The Anticancer Activity for the Bumetanide-Based Analogs via Targeting the Tumor-Associated Membrane-Bound Human Carbonic Anhydrase-IX Enzyme

 , , and
, , and 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Biological Evaluation
2.1.1. Carbonic Anhydrase Inhibition Assay
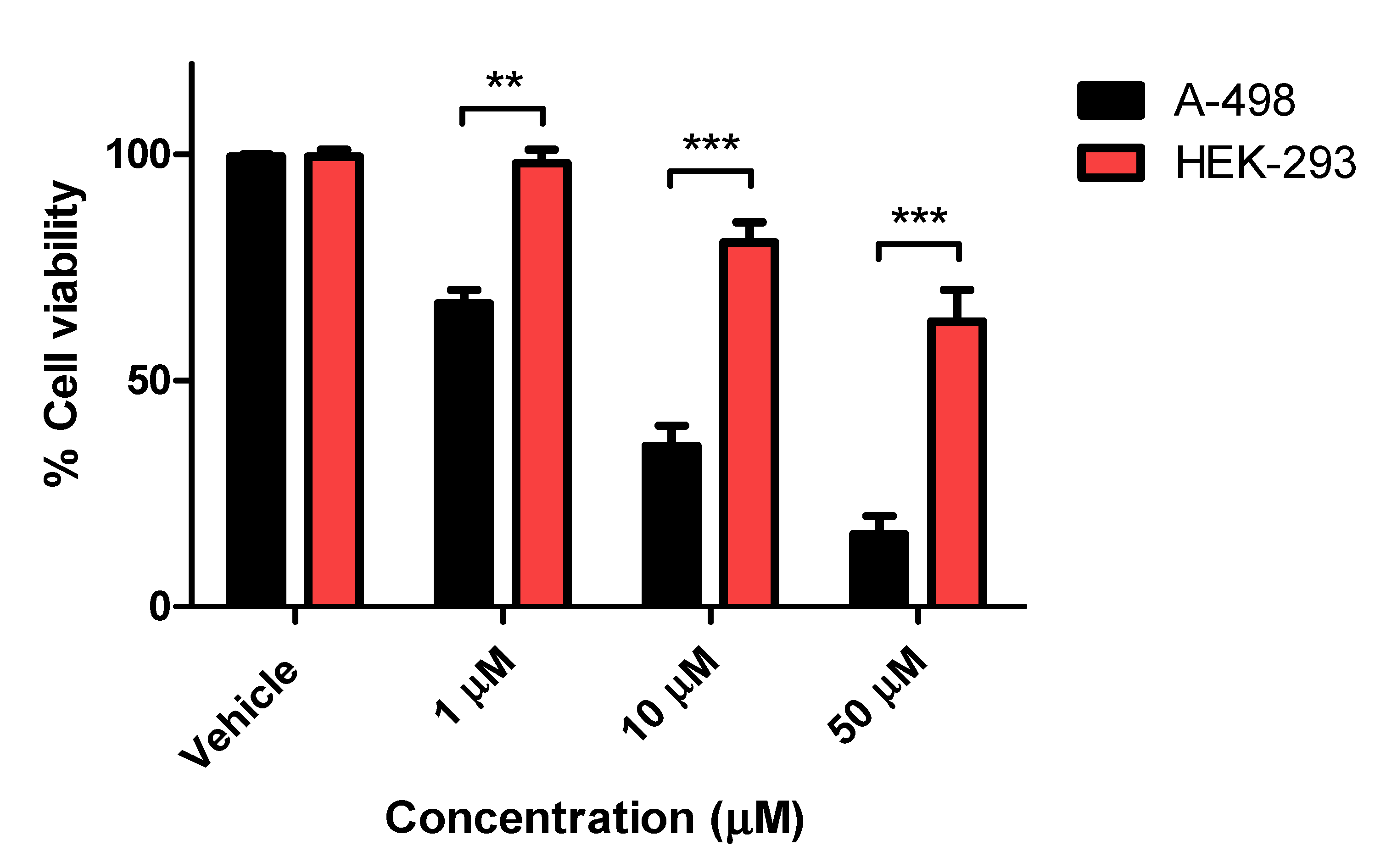
2.1.2. Cell Proliferation Assay
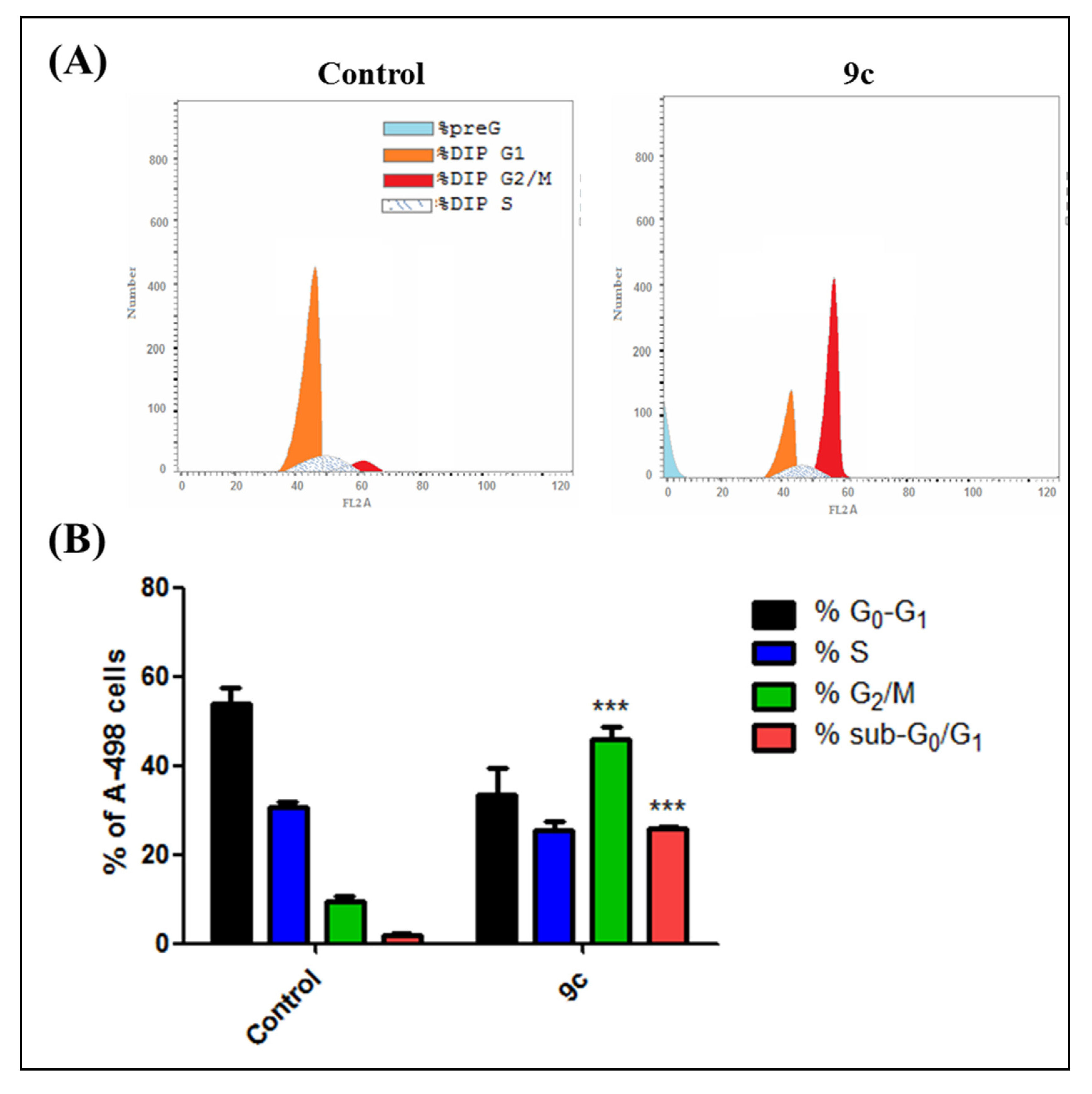
2.1.3. Cell Cycle Analysis and Apoptosis Rate
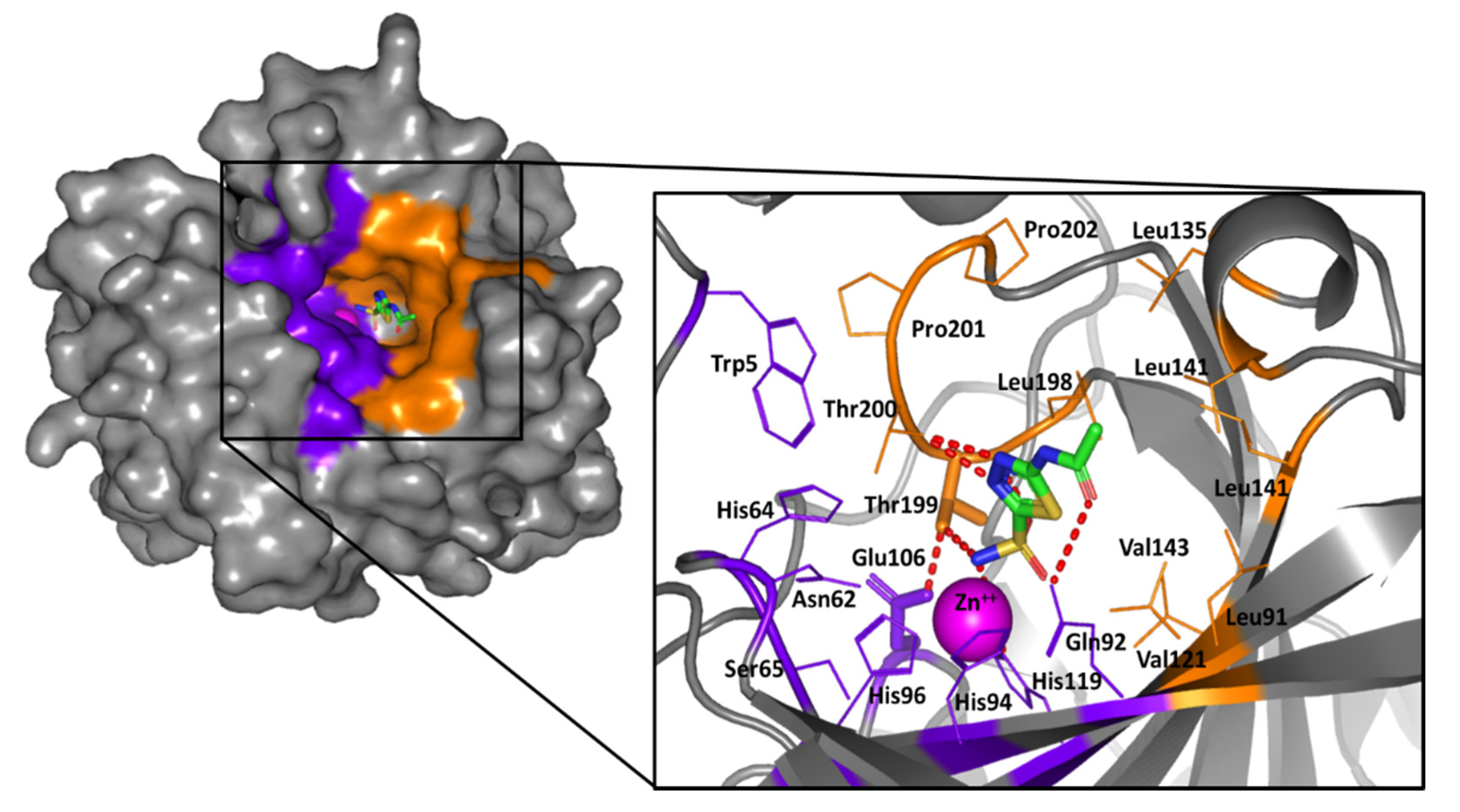
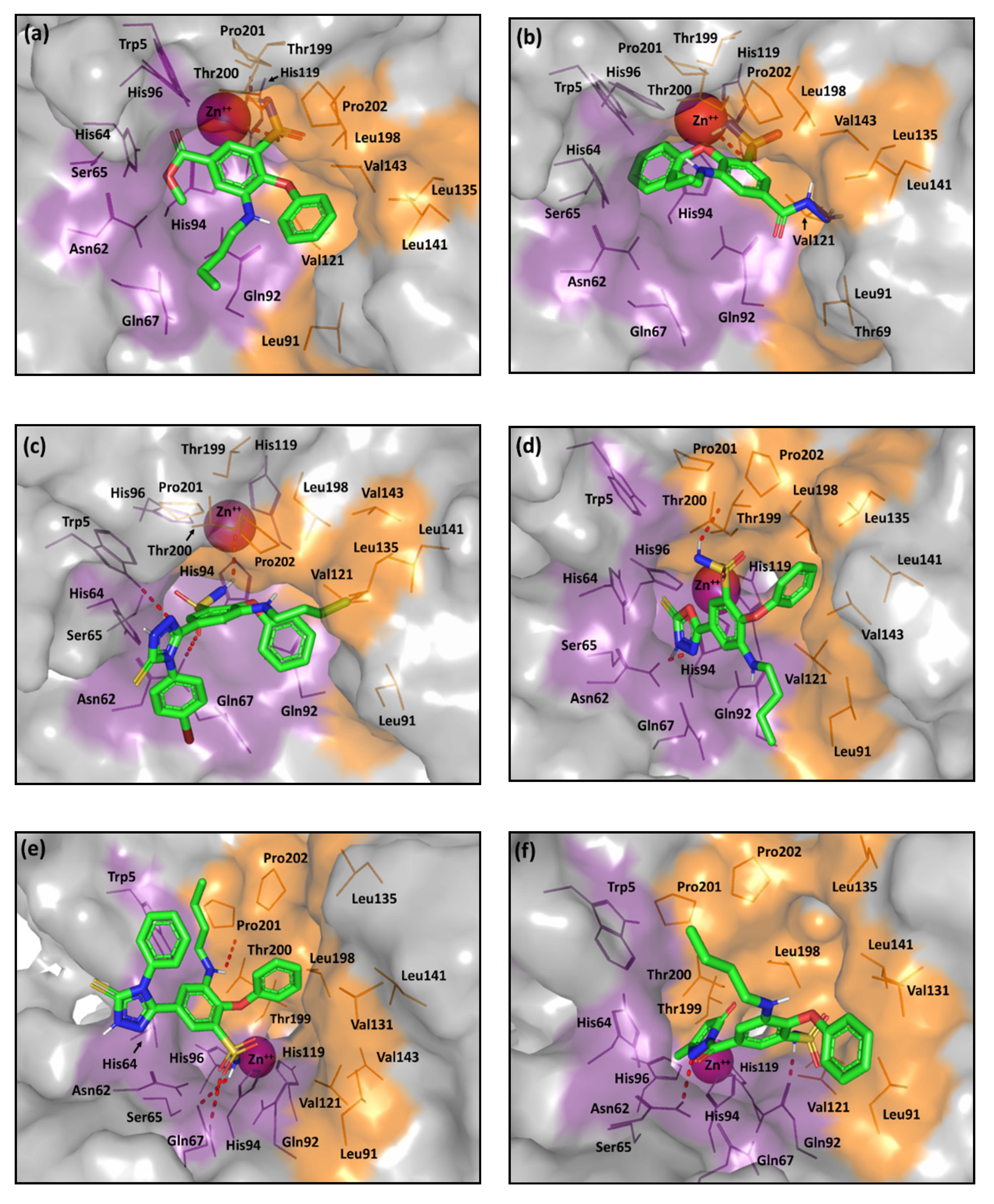
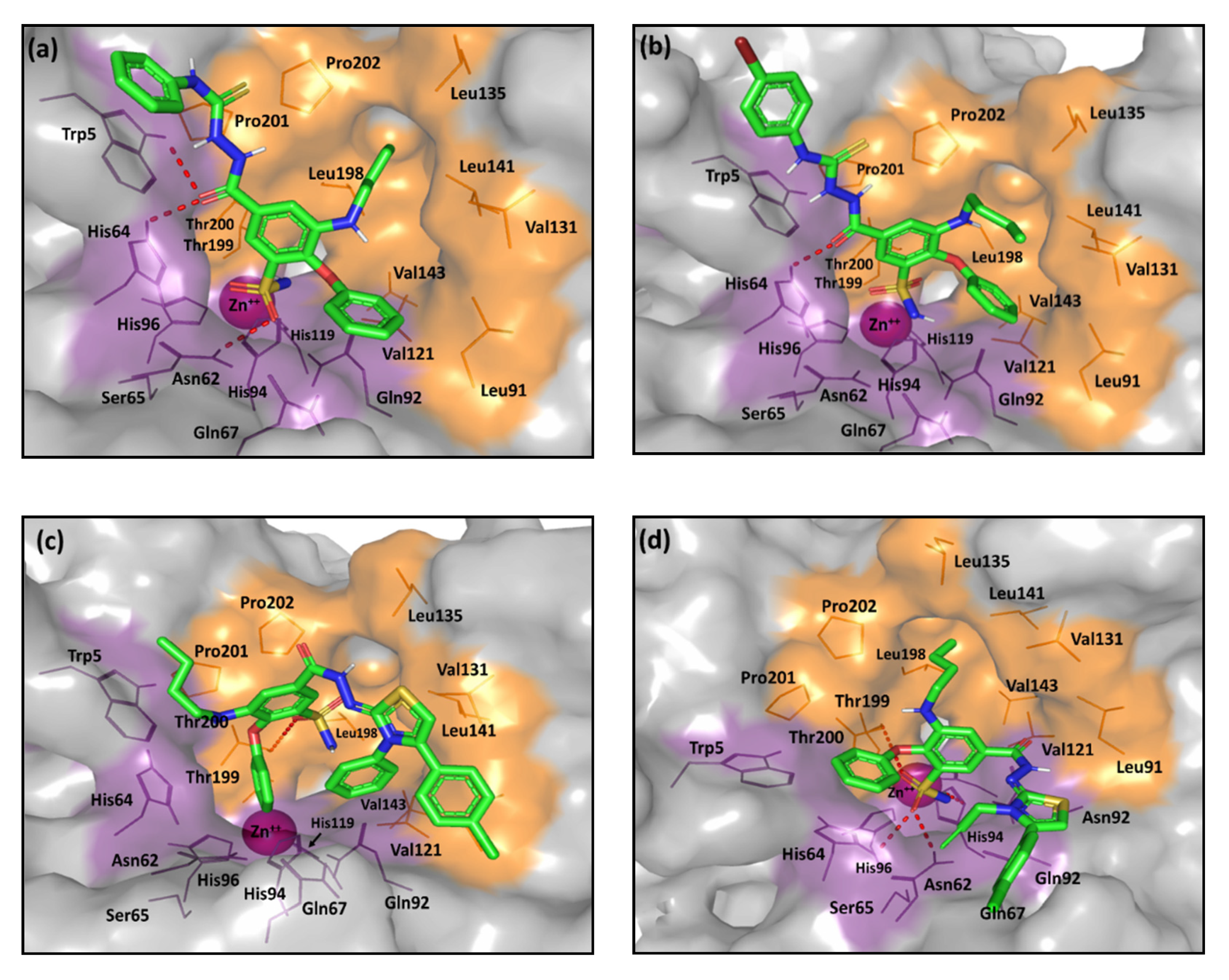
2.2. Computational Study
3. Materials and Methods
3.1. Carbonic Anhydrase Inhibition Assay
3.2. MTT Cytotoxicity Assay
3.3. Cell Cycle Analysis and Apoptosis Rate Investigation
3.4. Statistical Analysis
3.5. Molecular Docking Protocol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Collaborators, G.R.F. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1659–1724. [Google Scholar] [CrossRef] [Green Version]
- Stewart, B.W.; Wild, C.P. World Cancer Report 2014; International Agency for Research on Cancer: Lyon, France, 2014. [Google Scholar]
- Pucci, C.; Martinelli, C.; Ciofani, G. Innovative approaches for cancer treatment: Current perspectives and new challenges. Ecancermedicalscience 2019, 13, 961. [Google Scholar] [CrossRef] [PubMed]
- Urruticoechea, A.; Alemany, R.; Balart, J.; Villanueva, A.; Viñals, F.; Capellá, G. Recent advances in cancer therapy: An overview. Curr. Pharm. Des. 2010, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueff, J.; Rodrigues, A.S. Cancer Drug Resistance: A Brief Overview from a Genetic Viewpoint. Methods Mol. Biol. 2016, 1395, 1–18. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Borst, P. Cancer drug pan-resistance: Pumps, cancer stem cells, quiescence, epithelial to mesenchymal transition, blocked cell death pathways, persisters or what? Open Biol. 2012, 2, 120066. [Google Scholar] [CrossRef] [Green Version]
- Alfarouk, K.O.; Stock, C.M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to cancer chemotherapy: Failure in drug response from ADME to P-gp. Cancer Cell Int. 2015, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T. Carbonic anhydrase inhibitors: An editorial. Expert Opin. Ther. Pat. 2013, 23, 677–679. [Google Scholar] [CrossRef] [Green Version]
- Monti, S.M.; Supuran, C.T.; De Simone, G. Anticancer carbonic anhydrase inhibitors: A patent review (2008–2013). Expert Opin. Ther. Pat. 2013, 23, 737–749. [Google Scholar] [CrossRef]
- Gondi, G.; Mysliwietz, J.; Hulikova, A.; Jen, J.P.; Swietach, P.; Kremmer, E.; Zeidler, R. Antitumor efficacy of a monoclonal antibody that inhibits the activity of cancer-associated carbonic anhydrase XII. Cancer Res. 2013, 73, 6494–6503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Supuran, C.T. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med. Chem. 2011, 3, 1165–1180. [Google Scholar] [CrossRef] [PubMed]
- Sterling, D.; Reithmeier, R.A.; Casey, J.R. Carbonic anhydrase: In the driver’s seat for bicarbonate transport. JOP 2001, 2, 165–170. [Google Scholar]
- Purkerson, J.M.; Schwartz, G.J. The role of carbonic anhydrases in renal physiology. Kidney Int. 2007, 71, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Henry, R.P. Multiple roles of carbonic anhydrase in cellular transport and metabolism. Annu. Rev. Physiol. 1996, 58, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Furuki, T.; Sakurai, M.; Inoue, Y. The catalytic mechanism of carbonic anhydrase. Tanpakushitsu Kakusan Koso 1995, 40, 1835–1845. [Google Scholar]
- McKenna, R.; Frost, S.C. Overview of the carbonic anhydrase family. Subcell. Biochem. 2014, 75, 3–5. [Google Scholar] [CrossRef]
- McDonald, P.C.; Winum, J.Y.; Supuran, C.T.; Dedhar, S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget 2012, 3, 84–97. [Google Scholar] [CrossRef] [Green Version]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [Green Version]
- Supuran, C.T.; Winum, J.Y. Carbonic anhydrase IX inhibitors in cancer therapy: An update. Future Med. Chem. 2015, 7, 1407–1414. [Google Scholar] [CrossRef]
- Supuran, C.T.; Winum, J.Y. Designing carbonic anhydrase inhibitors for the treatment of breast cancer. Expert Opin. Drug Discov. 2015, 10, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Mboge, M.Y.; McKenna, R.; Frost, S.C. Advances in Anti-Cancer Drug Development Targeting Carbonic Anhydrase IX and XII. Top Anticancer Res. 2015, 5, 3–42. [Google Scholar]
- Mahon, B.P.; Pinard, M.A.; McKenna, R. Targeting carbonic anhydrase IX activity and expression. Molecules 2015, 20, 2323–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okuno, K.; Matsubara, T.; Nakamura, T.; Iino, T.; Kakimoto, T.; Asanuma, K.; Matsumine, A.; Sudo, A. Carbonic anhydrase IX enhances tumor cell proliferation and tumor progression in osteosarcoma. Onco Targets Ther. 2018, 11, 6879–6886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanica, L.; Gheorghiu, M.; Stan, M.; Polonschii, C.; David, S.; Bratu, D.; Dinischiotu, A.; Supuran, C.T.; Gheorghiu, E. Quantitative assessment of specific carbonic anhydrase inhibitors effect on hypoxic cells using electrical impedance assays. J. Enzyme Inhib. Med. Chem. 2017, 32, 1079–1090. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Inhibition of carbonic anhydrase IX as a novel anticancer mechanism. World J. Clin. Oncol. 2012, 3, 98–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinard, M.A.; Aggarwal, M.; Mahon, B.P.; Tu, C.; McKenna, R. A sucrose-binding site provides a lead towards an isoform-specific inhibitor of the cancer-associated enzyme carbonic anhydrase IX. Acta Crystallogr. F Struct. Biol. Commun. 2015, 71, 1352–1358. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, H.; Oosterwijk, E.; Selman, Y.; Mira, J.C.; Medrano, T.; Shiverick, K.T.; Frost, S.C. Antibody-specific detection of CAIX in breast and prostate cancers. Biochem. Biophys. Res. Commun. 2009, 386, 488–492. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Wang, Z.; Wu, J.; Jiang, C. The role of hypoxia inducible factor-1 in hepatocellular carcinoma. Biomed. Res. Int. 2014, 2014, 409272. [Google Scholar] [CrossRef]
- Chia, S.K.; Wykoff, C.C.; Watson, P.H.; Han, C.; Leek, R.D.; Pastorek, J.; Gatter, K.C.; Ratcliffe, P.; Harris, A.L. Prognostic significance of a novel hypoxia-regulated marker, carbonic anhydrase IX, in invasive breast carcinoma. J. Clin. Oncol. 2001, 19, 3660–3668. [Google Scholar] [CrossRef]
- Wykoff, C.C.; Beasley, N.; Watson, P.H.; Campo, L.; Chia, S.K.; English, R.; Pastorek, J.; Sly, W.S.; Ratcliffe, P.; Harris, A.L. Expression of the hypoxia-inducible and tumor-associated carbonic anhydrases in ductal carcinoma in situ of the breast. Am. J. Pathol. 2001, 158, 1011–1019. [Google Scholar] [CrossRef] [Green Version]
- Wykoff, C.C.; Beasley, N.J.; Watson, P.H.; Turner, K.J.; Pastorek, J.; Sibtain, A.; Wilson, G.D.; Turley, H.; Talks, K.L.; Maxwell, P.H.; et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000, 60, 7075–7083. [Google Scholar] [PubMed]
- Carta, F.; Supuran, C.T.; Scozzafava, A. Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med. Chem. 2014, 6, 1149–1165. [Google Scholar] [CrossRef]
- Winum, J.Y.; Supuran, C.T. Recent advances in the discovery of zinc-binding motifs for the development of carbonic anhydrase inhibitors. J. Enzym. Inhib. Med. Chem. 2015, 30, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. How many carbonic anhydrase inhibition mechanisms exist? J. Enzym. Inhib. Med. Chem. 2016, 31, 345–360. [Google Scholar] [CrossRef]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple binding modes of inhibitors to carbonic anhydrases: How to design specific drugs targeting 15 different isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [Green Version]
- Di Fiore, A.; Pedone, C.; D’Ambrosio, K.; Scozzafava, A.; De Simone, G.; Supuran, C.T. Carbonic anhydrase inhibitors: Valdecoxib binds to a different active site region of the human isoform II as compared to the structurally related cyclooxygenase II "selective" inhibitor celecoxib. Bioorg. Med. Chem. Lett. 2006, 16, 437–442. [Google Scholar] [CrossRef]
- Temperini, C.; Cecchi, A.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Sulfonamide diuretics revisited--old leads for new applications? Org. Biomol. Chem. 2008, 6, 2499–2506. [Google Scholar] [CrossRef] [Green Version]
- Carta, F.; Supuran, C.T. Diuretics with carbonic anhydrase inhibitory action: A patent and literature review (2005–2013). Expert Opin. Ther. Pat. 2013, 23, 681–691. [Google Scholar] [CrossRef] [Green Version]
- Temperini, C.; Cecchi, A.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Interaction of indapamide and related diuretics with 12 mammalian isozymes and X-ray crystallographic studies for the indapamide-isozyme II adduct. Bioorg. Med. Chem. Lett. 2008, 18, 2567–2573. [Google Scholar] [CrossRef]
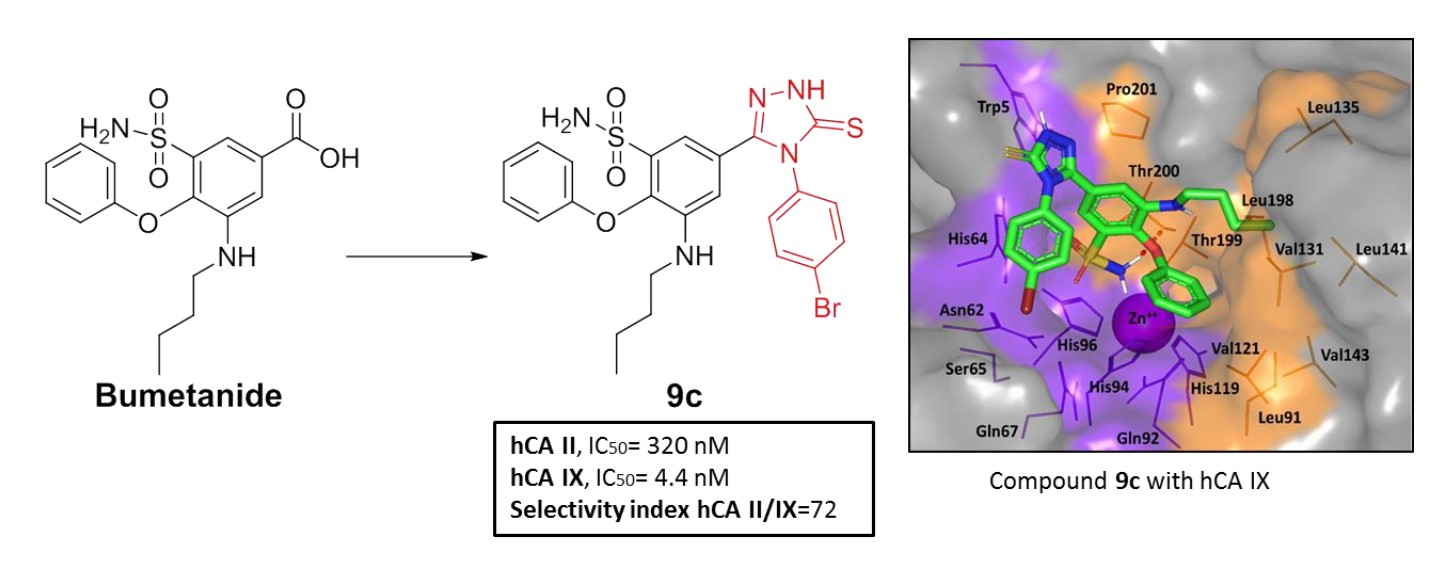
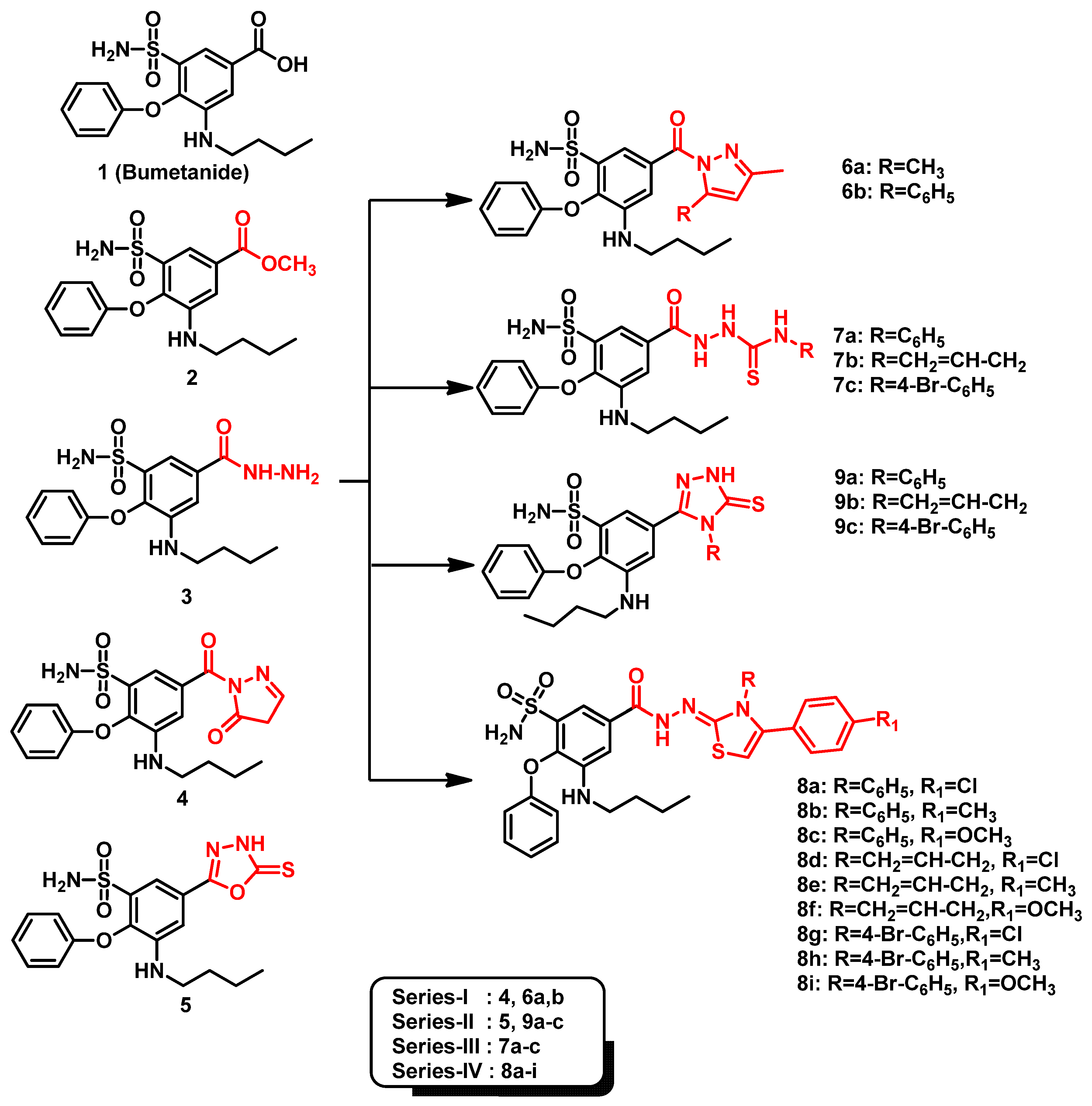
- Ibrahim, T.S.; Salem, I.M.; Mostafa, S.M.; El-Sabbagh, O.I.; ElKhamisi, M.K.; Hegazy, L.; Elgendy, B. Design, Synthesis, and Pharmacological Evaluation of Novel and Selective COX-2 Inhibitors Based on Bumetanide Scaffold. Bioorg. Chem. 2020, 100, 103878. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, J.A.; Cohen, S.M. Investigating the selectivity of metalloenzyme inhibitors. J. Med. Chem. 2013, 56, 7997–8007. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Cohen, S.M. Nucleophile recognition as an alternative inhibition mode for benzoic acid based carbonic anhydrase inhibitors. Chem. Commun. 2012, 48, 5259–5261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapegin, A.; Kalinin, S.; Angeli, A.; Supuran, C.T.; Krasavin, M. Unprotected primary sulfonamide group facilitates ring-forming cascade en route to polycyclic [1,4]oxazepine-based carbonic anhydrase inhibitors. Bioorg. Chem. 2018, 76, 140–146. [Google Scholar] [CrossRef]
- Nocentini, A.; Moi, D.; Balboni, G.; Onnis, V.; Supuran, C.T. Discovery of thiazolin-4-one-based aromatic sulfamates as a new class of carbonic anhydrase isoforms I, II, IV, and IX inhibitors. Bioorg. Chem. 2018, 77, 293–299. [Google Scholar] [CrossRef]
- Gul, H.I.; Yamali, C.; Sakagami, H.; Angeli, A.; Leitans, J.; Kazaks, A.; Tars, K.; Ozgun, D.O.; Supuran, C.T. New anticancer drug candidates sulfonamides as selective hCA IX or hCA XII inhibitors. Bioorg. Chem. 2018, 77, 411–419. [Google Scholar] [CrossRef]
- Mboge, M.Y.; Chen, Z.; Wolff, A.; Mathias, J.V.; Tu, C.; Brown, K.D.; Bozdag, M.; Carta, F.; Supuran, C.T.; McKenna, R.; et al. Selective inhibition of carbonic anhydrase IX over carbonic anhydrase XII in breast cancer cells using benzene sulfonamides: Disconnect between activity and growth inhibition. PLoS ONE 2018, 13, e0207417. [Google Scholar] [CrossRef] [Green Version]
- Boriack, P.A.; Christianson, D.W.; Kingery-Wood, J.; Whitesides, G.M. Secondary interactions significantly removed from the sulfonamide binding pocket of carbonic anhydrase II influence inhibitor binding constants. J. Med. Chem. 1995, 38, 2286–2291. [Google Scholar] [CrossRef]
- Aggarwal, M.; Kondeti, B.; McKenna, R. Insights towards sulfonamide drug specificity in α-carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1526–1533. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, M.; McKenna, R. Update on carbonic anhydrase inhibitors: A patent review (2008–2011). Expert Opin. Ther. Pat. 2012, 22, 903–915. [Google Scholar] [CrossRef]
- Singh, S.; Lomelino, C.L.; Mboge, M.Y.; Frost, S.C.; McKenna, R. Cancer Drug Development of Carbonic Anhydrase Inhibitors beyond the Active Site. Molecules 2018, 23, 1045. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.K.; Ludwig, P.A.; Christianson, D.W. Two-Site Binding of Phenol in the Active Site of Human Carbonic Anhydrase II: Structural Implications for Substrate Association. J. Am. Chem. Soc. 1994, 116, 3659–3660. [Google Scholar] [CrossRef]
- Carta, F.; Temperini, C.; Innocenti, A.; Scozzafava, A.; Kaila, K.; Supuran, C.T. Polyamines Inhibit Carbonic Anhydrases by Anchoring to the Zinc-Coordinated Water Molecule. J. Med. Chem. 2010, 53, 5511–5522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innocenti, A.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Interactions of phenols with the 12 catalytically active mammalian isoforms (CA I-XIV). Bioorg. Med. Chem. Lett. 2008, 18, 1583–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Innocenti, A.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Carbonic anhydrase inhibitors: Inhibition of mammalian isoforms I–XIV with a series of substituted phenols including paracetamol and salicylic acid. Bioorg. Med. Chem. 2008, 16, 7424–7428. [Google Scholar] [CrossRef]
- Bayram, E.; Senturk, M.; Kufrevioglu, O.I.; Supuran, C.T. In vitro inhibition of salicylic acid derivatives on human cytosolic carbonic anhydrase isozymes I and II. Bioorg. Med. Chem. 2008, 16, 9101–9105. [Google Scholar] [CrossRef]
- Sentürk, M.; Gülçin, I.; Daştan, A.; Küfrevioğlu, O.I.; Supuran, C.T. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg. Med. Chem. 2009, 17, 3207–3211. [Google Scholar] [CrossRef]
- Innocenti, A.; Beyza Öztürk Sarıkaya, S.; Gülçin, İ.; Supuran, C.T. Carbonic anhydrase inhibitors. Inhibition of mammalian isoforms I-XIV with a series of natural product polyphenols and phenolic acids. Bioorg. Med. Chem. 2010, 18, 2159–2164. [Google Scholar] [CrossRef]
- Davis, R.A.; Innocenti, A.; Poulsen, S.A.; Supuran, C.T. Carbonic anhydrase inhibitors. Identification of selective inhibitors of the human mitochondrial isozymes VA and VB over the cytosolic isozymes I and II from a natural product-based phenolic library. Bioorg. Med. Chem. 2010, 18, 14–18. [Google Scholar] [CrossRef]
- Davis, R.A.; Hofmann, A.; Osman, A.; Hall, R.A.; Mühlschlegel, F.A.; Vullo, D.; Innocenti, A.; Supuran, C.T.; Poulsen, S.A. Natural product-based phenols as novel probes for mycobacterial and fungal carbonic anhydrases. J. Med. Chem. 2011, 54, 1682–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carta, F.; Vullo, D.; Maresca, A.; Scozzafava, A.; Supuran, C.T. Mono-/dihydroxybenzoic acid esters and phenol pyridinium derivatives as inhibitors of the mammalian carbonic anhydrase isoforms I, II, VII, IX, XII and XIV. Bioorg. Med. Chem. 2013, 21, 1564–1569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tars, K.; Vullo, D.; Kazaks, A.; Leitans, J.; Lends, A.; Grandane, A.; Zalubovskis, R.; Scozzafava, A.; Supuran, C.T. Sulfocoumarins (1,2-Benzoxathiine-2,2-dioxides): A Class of Potent and Isoform-Selective Inhibitors of Tumor-Associated Carbonic Anhydrases. J. Med. Chem. 2013, 56, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Krasavin, M.; Shetnev, A.; Sharonova, T.; Baykov, S.; Tuccinardi, T.; Kalinin, S.; Angeli, A.; Supuran, C.T. Heterocyclic periphery in the design of carbonic anhydrase inhibitors: 1,2,4-Oxadiazol-5-yl benzenesulfonamides as potent and selective inhibitors of cytosolic hCA II and membrane-bound hCA IX isoforms. Bioorg. Chem. 2018, 76, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Buchieri, M.V.; Riafrecha, L.E.; Rodríguez, O.M.; Vullo, D.; Morbidoni, H.R.; Supuran, C.T.; Colinas, P.A. Inhibition of the β-carbonic anhydrases from Mycobacterium tuberculosis with C-cinnamoyl glycosides: Identification of the first inhibitor with anti-mycobacterial activity. Bioorg. Med. Chem. Lett. 2013, 23, 740–743. [Google Scholar] [CrossRef]
- Riafrecha, L.E.; Rodríguez, O.M.; Vullo, D.; Supuran, C.T.; Colinas, P.A. Synthesis of C-cinnamoyl glycosides and their inhibitory activity against mammalian carbonic anhydrases. Bioorg. Med. Chem. 2013, 21, 1489–1494. [Google Scholar] [CrossRef]
- Ekinci, D.; Karagoz, L.; Senturk, M.; Supuran, C.T. Carbonic anhydrase inhibitors: In vitro inhibition of α isoforms (hCA I, hCA II, bCA III, hCA IV) by flavonoids. J. Enzym. Inhib. Med. Chem. 2013, 28, 283–288. [Google Scholar] [CrossRef]
- Berridge, M.V.; Herst, P.M.; Tan, A.S. Tetrazolium dyes as tools in cell biology: New insights into their cellular reduction. In Biotechnology Annual Review; Elsevier: Amsterdam, The Netherlands, 2005; Volume 11, pp. 127–152. [Google Scholar]
- Cheng, A.; Best, S.A.; Merz, K.M.; Reynolds, C.H. GB/SA water model for the Merck molecular force field (MMFF). J. Mol. Graph. Model. 2000, 18, 273–282. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
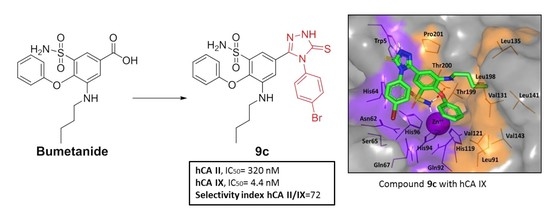
| Compound | hCA-II IC50(nM) b | hCA-IX IC50(nM) b | Selectivity Ratio c II/IX |
|---|---|---|---|
| 2 | 668.15 | 23.73 | 28.1 |
| 3 | 1614.82 | 7.87 | 205.1 |
| 4 | 1052.55 | 16.45 | 63.9 |
| 5 | 486.01 | 12.44 | 39.0 |
| 6a | N/D | 16.56 | |
| 6b | 858.26 | 5.43 | 158.9 |
| 7a | 1571.85 | 17.74 | 88.6 |
| 7b | 1344.25 | 14.39 | 93.4 |
| 7c | 1535.37 | 12.91 | 118.9 |
| 8a | 251.25 | 6.53 | 38.6 |
| 8b | 1318.38 | 12.97 | 101.6 |
| 8c | 473.66 | 12.60 | 37.5 |
| 8d | 5117.31 | 6.36 | 804.6 |
| 8e | 517.66 | 11.51 | 44.9 |
| 8f | 999.18 | 13.41 | 74.5 |
| 8g | 784.08 | 7.18 | 109.2 |
| 8h | 208.81 | 14.31 | 14.5 |
| 8i | 426.45 | 10.57 | 40.3 |
| 9a | 4061.34 | 5.31 | 764.8 |
| 9b | 205.21 | 7.25 | 28.3 |
| 9c | 320.08 | 4.42 | 72.4 |
| Acetazolamide | 17.1 | 31.6 | 0.5 |
| Compound | A-498 IC50 (μM) b | SCaBER IC50 (μM) b |
|---|---|---|
| Acetazolamide | 40.00 ± 0.37 | 40.63 ± 0.36 |
| 8a | 15.66 ± 0.63 | 28.36 ± 1.4 |
| 8h | 6.55 ± 0.26 | 78.04 ± 0.29 |
| 9b | 16.08 ± 1.4 | 78.07 ± 1.7 |
| 9c | 5.07 ± 0.11 | 8.80 ± 1.9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malebari, A.M.; Ibrahim, T.S.; Salem, I.M.; Salama, I.; Khayyat, A.N.; Mostafa, S.M.; El-Sabbagh, O.I.; Darwish, K.M. The Anticancer Activity for the Bumetanide-Based Analogs via Targeting the Tumor-Associated Membrane-Bound Human Carbonic Anhydrase-IX Enzyme. Pharmaceuticals 2020, 13, 252. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090252
Malebari AM, Ibrahim TS, Salem IM, Salama I, Khayyat AN, Mostafa SM, El-Sabbagh OI, Darwish KM. The Anticancer Activity for the Bumetanide-Based Analogs via Targeting the Tumor-Associated Membrane-Bound Human Carbonic Anhydrase-IX Enzyme. Pharmaceuticals. 2020; 13(9):252. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090252
Chicago/Turabian StyleMalebari, Azizah M., Tarek S. Ibrahim, Ibrahim M. Salem, Ismail Salama, Ahdab N. Khayyat, Samia M. Mostafa, Osama I. El-Sabbagh, and Khaled M. Darwish. 2020. "The Anticancer Activity for the Bumetanide-Based Analogs via Targeting the Tumor-Associated Membrane-Bound Human Carbonic Anhydrase-IX Enzyme" Pharmaceuticals 13, no. 9: 252. https://0-doi-org.brum.beds.ac.uk/10.3390/ph13090252