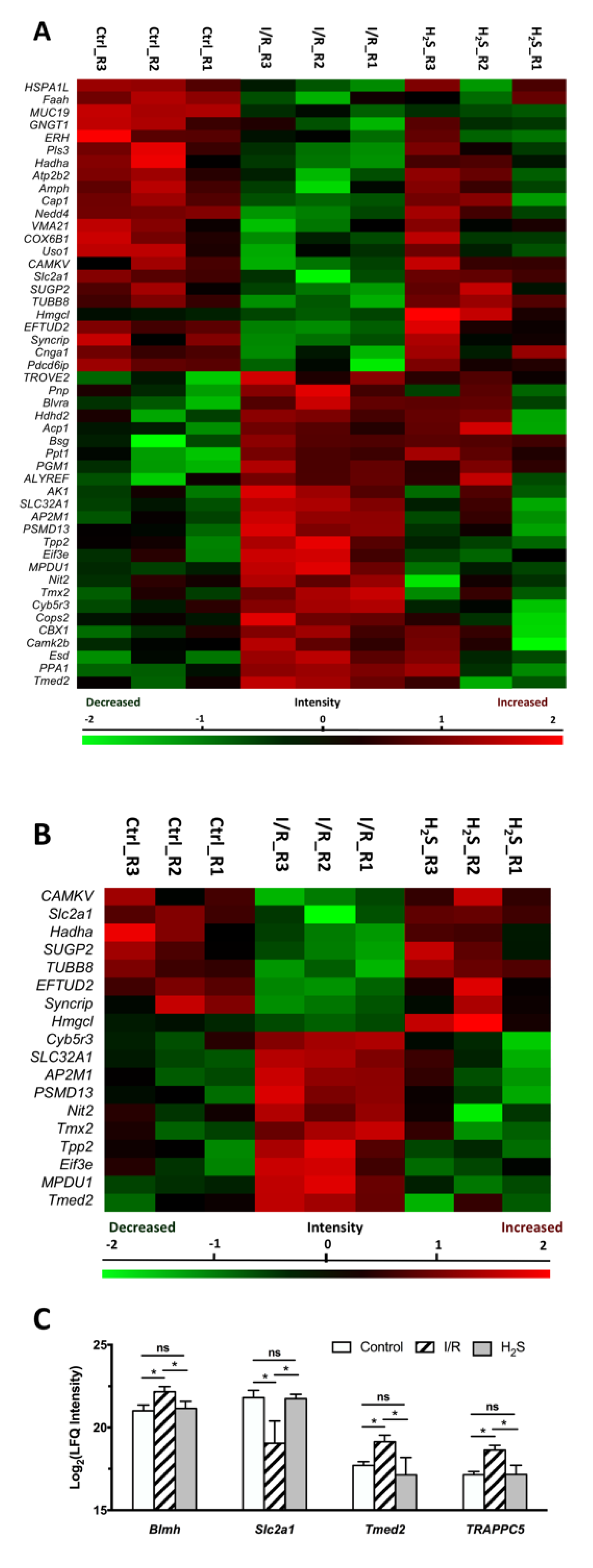
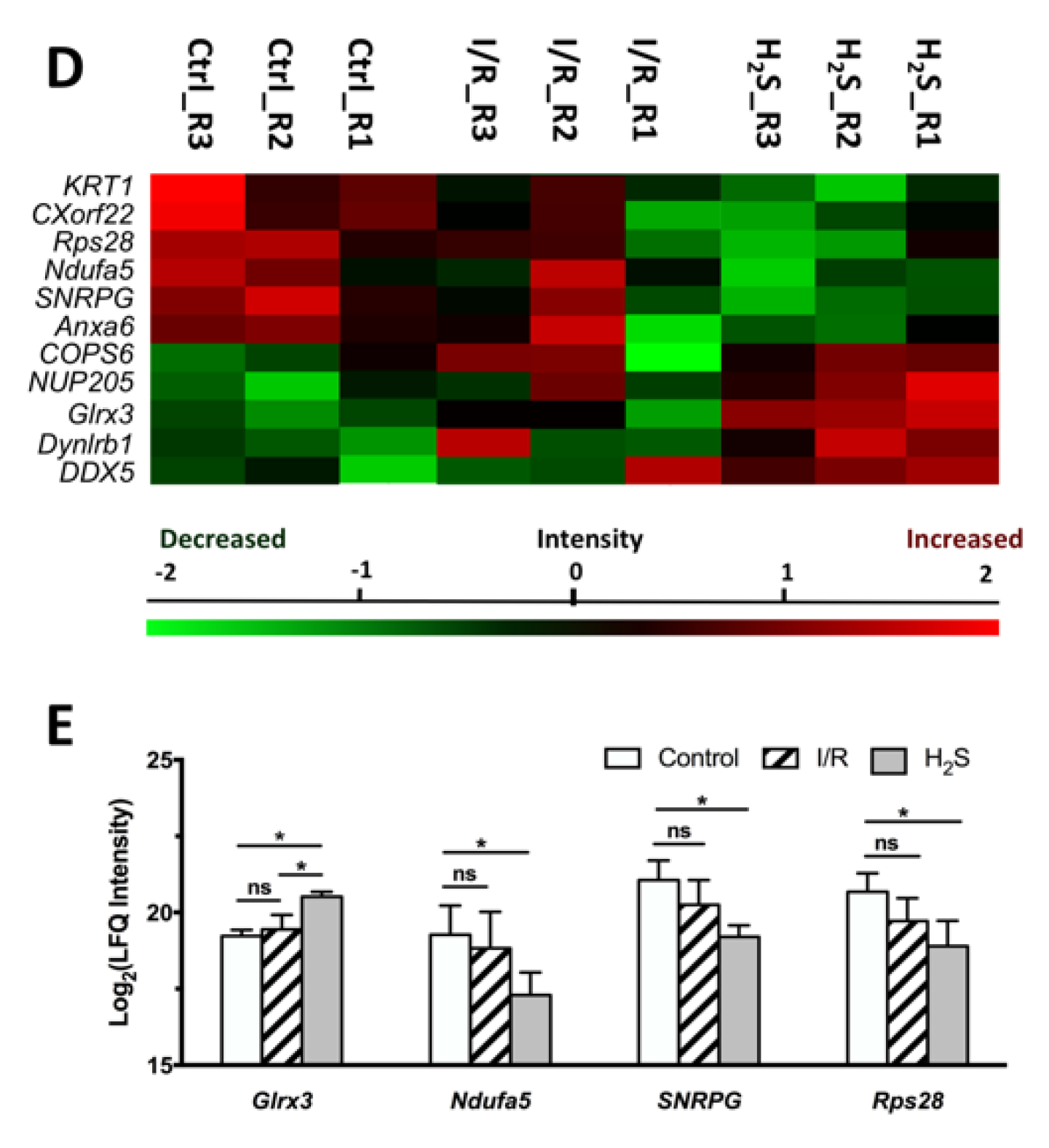
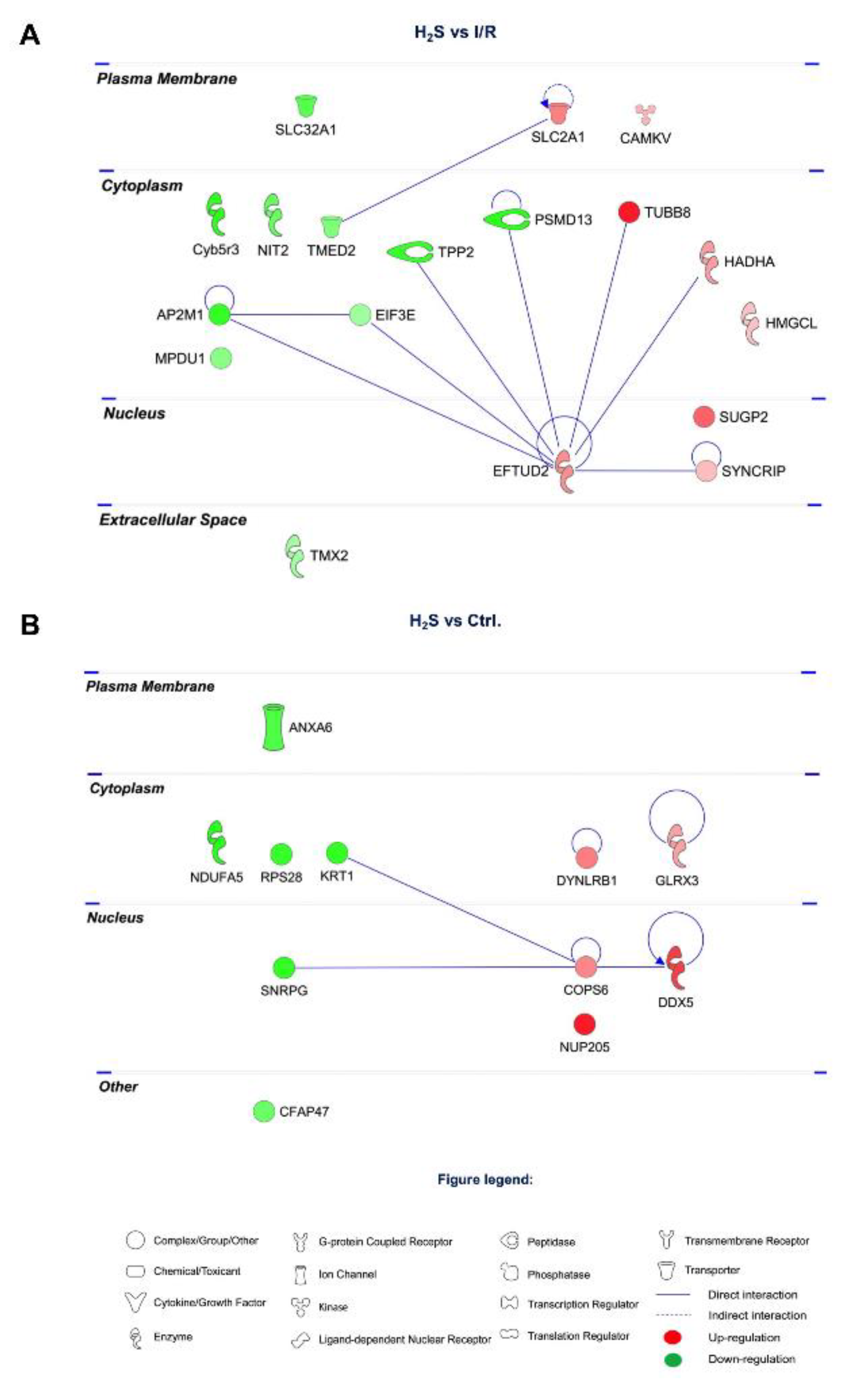
2.3. Differential Expression of Retinal Proteins between I/R and H2S Groups
2.4. Pathway Analysis of the Differentially Expressed Retinal Proteins in I/R and H2S Groups
3. Discussion
3.1. Changes in Iron Homeostasis and ROS Regulation
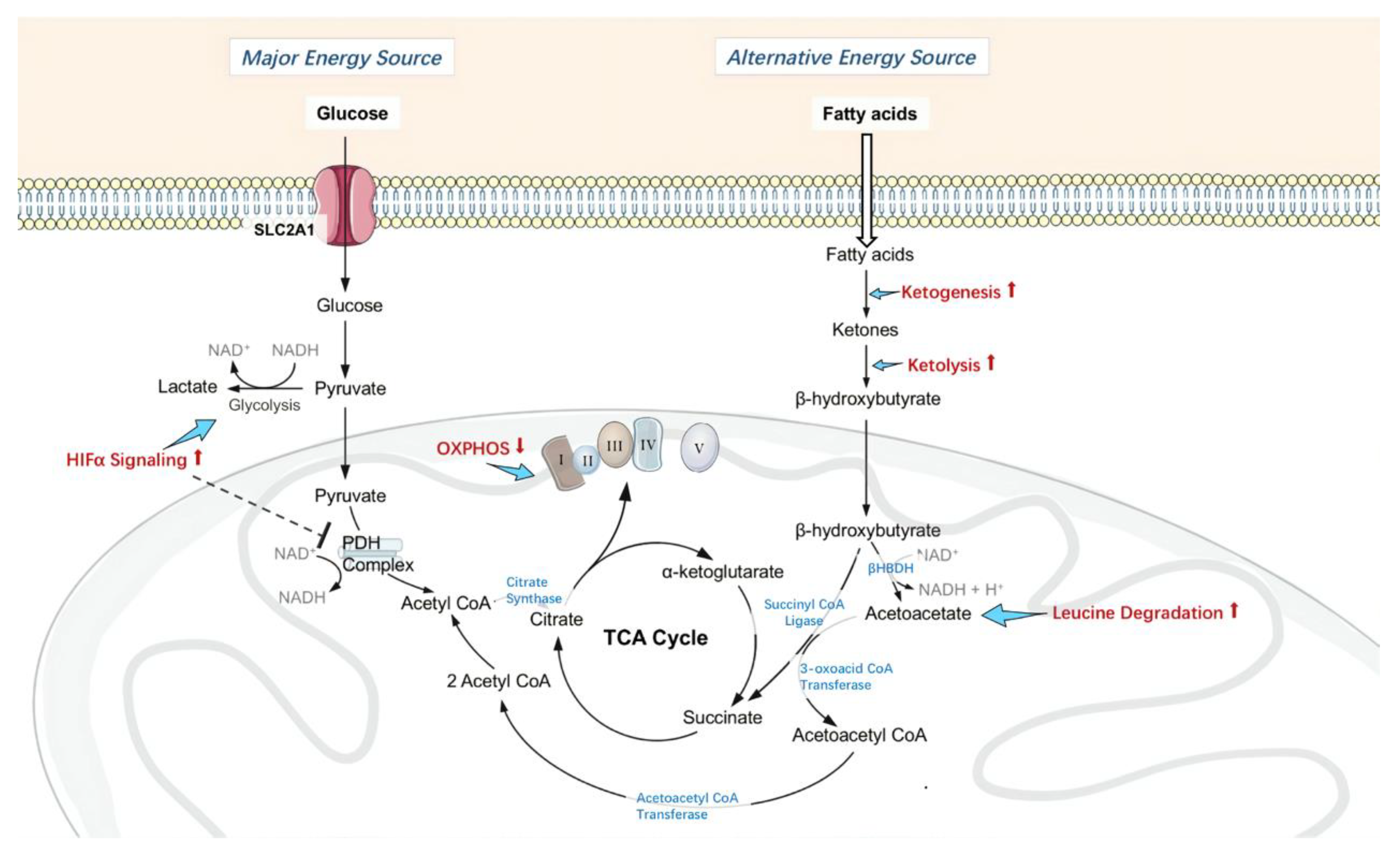
3.2. Changes in Retinal Metabolism, Mitochondrial Homeostasis and Function
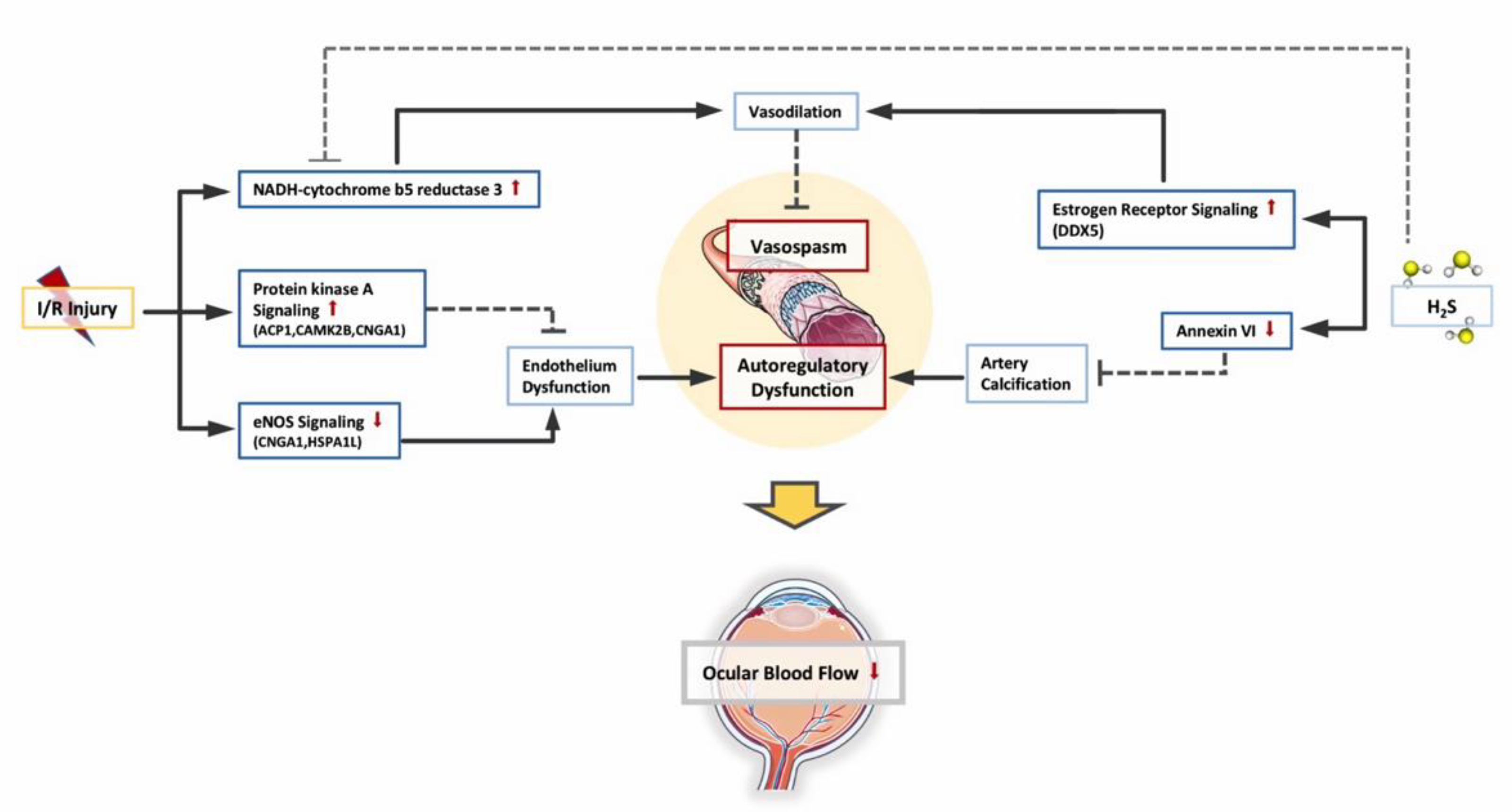
3.3. Changes in Retinal Vascular Function
3.4. Changes in GABA Receptor Signaling
3.5. Changes in DNA Repair
4. Materials and Methods
4.1. Animals
4.2. Intravitreal Injection of GYY4137
4.3. Ischemia-Reperfusion Injury Model
4.4. Preparation of Retinal Explants
4.5. Quantification of Retinal Ganglion Cells
4.6. Mass Spectrometry Sample Preparation
4.7. Bioinformatics, Functional Annotation and Pathways Analyses and Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global causes of blindness and distance vision impairment 1990–2020: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e1221–e1234. [Google Scholar] [CrossRef] [Green Version]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef]
- Keating, D.J. Mitochondrial dysfunction, oxidative stress, regulation of exocytosis and their relevance to neurodegenerative diseases. J. Neurochem. 2008, 104, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, R.; Jeong, N.Y.; Jung, J. Therapeutic importance of hydrogen sulfide in age-associated neurodegenerative diseases. Neural Regen. Res. 2020, 15, 653–662. [Google Scholar] [CrossRef]
- Snyder, S.H.; Jaffrey, S.R.; Zakhary, R. Nitric oxide and carbon monoxide: Parallel roles as neural messengers. Brain Res. Brain Res. Rev. 1998, 26, 167–175. [Google Scholar] [CrossRef]
- Verma, A.; Hirsch, D.J.; Glatt, C.E.; Ronnett, G.V.; Snyder, S.H. Carbon monoxide: A putative neural messenger. Science 1993, 259, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Bucolo, C.; Drago, F. Carbon monoxide and the eye: Implications for glaucoma therapy. Pharmacol. Ther. 2011, 130, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Flammer, J.; Haefliger, I.O. Isoproterenol, forskolin, and cAMP-induced nitric oxide production in pig ciliary processes. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1833–1837. [Google Scholar]
- Meyer, P.; Champion, C.; Schlotzer-Schrehardt, U.; Flammer, J.; Haefliger, I.O. Localization of nitric oxide synthase isoforms in porcine ocular tissues. Curr. Eye Res. 1999, 18, 375–380. [Google Scholar] [CrossRef]
- Fleischhauer, J.C.; Beny, J.L.; Flammer, J.; Haefliger, I.O. NO/cGMP pathway activation and membrane potential depolarization in pig ciliary epithelium. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1759–1763. [Google Scholar]
- Giuffrida, S.; Bucolo, C.; Drago, F. Topical application of a nitric oxide synthase inhibitor reduces intraocular pressure in rabbits with experimental glaucoma. J. Ocul. Pharmacol. Ther. 2003, 19, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, A.H.; Das, S.; Vora, S.; Gachie, E.; Kawai, S.; Manning, P.T.; Connor, J.R. A prodrug of a selective inhibitor of inducible nitric oxide synthase is neuroprotective in the rat model of glaucoma. J. Glaucoma 2002, 11, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, A.H.; Sawada, A.; Becker, B. Inhibition of nitric-oxide synthase 2 by aminoguanidine provides neuroprotection of retinal ganglion cells in a rat model of chronic glaucoma. Proc. Natl. Acad. Sci. USA 1999, 96, 9944–9948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maher, P.; Hanneken, A. Flavonoids protect retinal ganglion cells from oxidative stress-induced death. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4796–4803. [Google Scholar] [CrossRef]
- Patil, K.; Bellner, L.; Cullaro, G.; Gotlinger, K.H.; Dunn, M.W.; Schwartzman, M.L. Heme oxygenase-1 induction attenuates corneal inflammation and accelerates wound healing after epithelial injury. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3379–3386. [Google Scholar] [CrossRef] [PubMed]
- Stagni, E.; Privitera, M.G.; Bucolo, C.; Leggio, G.M.; Motterlini, R.; Drago, F. A water-soluble carbon monoxide-releasing molecule (CORM-3) lowers intraocular pressure in rabbits. Br. J. Ophthalmol. 2009, 93, 254–257. [Google Scholar] [CrossRef]
- Gadalla, M.M.; Snyder, S.H. Hydrogen sulfide as a gasotransmitter. J. Neurochem. 2010, 113, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H. Signaling molecules: Hydrogen sulfide and polysulfide. Antioxid. Redox Signal. 2015, 22, 362–376. [Google Scholar] [CrossRef] [Green Version]
- Biermann, J.; Lagreze, W.A.; Schallner, N.; Schwer, C.I.; Goebel, U. Inhalative preconditioning with hydrogen sulfide attenuated apoptosis after retinal ischemia/reperfusion injury. Mol. Vis. 2011, 17, 1275–1286. [Google Scholar]
- Si, Y.F.; Wang, J.; Guan, J.; Zhou, L.; Sheng, Y.; Zhao, J. Treatment with hydrogen sulfide alleviates streptozotocin-induced diabetic retinopathy in rats. Br. J. Pharmacol. 2013, 169, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Anders, F.; Thanos, S.; Mann, C.; Liu, A.; Grus, F.H.; Pfeiffer, N.; Prokosch-Willing, V. Hydrogen sulfide protects retinal ganglion cells against glaucomatous injury in vitro and in vivo. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5129–5141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaneko, Y.; Kimura, Y.; Kimura, H.; Niki, I. L-Cysteine inhibits insulin release from the pancreatic beta-cell: Possible involvement of metabolic production of hydrogen sulfide, a novel gasotransmitter. Diabetes 2006, 55, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Bhatia, M. Hydrogen sulfide: A novel mediator of leukocyte activation. Immunopharmacol. Immunotoxicol. 2008, 30, 631–645. [Google Scholar] [CrossRef]
- Liu, H.; Mercieca, K.; Anders, F.; Prokosch, V. Hydrogen sulfide and beta-synuclein are involved and interlinked in the aging glaucomatous retina. J. Ophthalmol. 2020, 2020, 8642135. [Google Scholar] [CrossRef] [Green Version]
- Hosoya, K.; Nakamura, G.; Akanuma, S.; Tomi, M.; Tachikawa, M. Dehydroascorbic acid uptake and intracellular ascorbic acid accumulation in cultured Muller glial cells (TR-MUL). Neurochem. Int. 2008, 52, 1351–1357. [Google Scholar] [CrossRef] [PubMed]
- Spoelstra-de Man, A.M.E.; Elbers, P.W.G.; Oudemans-Van Straaten, H.M. Vitamin C: Should we supplement? Curr. Opin. Crit. Care 2018, 24, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Krauss, A.; Ferrada, L.; Astuya, A.; Salazar, K.; Cisternas, P.; Martinez, F.; Ramirez, E.; Nualart, F. Dehydroascorbic acid promotes cell death in neurons under oxidative stress: A protective role for astrocytes. Mol. Neurobiol. 2016, 53, 5847–5863. [Google Scholar] [CrossRef]
- Gozzelino, R. The pathophysiology of heme in the brain. Curr. Alzheimer Res. 2016, 13, 174–184. [Google Scholar] [CrossRef]
- Zhang, D.L.; Ghosh, M.C.; Rouault, T.A. The physiological functions of iron regulatory proteins in iron homeostasis—An update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef] [Green Version]
- Gutteridge, J.M.; Halliwell, B. Free radicals and antioxidants in the year 2000. A historical look to the future. Ann. N. Y. Acad. Sci. 2000, 899, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R.; Arosio, P. Iron homeostasis in health and disease. Int. J. Mol. Sci. 2016, 17, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alavi, F.S.; Gheidi, M.; Zahedi, M.; Safari, N.; Ryde, U. A novel mechanism of heme degradation to biliverdin studied by QM/MM and QM calculations. Dalton Trans. 2018, 47, 8283–8291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapitulnik, J.; Maines, M.D. Pleiotropic functions of biliverdin reductase: Cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol. Sci. 2009, 30, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R. Antioxidant activities of bile pigments. Antioxid. Redox Signal. 2004, 6, 841–849. [Google Scholar] [CrossRef]
- Barone, E.; Trombino, S.; Cassano, R.; Sgambato, A.; De Paola, B.; Di Stasio, E.; Picci, N.; Preziosi, P.; Mancuso, C. Characterization of the S-denitrosylating activity of bilirubin. J. Cell. Mol. Med. 2009, 13, 2365–2375. [Google Scholar] [CrossRef]
- Chen, W.; Maghzal, G.J.; Ayer, A.; Suarna, C.; Dunn, L.L.; Stocker, R. Absence of the biliverdin reductase-a gene is associated with increased endogenous oxidative stress. Free Radic. Biol. Med. 2018, 115, 156–165. [Google Scholar] [CrossRef]
- Sharma, N.; Tramutola, A.; Lanzillotta, C.; Arena, A.; Blarzino, C.; Cassano, T.; Butterfield, D.A.; Di Domenico, F.; Perluigi, M.; Barone, E. Loss of biliverdin reductase-A favors Tau hyper-phosphorylation in Alzheimer’s disease. Neurobiol. Dis. 2019, 125, 176–189. [Google Scholar] [CrossRef]
- O’Brien, L.; Hosick, P.A.; John, K.; Stec, D.E.; Hinds, T.D., Jr. Biliverdin reductase isozymes in metabolism. Trends Endocrinol. Metab. TEM 2015, 26, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Tudor, C.; Lerner-Marmarosh, N.; Engelborghs, Y.; Gibbs, P.E.; Maines, M.D. Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem. J. 2008, 413, 405–416. [Google Scholar] [CrossRef] [Green Version]
- Durgin, B.G.; Hahn, S.A.; Schmidt, H.M.; Miller, M.P.; Hafeez, N.; Mathar, I.; Freitag, D.; Sandner, P.; Straub, A.C. Loss of smooth muscle CYB5R3 amplifies angiotensin II-induced hypertension by increasing sGC heme oxidation. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Haunhorst, P.; Hanschmann, E.M.; Brautigam, L.; Stehling, O.; Hoffmann, B.; Muhlenhoff, U.; Lill, R.; Berndt, C.; Lillig, C.H. Crucial function of vertebrate glutaredoxin 3 (PICOT) in iron homeostasis and hemoglobin maturation. Mol. Biol. Cell 2013, 24, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Aslund, F.; Ehn, B.; Miranda-Vizuete, A.; Pueyo, C.; Holmgren, A. Two additional glutaredoxins exist in Escherichia coli: Glutaredoxin 3 is a hydrogen donor for ribonucleotide reductase in a thioredoxin/glutaredoxin 1 double mutant. Proc. Natl. Acad. Sci. USA 1994, 91, 9813–9817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta 2008, 1780, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Li, J.; Chain, C.Y.; Pasquevich, G.A.; Pasquevich, A.F.; Cowan, J.A. Glutathione complexed Fe-S centers. J. Am. Chem. Soc. 2012, 134, 10745–10748. [Google Scholar] [CrossRef] [Green Version]
- Netz, D.J.; Stith, C.M.; Stumpfig, M.; Kopf, G.; Vogel, D.; Genau, H.M.; Stodola, J.L.; Lill, R.; Burgers, P.M.; Pierik, A.J. Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes. Nat. Chem. Biol. 2011, 8, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Rudolf, J.; Makrantoni, V.; Ingledew, W.J.; Stark, M.J.; White, M.F. The DNA repair helicases XPD and FancJ have essential iron-sulfur domains. Mol. Cell 2006, 23, 801–808. [Google Scholar] [CrossRef]
- Kispal, G.; Sipos, K.; Lange, H.; Fekete, Z.; Bedekovics, T.; Janaky, T.; Bassler, J.; Aguilar Netz, D.J.; Balk, J.; Rotte, C.; et al. Biogenesis of cytosolic ribosomes requires the essential iron-sulphur protein Rli1p and mitochondria. EMBO J. 2005, 24, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Maio, N.; Rouault, T.A. Iron-Sulfur cluster biogenesis in mammalian cells: New insights into the molecular mechanisms of cluster delivery. Biochim. Biophys. Acta 2015, 1853, 1493–1512. [Google Scholar] [CrossRef] [Green Version]
- Wachnowsky, C.; Fidai, I.; Cowan, J.A. Cytosolic iron-sulfur cluster transfer-a proposed kinetic pathway for reconstitution of glutaredoxin 3. FEBS Lett. 2016, 590, 4531–4540. [Google Scholar] [CrossRef] [Green Version]
- Muhlenhoff, U.; Molik, S.; Godoy, J.R.; Uzarska, M.A.; Richter, N.; Seubert, A.; Zhang, Y.; Stubbe, J.; Pierrel, F.; Herrero, E.; et al. Cytosolic monothiol glutaredoxins function in intracellular iron sensing and trafficking via their bound iron-sulfur cluster. Cell Metab. 2010, 12, 373–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwema, T.; Picciocchi, A.; Traore, D.A.; Ferrer, J.L.; Chauvat, F.; Jacquamet, L. Structural basis for delivery of the intact [Fe2S2] cluster by monothiol glutaredoxin. Biochemistry 2009, 48, 6041–6043. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Rao, B.; Wachnowsky, C.; Cowan, J.A. Cluster exchange reactivity of [2Fe-2S] cluster-bridged complexes of BOLA3 with monothiol glutaredoxins. Met. Integr. Biometal Sci. 2018, 10, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Li, B.; Zhang, Z.; Wang, Q.; Qiao, T.; Li, K. Human glutaredoxin 3 can bind and effectively transfer [4Fe-4S] cluster to apo-iron regulatory protein 1. Biochem. Biophys. Res. Commun. 2015, 465, 620–624. [Google Scholar] [CrossRef]
- Li, H.; Mapolelo, D.T.; Dingra, N.N.; Naik, S.G.; Lees, N.S.; Hoffman, B.M.; Riggs-Gelasco, P.J.; Huynh, B.H.; Johnson, M.K.; Outten, C.E. The yeast iron regulatory proteins Grx3/4 and Fra2 form heterodimeric complexes containing a [2Fe-2S] cluster with cysteinyl and histidyl ligation. Biochemistry 2009, 48, 9569–9581. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.Q.; Cui, H.; Yu, Y.B.; Shi, H.Q.; Zhou, Y.; Liu, M.J. MicroRNA-141-3p inhibits retinal neovascularization and retinal ganglion cell apoptosis in glaucoma mice through the inactivation of Docking protein 5-dependent mitogen-activated protein kinase signaling pathway. J. Cell. Physiol. 2019, 234, 8873–8887. [Google Scholar] [CrossRef]
- Wu, B.; Teng, H.; Zhang, L.; Li, H.; Li, J.; Wang, L.; Li, H. Interaction of hydrogen sulfide with oxygen sensing under hypoxia. Oxid. Med. Cell. Longev. 2015, 2015, 758678. [Google Scholar] [CrossRef] [Green Version]
- Greco, V.; Spalloni, A.; Corasolla Carregari, V.; Pieroni, L.; Persichilli, S.; Mercuri, N.B.; Urbani, A.; Longone, P. Proteomics and toxicity analysis of spinal-cord primary cultures upon hydrogen sulfide treatment. Antioxidants 2018, 7, 87. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Pan, L.; Zhuo, Y.; Gong, Q.; Rose, P.; Zhu, Y. Hypoxia-Inducible factor-1alpha is involved in the pro-angiogenic effect of hydrogen sulfide under hypoxic stress. Biol. Pharm. Bull. 2010, 33, 1550–1554. [Google Scholar] [CrossRef] [Green Version]
- Kai, S.; Tanaka, T.; Daijo, H.; Harada, H.; Kishimoto, S.; Suzuki, K.; Takabuchi, S.; Takenaga, K.; Fukuda, K.; Hirota, K. Hydrogen sulfide inhibits hypoxia- but not anoxia-induced hypoxia-inducible factor 1 activation in a von hippel-lindau- and mitochondria-dependent manner. Antioxid. Redox Signal. 2012, 16, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Li, J.; Yu, Z.; Rao, H.; Wang, S.; Lan, H. HMGB1 promotes HLF-1 proliferation and ECM production through activating HIF1-alpha-regulated aerobic glycolysis. Pulm. Pharmacol. Ther. 2017, 45, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rak, M.; Rustin, P. Supernumerary subunits NDUFA3, NDUFA5 and NDUFA12 are required for the formation of the extramembrane arm of human mitochondrial complex I. FEBS Lett. 2014, 588, 1832–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peralta, S.; Torraco, A.; Wenz, T.; Garcia, S.; Diaz, F.; Moraes, C.T. Partial complex I deficiency due to the CNS conditional ablation of Ndufa5 results in a mild chronic encephalopathy but no increase in oxidative damage. Hum. Mol. Genet. 2014, 23, 1399–1412. [Google Scholar] [CrossRef] [Green Version]
- Nie, H.; Yu, X.; He, H.; Zhou, L.; Li, Q.; Song, C.; Wang, D.; Ren, T.; Chen, Z.; Huang, H.; et al. Hepatocyte miR-33a mediates mitochondrial dysfunction and hepatosteatosis by suppressing NDUFA5. J. Cell. Mol. Med. 2018, 22, 6285–6293. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Fiskum, G.; Schubert, D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 2002, 80, 780–787. [Google Scholar] [CrossRef]
- Kudin, A.P.; Bimpong-Buta, N.Y.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [Green Version]
- Flones, I.H.; Fernandez-Vizarra, E.; Lykouri, M.; Brakedal, B.; Skeie, G.O.; Miletic, H.; Lilleng, P.K.; Alves, G.; Tysnes, O.B.; Haugarvoll, K.; et al. Neuronal complex I deficiency occurs throughout the Parkinson’s disease brain, but is not associated with neurodegeneration or mitochondrial DNA damage. Acta Neuropathol. 2018, 135, 409–425. [Google Scholar] [CrossRef] [Green Version]
- Burwell, L.S.; Brookes, P.S. Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxid. Redox Signal. 2008, 10, 579–599. [Google Scholar] [CrossRef]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev. Cell 2017, 40, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Papandreou, I.; Cairns, R.A.; Fontana, L.; Lim, A.L.; Denko, N.C. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006, 3, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasior, M.; Rogawski, M.A.; Hartman, A.L. Neuroprotective and disease-modifying effects of the ketogenic diet. Behav. Pharmacol. 2006, 17, 431–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Auwera, I.; Wera, S.; Van Leuven, F.; Henderson, S.T. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr. Metab. 2005, 2, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.V.; Gordon, M.N.; Connor, K.E.; Good, R.A.; Engelman, R.W.; Mason, J.; Morgan, D.G.; Morgan, T.E.; Finch, C.E. Caloric restriction attenuates Abeta-deposition in Alzheimer transgenic models. Neurobiol. Aging 2005, 26, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.P.; Calon, F.; Morihara, T.; Yang, F.; Teter, B.; Ubeda, O.; Salem, N., Jr.; Frautschy, S.A.; Cole, G.M. A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse model. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 3032–3040. [Google Scholar] [CrossRef] [Green Version]
- Holmer, H.K.; Keyghobadi, M.; Moore, C.; Menashe, R.A.; Meshul, C.K. Dietary restriction affects striatal glutamate in the MPTP-induced mouse model of nigrostriatal degeneration. Synapse 2005, 57, 100–112. [Google Scholar] [CrossRef]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.C.; Yan, S.D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-Beta-Hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef] [Green Version]
- Prins, M.L.; Fujima, L.S.; Hovda, D.A. Age-Dependent reduction of cortical contusion volume by ketones after traumatic brain injury. J. Neurosci. Res. 2005, 82, 413–420. [Google Scholar] [CrossRef]
- Cahill, G.F., Jr.; Veech, R.L. Ketoacids? Good medicine? Trans. Am. Clin. Climatol. Assoc. 2003, 114, 149–161; discussion 162–163. [Google Scholar]
- Kashiwaya, Y.; Sato, K.; Tsuchiya, N.; Thomas, S.; Fell, D.A.; Veech, R.L.; Passonneau, J.V. Control of glucose utilization in working perfused rat heart. J. Biol. Chem. 1994, 269, 25502–25514. [Google Scholar]
- Sato, K.; Kashiwaya, Y.; Keon, C.A.; Tsuchiya, N.; King, M.T.; Radda, G.K.; Chance, B.; Clarke, K.; Veech, R.L. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1995, 9, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.; Arend, O.; Sponsel, W.E.; Schulte, K.; Cantor, L.B.; Reim, M. Retinal hemodynamics using scanning laser ophthalmoscopy and hemorheology in chronic open-angle glaucoma. Ophthalmology 1993, 100, 1561–1566. [Google Scholar] [CrossRef]
- Harju, M.; Vesti, E. Blood flow of the optic nerve head and peripapillary retina in exfoliation syndrome with unilateral glaucoma or ocular hypertension. Graefe’s Arch. Clin. Exp. Ophthalmol. 2001, 239, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.Q.; Vaegan; Millar, T.J.; Beaumont, P.; Sarks, S. Widespread choroidal insufficiency in primary open-angle glaucoma. J. Glaucoma 1997, 6, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Flammer, J.; Mozaffarieh, M. Autoregulation, a balancing act between supply and demand. Can. J. Ophthalmol. 2008, 43, 317–321. [Google Scholar] [CrossRef]
- Galassi, F.; Giambene, B.; Varriale, R. Systemic vascular dysregulation and retrobulbar hemodynamics in normal-tension glaucoma. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4467–4471. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Wang, R. H(2)S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H474–H480. [Google Scholar] [CrossRef]
- Siragusa, M.; Fleming, I. The eNOS signalosome and its link to endothelial dysfunction. Pflug. Arch. Eur. J. Physiol. 2016, 468, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Sessa, W.C. eNOS at a glance. J. Cell Sci. 2004, 117, 2427–2429. [Google Scholar] [CrossRef] [Green Version]
- Tasken, K.; Aandahl, E.M. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol. Rev. 2004, 84, 137–167. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.; Hartel, F.V.; Arshad, M.; Gunduz, D.; Abdallah, Y.; Sauer, H.; Piper, H.M.; Noll, T. cAMP/PKA antagonizes thrombin-induced inactivation of endothelial myosin light chain phosphatase: Role of CPI-17. Cardiovasc. Res. 2010, 87, 375–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, C.; Osborne, L.D.; Guilluy, C.; Chen, Z.; O’Brien, E.T., 3rd; Reader, J.S.; Burridge, K.; Superfine, R.; Tzima, E. Haemodynamic and extracellular matrix cues regulate the mechanical phenotype and stiffness of aortic endothelial cells. Nat. Commun. 2014, 5, 3984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldfinger, L.E.; Tzima, E.; Stockton, R.; Kiosses, W.B.; Kinbara, K.; Tkachenko, E.; Gutierrez, E.; Groisman, A.; Nguyen, P.; Chien, S.; et al. Localized alpha4 integrin phosphorylation directs shear stress-induced endothelial cell alignment. Circ. Res. 2008, 103, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakre, M.M.; Zhu, Y.; Yin, H.; Burton, D.W.; Terkeltaub, R.; Deftos, L.J.; Varner, J.A. Parathyroid hormone-related peptide is a naturally occurring, protein kinase A-dependent angiogenesis inhibitor. Nat. Med. 2002, 8, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Garmy-Susini, B.; Avraamides, C.J.; Stoletov, K.; Klemke, R.L.; Varner, J.A. A PKA-Csk-pp60Src signaling pathway regulates the switch between endothelial cell invasion and cell-cell adhesion during vascular sprouting. Blood 2010, 116, 5773–5783. [Google Scholar] [CrossRef] [Green Version]
- Plitzko, B.; Havemeyer, A.; Bork, B.; Bittner, F.; Mendel, R.; Clement, B. Defining the role of the NADH-Cytochrome-b5 Reductase 3 in the mitochondrial amidoxime reducing component enzyme system. Drug Metab. Dispos. Biol. Fate Chem. 2016, 44, 1617–1621. [Google Scholar] [CrossRef]
- Wood, K.C.; Durgin, B.G.; Schmidt, H.M.; Hahn, S.A.; Baust, J.J.; Bachman, T.; Vitturi, D.A.; Ghosh, S.; Ofori-Acquah, S.F.; Mora, A.L.; et al. Smooth muscle cytochrome b5 reductase 3 deficiency accelerates pulmonary hypertension development in sickle cell mice. Blood Adv. 2019, 3, 4104–4116. [Google Scholar] [CrossRef]
- Rahaman, M.M.; Nguyen, A.T.; Miller, M.P.; Hahn, S.A.; Sparacino-Watkins, C.; Jobbagy, S.; Carew, N.T.; Cantu-Medellin, N.; Wood, K.C.; Baty, C.J.; et al. Cytochrome b5 Reductase 3 Modulates Soluble Guanylate Cyclase Redox State and cGMP Signaling. Circ. Res. 2017, 121, 137–148. [Google Scholar] [CrossRef]
- Fukuto, J.M.; Carrington, S.J.; Tantillo, D.J.; Harrison, J.G.; Ignarro, L.J.; Freeman, B.A.; Chen, A.; Wink, D.A. Small molecule signaling agents: The integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species. Chem. Res. Toxicol. 2012, 25, 769–793. [Google Scholar] [CrossRef]
- Samaan, S.; Tranchevent, L.C.; Dardenne, E.; Polay Espinoza, M.; Zonta, E.; Germann, S.; Gratadou, L.; Dutertre, M.; Auboeuf, D. The Ddx5 and Ddx17 RNA helicases are cornerstones in the complex regulatory array of steroid hormone-signaling pathways. Nucleic Acids Res. 2014, 42, 2197–2207. [Google Scholar] [CrossRef]
- Dewundara, S.S.; Wiggs, J.L.; Sullivan, D.A.; Pasquale, L.R. Is estrogen a therapeutic target for glaucoma? Semin. Ophthalmol. 2016, 31, 140–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, P.; Harris, A.; Toris, C.; Tobe, L.; Lang, M.; Belamkar, A.; Ng, A.; Verticchio Vercellin, A.C.; Mathew, S.; Siesky, B. Effects of sex hormones on ocular blood flow and intraocular pressure in primary open-angle glaucoma: A review. J. Glaucoma 2018, 27, 1037–1041. [Google Scholar] [CrossRef] [Green Version]
- Wickham, L.A.; Gao, J.; Toda, I.; Rocha, E.M.; Ono, M.; Sullivan, D.A. Identification of androgen, estrogen and progesterone receptor mRNAs in the eye. Acta Ophthalmol. Scand. 2000, 78, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Kobayashi, H.; Ueda, M.; Honda, Y. Estrogen receptor expression in bovine and rat retinas. Investig. Ophthalmol. Vis. Sci. 1998, 39, 2105–2110. [Google Scholar]
- Chakrabarti, M.; Haque, A.; Banik, N.L.; Nagarkatti, P.; Nagarkatti, M.; Ray, S.K. Estrogen receptor agonists for attenuation of neuroinflammation and neurodegeneration. Brain Res. Bull. 2014, 109, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Ma, X.; Zhao, Q.; Li, Y.; Xing, Y.; Deng, Q.; Shen, Y. The neuroprotective effects of novel estrogen receptor GPER1 in mouse retinal ganglion cell degeneration. Exp. Eye Res. 2019, 189, 107826. [Google Scholar] [CrossRef]
- Sarzi, E.; Seveno, M.; Angebault, C.; Milea, D.; Ronnback, C.; Quiles, M.; Adrian, M.; Grenier, J.; Caignard, A.; Lacroux, A.; et al. Increased steroidogenesis promotes early-onset and severe vision loss in females with OPA1 dominant optic atrophy. Hum. Mol. Genet. 2016, 25, 2539–2551. [Google Scholar] [CrossRef] [Green Version]
- Pisano, A.; Preziuso, C.; Iommarini, L.; Perli, E.; Grazioli, P.; Campese, A.F.; Maresca, A.; Montopoli, M.; Masuelli, L.; Sadun, A.A.; et al. Targeting estrogen receptor beta as preventive therapeutic strategy for Leber’s hereditary optic neuropathy. Hum. Mol. Genet. 2015, 24, 6921–6931. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, Y.; Zhang, Y.; Kam, W.R.; Pasquale, L.R.; Sullivan, D.A. Impact of aromatase absence on murine intraocular pressure and retinal ganglion cells. Sci. Rep. 2018, 8, 3280. [Google Scholar] [CrossRef]
- Feola, A.J.; Fu, J.; Allen, R.; Yang, V.; Campbell, I.C.; Ottensmeyer, A.; Ethier, C.R.; Pardue, M. Menopause exacerbates visual dysfunction in experimental glaucoma. Exp. Eye Res. 2019, 186, 107706. [Google Scholar] [CrossRef]
- Shah, A.; Schiffmacher, A.T.; Taneyhill, L.A. Annexin A6 controls neuronal membrane dynamics throughout chick cranial sensory gangliogenesis. Dev. Biol. 2017, 425, 85–99. [Google Scholar] [CrossRef]
- Klee, C.B. Ca2+-dependent phospholipid- (and membrane-) binding proteins. Biochemistry 1988, 27, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Kapustin, A.N.; Shanahan, C.M. Emerging roles for vascular smooth muscle cell exosomes in calcification and coagulation. J. Physiol. 2016, 594, 2905–2914. [Google Scholar] [CrossRef]
- Hubner, C.A.; Schroeder, B.C.; Ehmke, H. Regulation of vascular tone and arterial blood pressure: Role of chloride transport in vascular smooth muscle. Pflug. Arch. Eur. J. Physiol. 2015, 467, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Chang, Y.; Kim, C.W.; Kwon, M.J.; Choi, Y.; Ahn, J.; Kim, J.M.; Kim, H.S.; Shin, H.; Ryu, S. Intraocular pressure and coronary artery calcification in asymptomatic men and women. Br. J. Ophthalmol. 2015, 99, 932–936. [Google Scholar] [CrossRef]
- Borden, L.A. GABA transporter heterogeneity: Pharmacology and cellular localization. Neurochem. Int. 1996, 29, 335–356. [Google Scholar] [CrossRef]
- Crooks, J.; Kolb, H. Localization of GABA, glycine, glutamate and tyrosine hydroxylase in the human retina. J. Comp. Neurol. 1992, 315, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Hilton, E.J.; Hosking, S.L.; Betts, T. The effect of antiepileptic drugs on visual performance. Seizure 2004, 13, 113–128. [Google Scholar] [CrossRef] [Green Version]
- Sakaew, W.; Tachow, A.; Thoungseabyoun, W.; Khrongyut, S.; Rawangwong, A.; Polsan, Y.; Masahiko, W.; Kondo, H.; Hipkaeo, W. Expression and localization of VIAAT in distal uriniferous tubular epithelium of mouse. Ann. Anat. Anat. Anz. 2019, 222, 21–27. [Google Scholar] [CrossRef]
- Kononenko, N.L.; Puchkov, D.; Classen, G.A.; Walter, A.M.; Pechstein, A.; Sawade, L.; Kaempf, N.; Trimbuch, T.; Lorenz, D.; Rosenmund, C.; et al. Clathrin/AP-2 mediate synaptic vesicle reformation from endosome-like vacuoles but are not essential for membrane retrieval at central synapses. Neuron 2014, 82, 981–988. [Google Scholar] [CrossRef] [Green Version]
- Kononenko, N.L.; Classen, G.A.; Kuijpers, M.; Puchkov, D.; Maritzen, T.; Tempes, A.; Malik, A.R.; Skalecka, A.; Bera, S.; Jaworski, J.; et al. Retrograde transport of TrkB-containing autophagosomes via the adaptor AP-2 mediates neuronal complexity and prevents neurodegeneration. Nat. Commun. 2017, 8, 14819. [Google Scholar] [CrossRef] [PubMed]
- Helbig, I.; Lopez-Hernandez, T.; Shor, O.; Galer, P.; Ganesan, S.; Pendziwiat, M.; Rademacher, A.; Ellis, C.A.; Humpfer, N.; Schwarz, N.; et al. A recurrent missense variant in AP2M1 impairs clathrin-mediated endocytosis and causes developmental and epileptic encephalopathy. Am. J. Hum. Genet. 2019, 104, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kittler, J.T.; Chen, G.; Honing, S.; Bogdanov, Y.; McAinsh, K.; Arancibia-Carcamo, I.L.; Jovanovic, J.N.; Pangalos, M.N.; Haucke, V.; Yan, Z.; et al. Phospho-dependent binding of the clathrin AP2 adaptor complex to GABAA receptors regulates the efficacy of inhibitory synaptic transmission. Proc. Natl. Acad. Sci. USA 2005, 102, 14871–14876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froemke, R.C. Plasticity of cortical excitatory-inhibitory balance. Annu. Rev. Neurosci. 2015, 38, 195–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.R.; Muir, J.; Rao, Y.; Browarski, M.; Gruenig, M.C.; Sheehan, D.F.; Haucke, V.; Kittler, J.T. Stabilization of GABA(A) receptors at endocytic zones is mediated by an AP2 binding motif within the GABA(A) receptor beta3 subunit. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 2485–2498. [Google Scholar] [CrossRef] [Green Version]
- Neal, M.J.; Shah, M.A. Development of tolerance to the effects of vigabatrin (gamma-vinyl-GABA) on GABA release from rat cerebral cortex, spinal cord and retina. Br. J. Pharmacol. 1990, 100, 324–328. [Google Scholar] [CrossRef]
- Hosking, S.L.; Hilton, E.J. Neurotoxic effects of GABA-transaminase inhibitors in the treatment of epilepsy: Ocular perfusion and visual performance. Ophthalmic Physiol. Opt. J. Br. Coll. Ophthalmic Opt. 2002, 22, 440–447. [Google Scholar] [CrossRef]
- Hilton, E.J.; Hosking, S.L.; Betts, T. Epilepsy patients treated with antiepileptic drug therapy exhibit compromised ocular perfusion characteristics. Epilepsia 2002, 43, 1346–1350. [Google Scholar] [CrossRef] [Green Version]
- Hammond, E.J.; Wilder, B.J. Gamma-vinyl GABA. Gen. Pharmacol. 1985, 16, 441–447. [Google Scholar] [CrossRef]
- Wild, J.M.; Martinez, C.; Reinshagen, G.; Harding, G.F. Characteristics of a unique visual field defect attributed to vigabatrin. Epilepsia 1999, 40, 1784–1794. [Google Scholar] [CrossRef]
- Fujiwara, M.; Muramatsu, I. Gamma-Aminobutyric acid receptor on vascular smooth muscle of dog cerebral arteries. Br. J. Pharmacol. 1975, 55, 561–562. [Google Scholar] [CrossRef] [PubMed]
- Edvinsson, L.; Krause, D.N. Pharmacological characterization of GABA receptors mediating vasodilation of verebral arteries in vitro. Brain Res. 1979, 173, 89–97. [Google Scholar] [CrossRef]
- Hinds, K.; Monaghan, K.P.; Frolund, B.; McGeown, J.G.; Curtis, T.M. GABAergic control of arteriolar diameter in the rat retina. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6798–6805. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.W.; Dubiel, W.; Wei, N.; Hofmann, K.; Mundt, K.; Colicelli, J.; Kato, J.; Naumann, M.; Segal, D.; Seeger, M.; et al. Unified nomenclature for the COP9 signalosome and its subunits: An essential regulator of development. Trends Genet. TIG 2000, 16, 202–203. [Google Scholar] [CrossRef]
- Boussiotis, V.A.; Freeman, G.J.; Taylor, P.A.; Berezovskaya, A.; Grass, I.; Blazar, B.R.; Nadler, L.M. p27kip1 functions as an anergy factor inhibiting interleukin 2 transcription and clonal expansion of alloreactive human and mouse helper T lymphocytes. Nat. Med. 2000, 6, 290–297. [Google Scholar] [CrossRef]
- Doronkin, S.; Djagaeva, I.; Beckendorf, S.K. The COP9 signalosome promotes degradation of Cyclin E during early Drosophila oogenesis. Dev. Cell 2003, 4, 699–710. [Google Scholar] [CrossRef] [Green Version]
- Yan, J.; Walz, K.; Nakamura, H.; Carattini-Rivera, S.; Zhao, Q.; Vogel, H.; Wei, N.; Justice, M.J.; Bradley, A.; Lupski, J.R. COP9 signalosome subunit 3 is essential for maintenance of cell proliferation in the mouse embryonic epiblast. Mol. Cell. Biol. 2003, 23, 6798–6808. [Google Scholar] [CrossRef] [Green Version]
- Hetfeld, B.K.; Peth, A.; Sun, X.M.; Henklein, P.; Cohen, G.M.; Dubiel, W. The COP9 signalosome-mediated deneddylation is stimulated by caspases during apoptosis. Apoptosis Int. J. Program. Cell Death 2008, 13, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Da Silva Correia, J.; Miranda, Y.; Leonard, N.; Ulevitch, R.J. The subunit CSN6 of the COP9 signalosome is cleaved during apoptosis. J. Biol. Chem. 2007, 282, 12557–12565. [Google Scholar] [CrossRef] [Green Version]
- De Laat, W.L.; Jaspers, N.G.; Hoeijmakers, J.H. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999, 13, 768–785. [Google Scholar] [CrossRef] [Green Version]
- Scharer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Du, W.; Acklin, S.M.; Jin, S.; Xia, F. SIRT2 protects peripheral neurons from cisplatin-induced injury by enhancing nucleotide excision repair. J. Clin. Investig. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros, L.F.; San Martin, A.; Ruminot, I.; Sandoval, P.Y.; Fernandez-Moncada, I.; Baeza-Lehnert, F.; Arce-Molina, R.; Contreras-Baeza, Y.; Cortes-Molina, F.; Galaz, A.; et al. Near-critical GLUT1 and Neurodegeneration. J. Neurosci. Res. 2017, 95, 2267–2274. [Google Scholar] [CrossRef] [Green Version]
- Perumal, N.; Funke, S.; Pfeiffer, N.; Grus, F.H. Proteomics analysis of human tears from aqueous-deficient and evaporative dry eye patients. Sci. Rep. 2016, 6, 29629. [Google Scholar] [CrossRef]
- Perumal, N.; Funke, S.; Pfeiffer, N.; Grus, F.H. Characterization of lacrimal proline-rich protein 4 (PRR4) in human tear proteome. Proteomics 2014, 14, 1698–1709. [Google Scholar] [CrossRef] [PubMed]
- Flammer, J.; Orgul, S.; Costa, V.P.; Orzalesi, N.; Krieglstein, G.K.; Serra, L.M.; Renard, J.P.; Stefansson, E. The impact of ocular blood flow in glaucoma. Prog. Retin. Eye Res. 2002, 21, 359–393. [Google Scholar] [CrossRef]
- Osborne, N.N.; Melena, J.; Chidlow, G.; Wood, J.P. A hypothesis to explain ganglion cell death caused by vascular insults at the optic nerve head: Possible implication for the treatment of glaucoma. Br. J. Ophthalmol. 2001, 85, 1252–1259. [Google Scholar] [CrossRef] [Green Version]
- Golubnitschaja-Labudova, O.; Liu, R.; Decker, C.; Zhu, P.; Haefliger, I.O.; Flammer, J. Altered gene expression in lymphocytes of patients with normal-tension glaucoma. Curr. Eye Res. 2000, 21, 867–876. [Google Scholar] [CrossRef]
- Flammer, J. Glaucomatous optic neuropathy: A reperfusion injury. Klin. Mon. Fur Augenheilkd. 2001, 218, 290–291. [Google Scholar] [CrossRef]
- Berkowitz, B.A.; Lukaszew, R.A.; Mullins, C.M.; Penn, J.S. Impaired hyaloidal circulation function and uncoordinated ocular growth patterns in experimental retinopathy of prematurity. Investig. Ophthalmol. Vis. Sci. 1998, 39, 391–396. [Google Scholar]
- Nadal-Nicolas, F.M.; Jimenez-Lopez, M.; Sobrado-Calvo, P.; Nieto-Lopez, L.; Canovas-Martinez, I.; Salinas-Navarro, M.; Vidal-Sanz, M.; Agudo, M. Brn3a as a marker of retinal ganglion cells: Qualitative and quantitative time course studies in naive and optic nerve-injured retinas. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3860–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadal-Nicolas, F.M.; Sobrado-Calvo, P.; Jimenez-Lopez, M.; Vidal-Sanz, M.; Agudo-Barriuso, M. Long-Term effect of optic nerve axotomy on the retinal ganglion cell layer. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6095–6112. [Google Scholar] [CrossRef] [PubMed]
- Nadal-Nicolas, F.M.; Salinas-Navarro, M.; Jimenez-Lopez, M.; Sobrado-Calvo, P.; Villegas-Perez, M.P.; Vidal-Sanz, M.; Agudo-Barriuso, M. Displaced retinal ganglion cells in albino and pigmented rats. Front. Neuroanat. 2014, 8, 99. [Google Scholar] [CrossRef] [Green Version]
- Anders, F.; Teister, J.; Liu, A.; Funke, S.; Grus, F.H.; Thanos, S.; von Pein, H.D.; Pfeiffer, N.; Prokosch, V. Intravitreal injection of beta-crystallin B2 improves retinal ganglion cell survival in an experimental animal model of glaucoma. PLoS ONE 2017, 12, e0175451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manicam, C.; Perumal, N.; Pfeiffer, N.; Grus, F.H.; Gericke, A. First insight into the proteome landscape of the porcine short posterior ciliary arteries: Key signalling pathways maintaining physiologic functions. Sci. Rep. 2016, 6, 38298. [Google Scholar] [CrossRef] [PubMed]
- Manicam, C.; Perumal, N.; Wasielica-Poslednik, J.; Ngongkole, Y.C.; Tschabunin, A.; Sievers, M.; Lisch, W.; Pfeiffer, N.; Grus, F.H.; Gericke, A. Proteomics unravels the regulatory mechanisms in human tears following acute renouncement of contact lens use: A comparison between hard and soft lenses. Sci. Rep. 2018, 8, 11526. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein IDs | Gene Names | p-Value | Abundance |
|---|---|---|---|
| Q15029 | EFTUD2 | 0.0006042 | low |
| Q62940 | Nedd4 | 0.00061229 | low |
| Q08163 | Cap1 | 0.00086462 | low |
| Q7Z5P9 | MUC19 | 0.00091244 | low |
| Q9H598 | SLC32A1 | 0.00091751 | high |
| Q15181 | PPA1 | 0.0016681 | high |
| Q3ZCM7 | TUBB8 | 0.0031885 | low |
| Q96CW1 | AP2M1 | 0.00352499 | high |
| O75352 | MPDU1 | 0.00377415 | high |
| P97519 | Hmgcl | 0.00470492 | low |
| P34931 | HSPA1L | 0.00595437 | low |
| P20070 | Cyb5r3 | 0.00814946 | high |
| P36871 | PGM1 | 0.00912329 | high |
| B0BNE5 | Esd | 0.0096494 | high |
| Q9UNM6 | PSMD13 | 0.0112771 | high |
| Q5XIK2 | Tmx2 | 0.0124046 | high |
| P46844 | BLVRA | 0.0135271 | high |
| Q63524 | Tmed2 | 0.0150562 | high |
| Q63598 | Pls3 | 0.0158343 | low |
| Q62927 | Cnga1 | 0.0175332 | low |
| Q8IX01 | SUGP2 | 0.019042 | low |
| P97612 | Faah | 0.0195873 | low |
| P61203 | Cops2 | 0.0208293 | high |
| Q497B0 | Nit2 | 0.0211131 | high |
| P39069 | Ak1 | 0.0216897 | high |
| P11506 | Atp2b2 | 0.0227347 | low |
| Q7TP47 | Syncrip | 0.0239965 | low |
| Q8NCB2 | CAMKV | 0.0247766 | low |
| P14854 | COX6B1 | 0.0270203 | low |
| P11167 | Slc2a1 | 0.0276784 | low |
| P45479 | Ppt1 | 0.0279009 | high |
| Q9QZA2 | Pdcd6ip | 0.0299058 | low |
| P41498 | Acp1 | 0.032686 | high |
| P10155 | TROVE2 | 0.0333496 | high |
| Q64428 | Hadha | 0.0337918 | low |
| P83916 | CBX1 | 0.0342579 | high |
| Q64560 | Tpp2 | 0.0352463 | high |
| P26453 | Bsg | 0.0366156 | high |
| Q641X8 | Eif3e | 0.0387402 | high |
| Q6AYR6 | Hdhd2 | 0.0399141 | high |
| P08413 | Camk2b | 0.0416695 | high |
| O08838 | Amph | 0.0425382 | low |
| Q86V81 | ALYREF | 0.0427918 | high |
| P63211 | GNGT1 | 0.043069 | low |
| P84090 | ERH | 0.0437723 | low |
| P41542 | Uso1 | 0.0456321 | low |
| Q3ZAQ7 | VMA21 | 0.0494713 | low |
| P85973 | Pnp | 0.0496038 | high |
| Protein Class | Gene Name | p-Value | Abundance |
|---|---|---|---|
| Cytoskeletal Protein | TUBB8 | 0.0031885 | low |
| Membrane Traffic Protein | AP2M1 | 0.00352499 | high |
| Tmed2 | 0.0150562 | high | |
| Metabolite Interconversion Enzyme | Hmgcl | 0.00470492 | low |
| Nit2 | 0.0211131 | high | |
| Hadha | 0.0337918 | low | |
| Nucleic Acid Binding Protein | SUGP2 | 0.019042 | low |
| Syncrip | 0.0239965 | low | |
| Protein Modifying Enzyme | PSMD13 | 0.0112771 | high |
| CAMKV | 0.0247766 | low | |
| Tpp2 | 0.0352463 | high | |
| Translational Protein | EFTUD2 | 0.0006042 | low |
| Eif3e | 0.0387402 | high | |
| Other Protein | SLC32A1 | 0.00091751 | high |
| MPDU1 | 0.00377415 | high | |
| Cyb5r3 | 0.00814946 | high | |
| Tmx2 | 0.0124046 | high | |
| Slc2a1 | 0.0276784 | low |
| Protein Class | Gene Name | p-Value | Abundance |
|---|---|---|---|
| Calcium-binding Protein | Anxa6 | 0.0245515 | low |
| Cytoskeletal Protein | Dynlrb1 | 0.0227117 | high |
| Metabolite Interconversion Enzyme | Glrx3 | 0.0010183 | high |
| Ndufa5 | 0.0473985 | low | |
| Nucleic Acid Binding Protein | SNRPG | 0.0125193 | low |
| Translational Protein | Rps28 | 0.0406697 | low |
| COPS6 | 0.0463107 | high | |
| Other Proteins | DDX5 | 0.0242271 | high |
| KRT1 | 0.0326712 | low | |
| NUP205 | 0.0335003 | high | |
| CXorf22 | 0.0419458 | low |
| Canonical Pathways | −log(p-Value) | Changes | Molecules | |
|---|---|---|---|---|
| Mitochondrial Homeostasis and Function | Ketogenesis | 3.62 | Downregulated | HADHA, HMGCL |
| Protein Ubiquitination Pathway | 1.7 | Upregulated | HSPA1L, PSMD13, USO1 | |
| Leucine Degradation I | 1.72 | Downregulated | HMGCL | |
| Ketolysis | 1.68 | Downregulated | HADHA | |
| Neuronal Calcium Dysregulation | Calcium Transport I | 1.68 | Upregulated | ATP2B2 |
| Cytotoxicity Regulation | Formaldehyde Oxidation II (Glutathione-dependent) | 2.37 | Downregulated | ESD |
| Anandamide Degradation | 2.2 | Upregulated | FAAH | |
| Purine Ribonucleosides Degradation to Ribose-1-phosphate | 1.83 | Downregulated | PNP | |
| Reactive Oxygen Species (ROS) Scavenging | Heme Degradation | 2.07 | Upregulated | BLVRA |
| Vitamin-C Transport | 1.31 | Downregulated | SLC2A1 | |
| Neural Transduction | Phototransduction Pathway | 2.25 | Downregulated | CNGA1, GNGT1 |
| GABA Receptor Signaling | 1.76 | Upregulated | AP2M1, SLC32A1 | |
| Vascular Function | eNOS Signaling | 1.35 | Downregulated | CNGA1, HSPA1L |
| Protein Kinase A Signaling | 1.29 | Upregulated | ACP1, CAMK2B, CNGA1 |
| Canonical Pathways | −log(p-Value) | Changes | Molecules | |
|---|---|---|---|---|
| Mitochondrial Homeostasis and Function | Ketogenesis | 3.78 | Upregulated | HADHA, HMGCL |
| Leucine Degradation I | 1.8 | Upregulated | HMGCL | |
| Ketolysis | 1.76 | Upregulated | HADHA | |
| Neural Transduction | GABA Receptor Signaling | 1.91 | Downregulated | AP2M1, SLC32A1 |
| ROS Regulation | Vitamin-C Transport | 1.38 | Upregulated | SLC2A1 |
| HIF1α Signaling | 0.742 | Upregulated | SLC2A1 | |
| Canonical Pathways | −log(p-Value) | Changes | Molecules | |
| NER pathway | 1.18 | Upregulated | COPS6 | |
| Oxidative Phosphorylation | 1.16 | Downregulated | NDUFA5 | |
| Estrogen Receptor Signaling | 1.04 | Upregulated | DDX5 | |
| Corticotropin Releasing Hormone Signaling | 1.02 | - | KRT1 | |
| Phagosome Maturation | 0.708 | - | DYNLRB1 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, H.; Perumal, N.; Manicam, C.; Mercieca, K.; Prokosch, V. Proteomics Reveals the Potential Protective Mechanism of Hydrogen Sulfide on Retinal Ganglion Cells in an Ischemia/Reperfusion Injury Animal Model. Pharmaceuticals 2020, 13, 213. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090213
Liu H, Perumal N, Manicam C, Mercieca K, Prokosch V. Proteomics Reveals the Potential Protective Mechanism of Hydrogen Sulfide on Retinal Ganglion Cells in an Ischemia/Reperfusion Injury Animal Model. Pharmaceuticals. 2020; 13(9):213. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090213
Chicago/Turabian StyleLiu, Hanhan, Natarajan Perumal, Caroline Manicam, Karl Mercieca, and Verena Prokosch. 2020. "Proteomics Reveals the Potential Protective Mechanism of Hydrogen Sulfide on Retinal Ganglion Cells in an Ischemia/Reperfusion Injury Animal Model" Pharmaceuticals 13, no. 9: 213. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090213