
Soft Coral-Derived Dihydrosinularin Exhibits Antiproliferative Effects Associated with Apoptosis and DNA Damage in Oral Cancer Cells

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
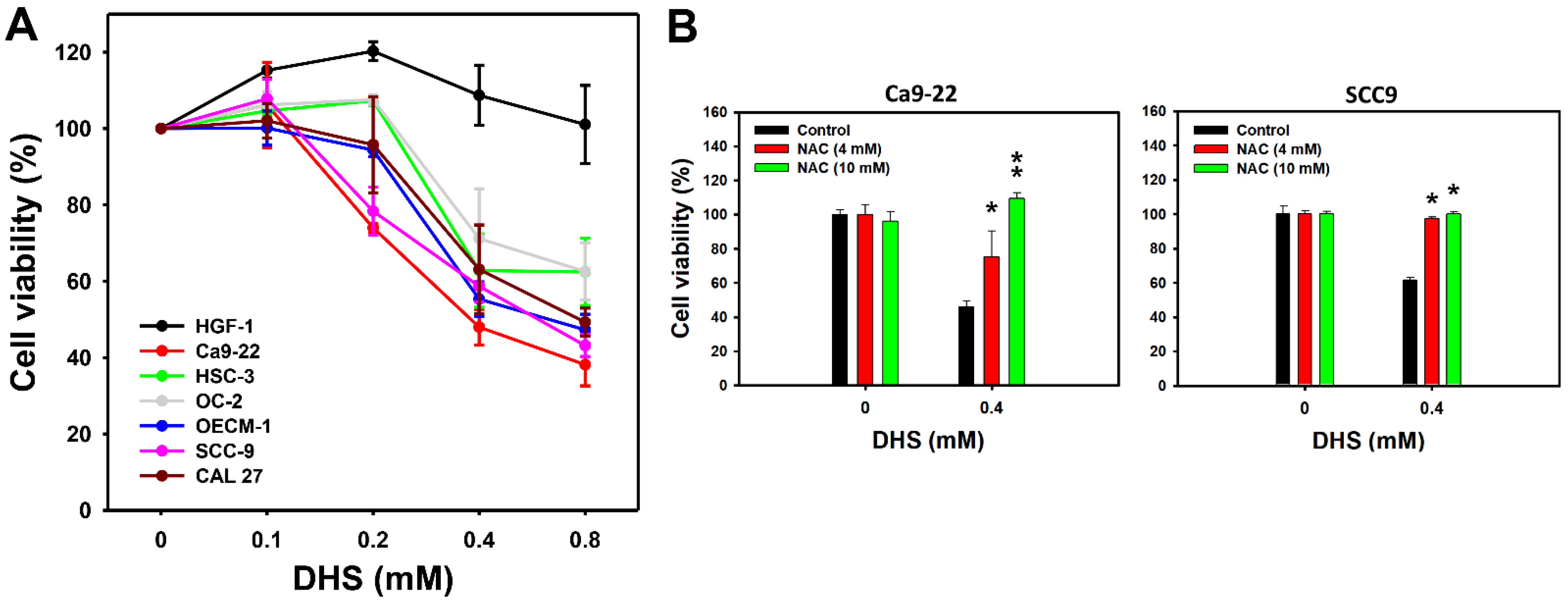
2.1. DHS Kills Oral Cancer Cells Alleviated by NAC
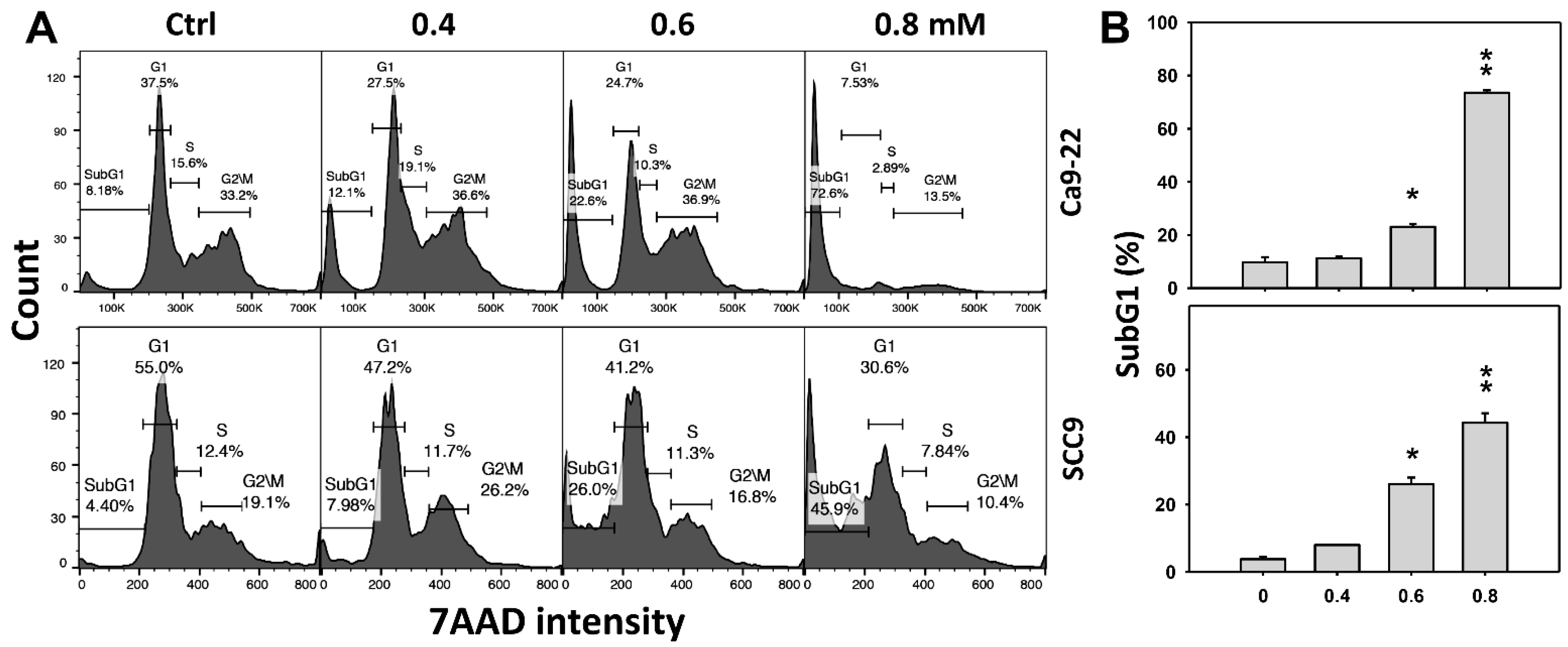
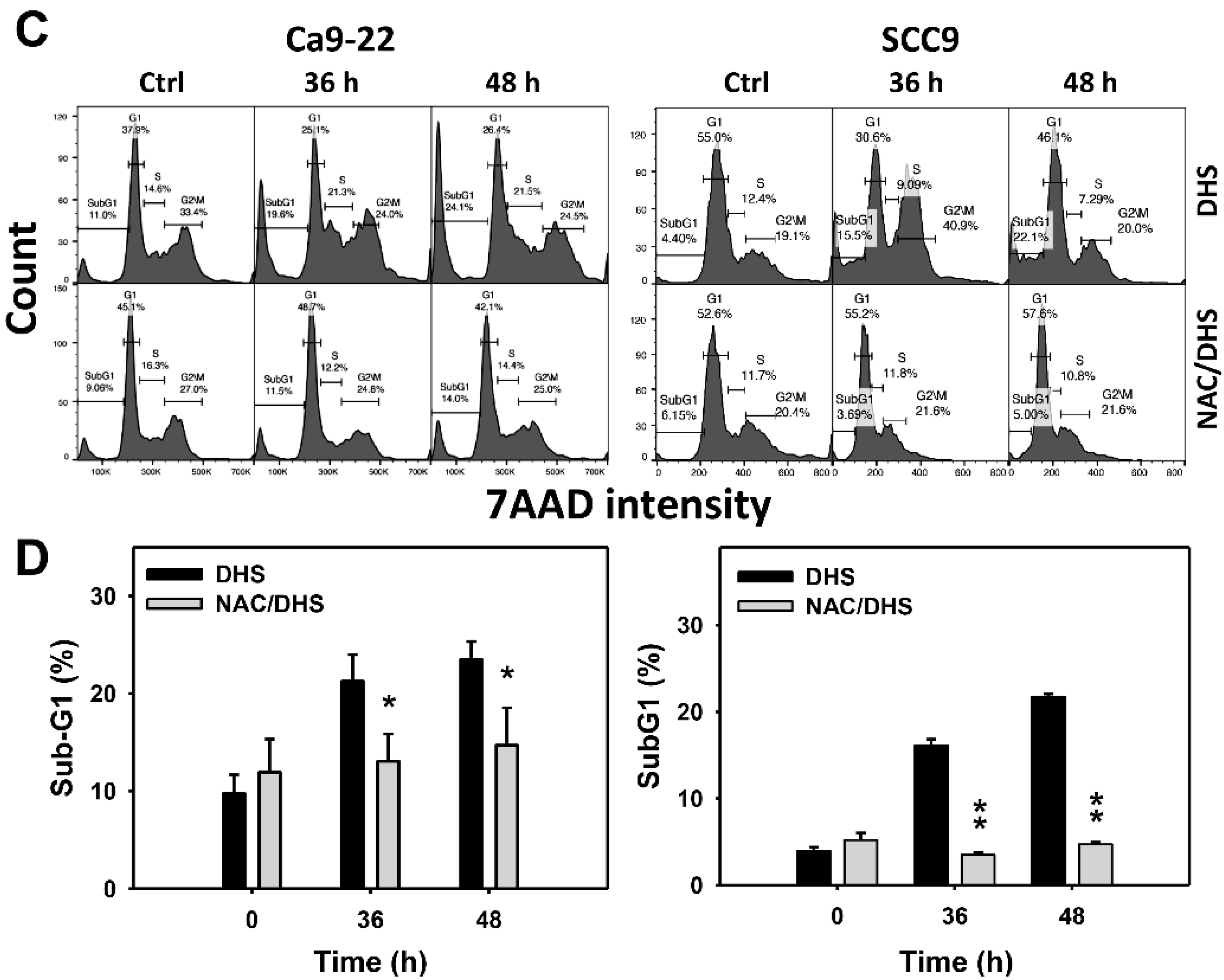
2.2. DHS Accumulates SubG1 Phase of Oral Cancer Cells Alleviated by NAC
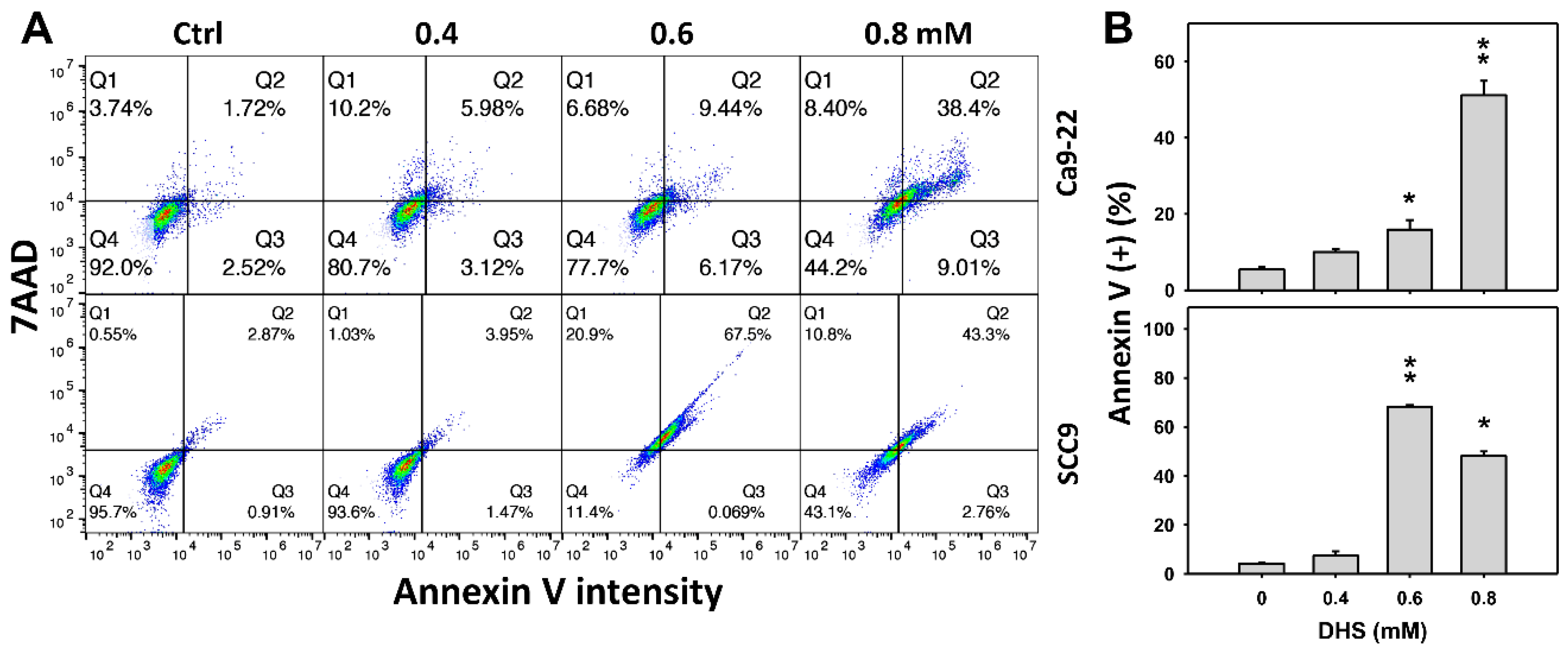
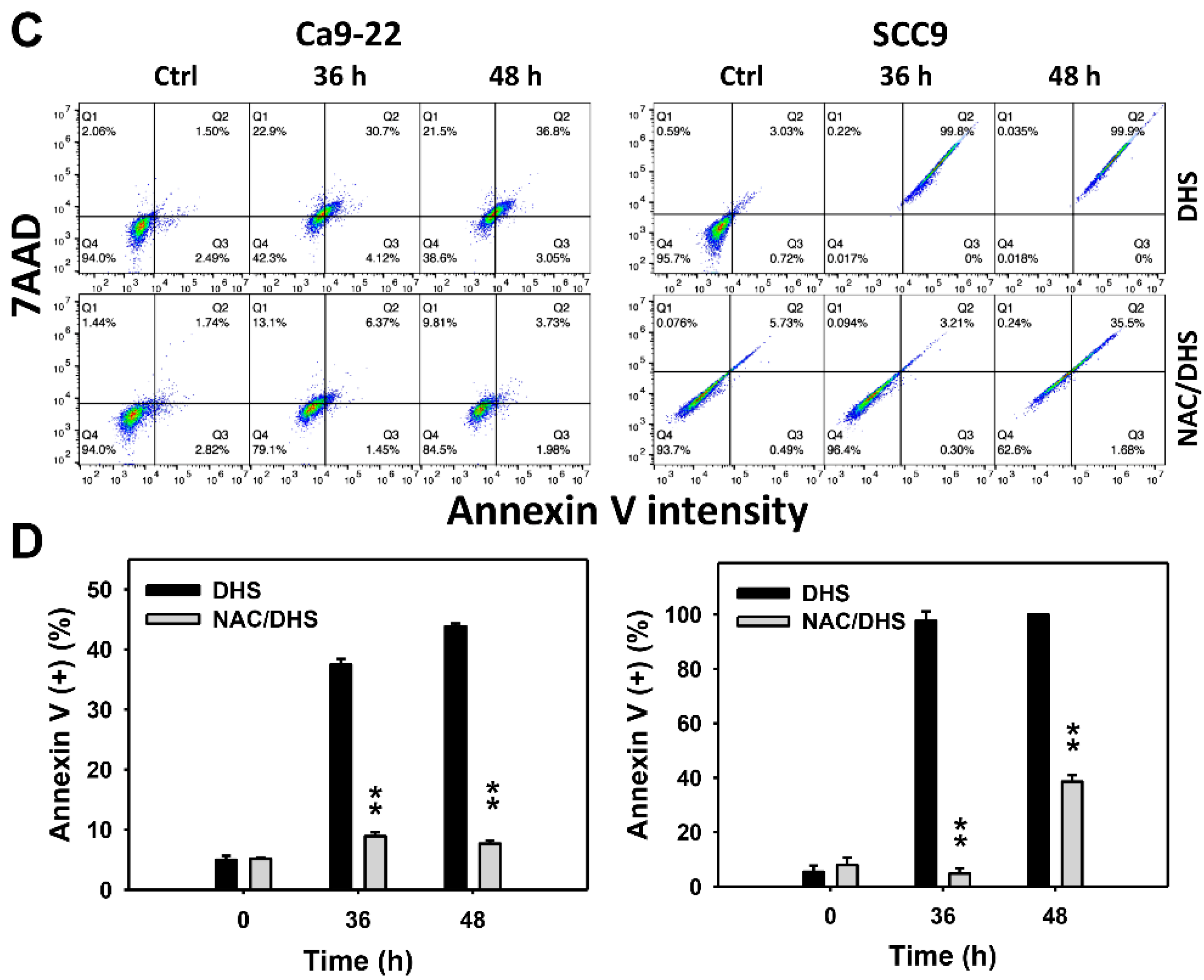
2.3. DHS Induces Annexin V-Detected Apoptosis of Oral Cancer Cells Alleviated by NAC
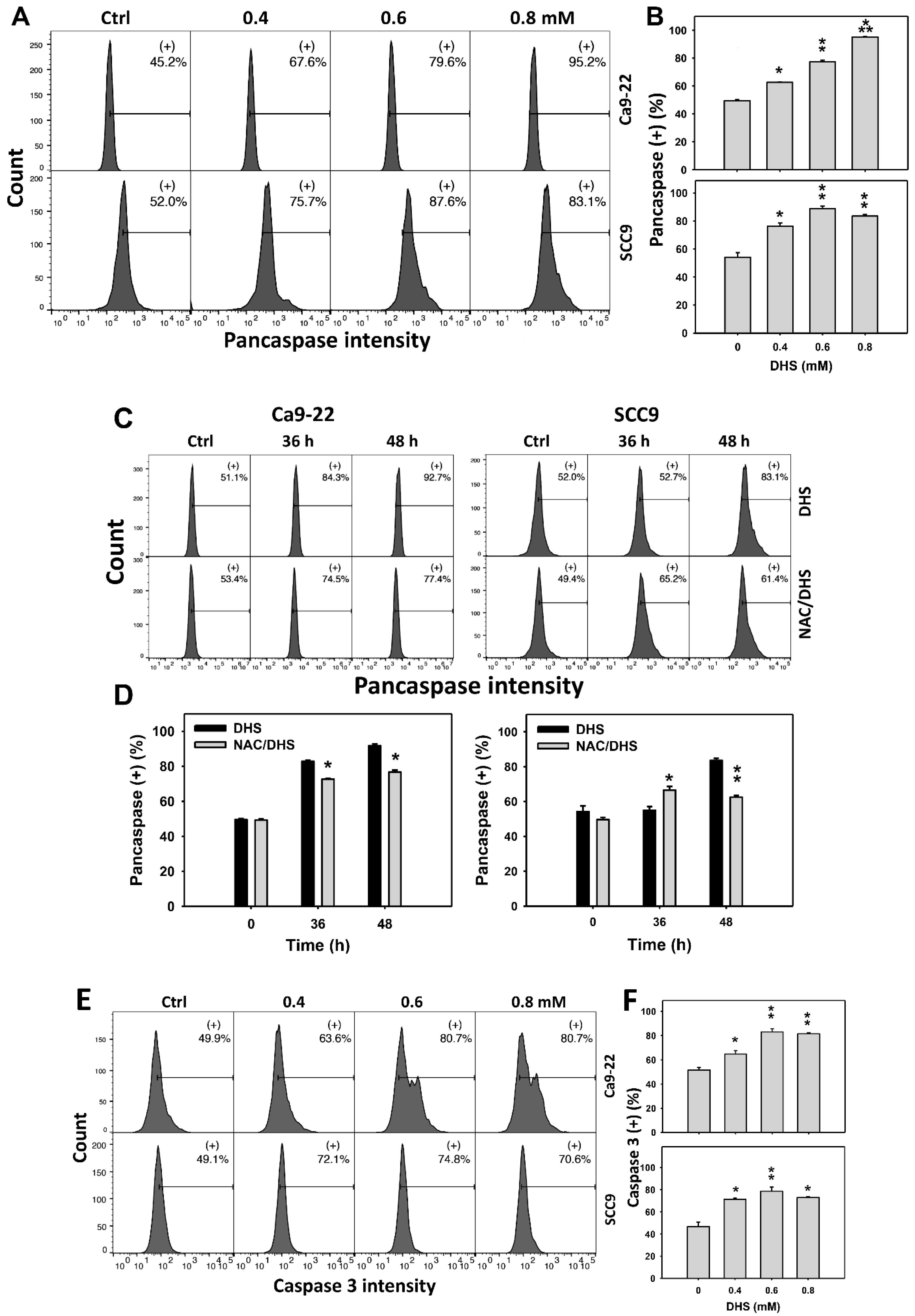
2.4. Alleviation by NAC of Apoptosis of Oral Cancer Cells as Detected According to DHS-Induced Caspase Activation
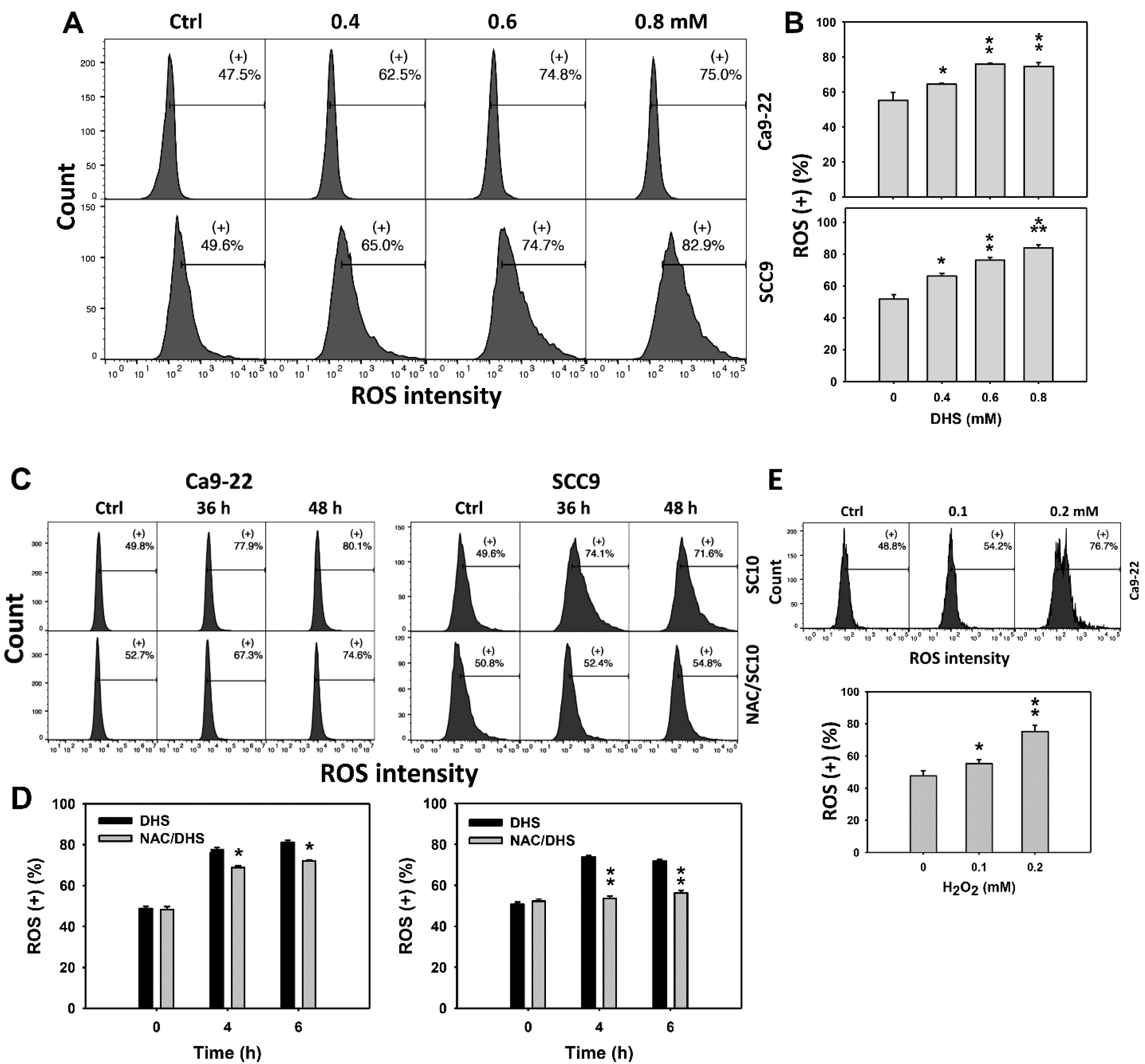
2.5. DHS Induces Reactive Oxygen Species (ROS) Stress of Oral Cancer Cells Alleviated by NAC
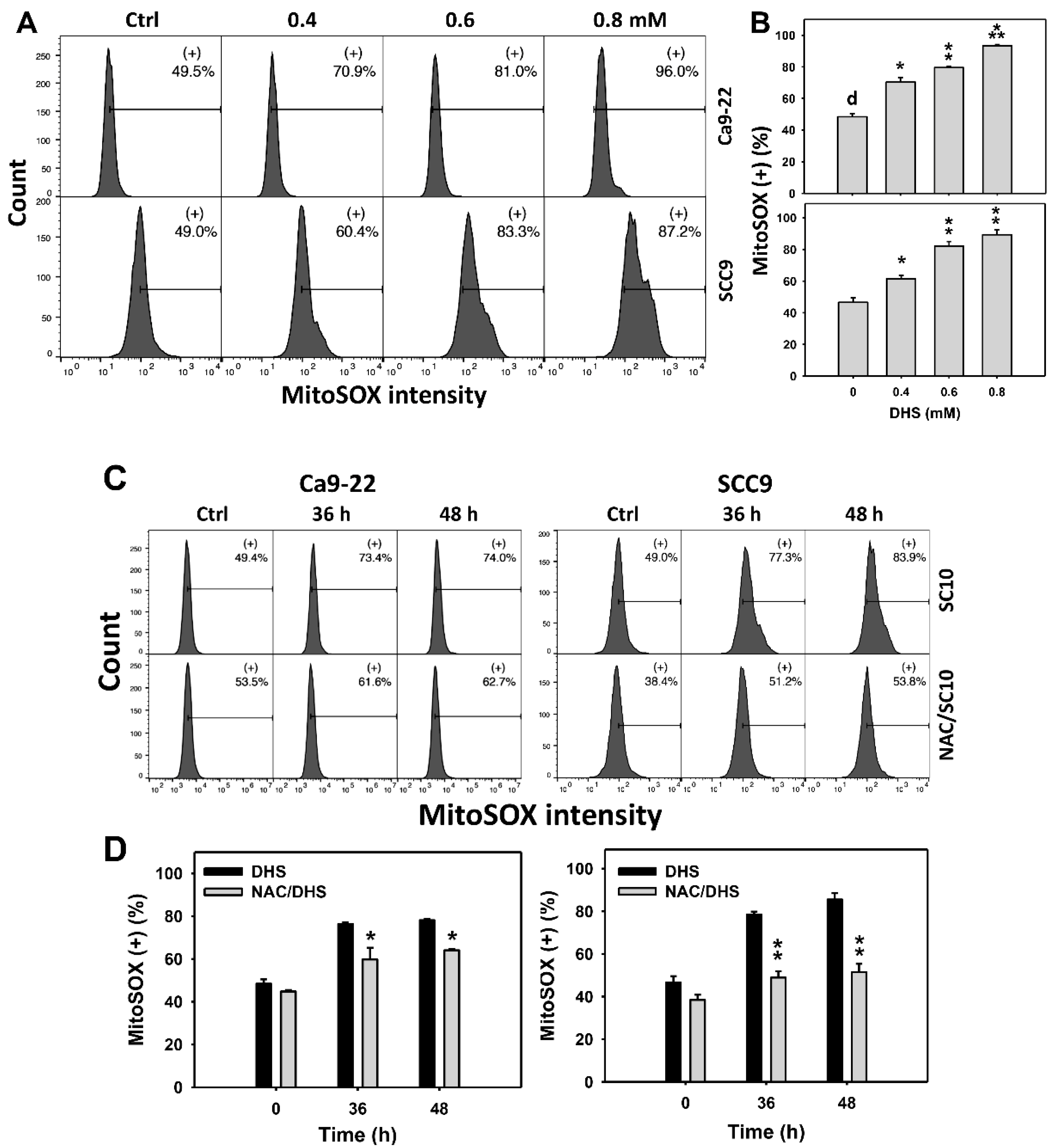
2.6. DHS Induces MitoSOX Stress of Oral Cancer Cells Alleviated by NAC
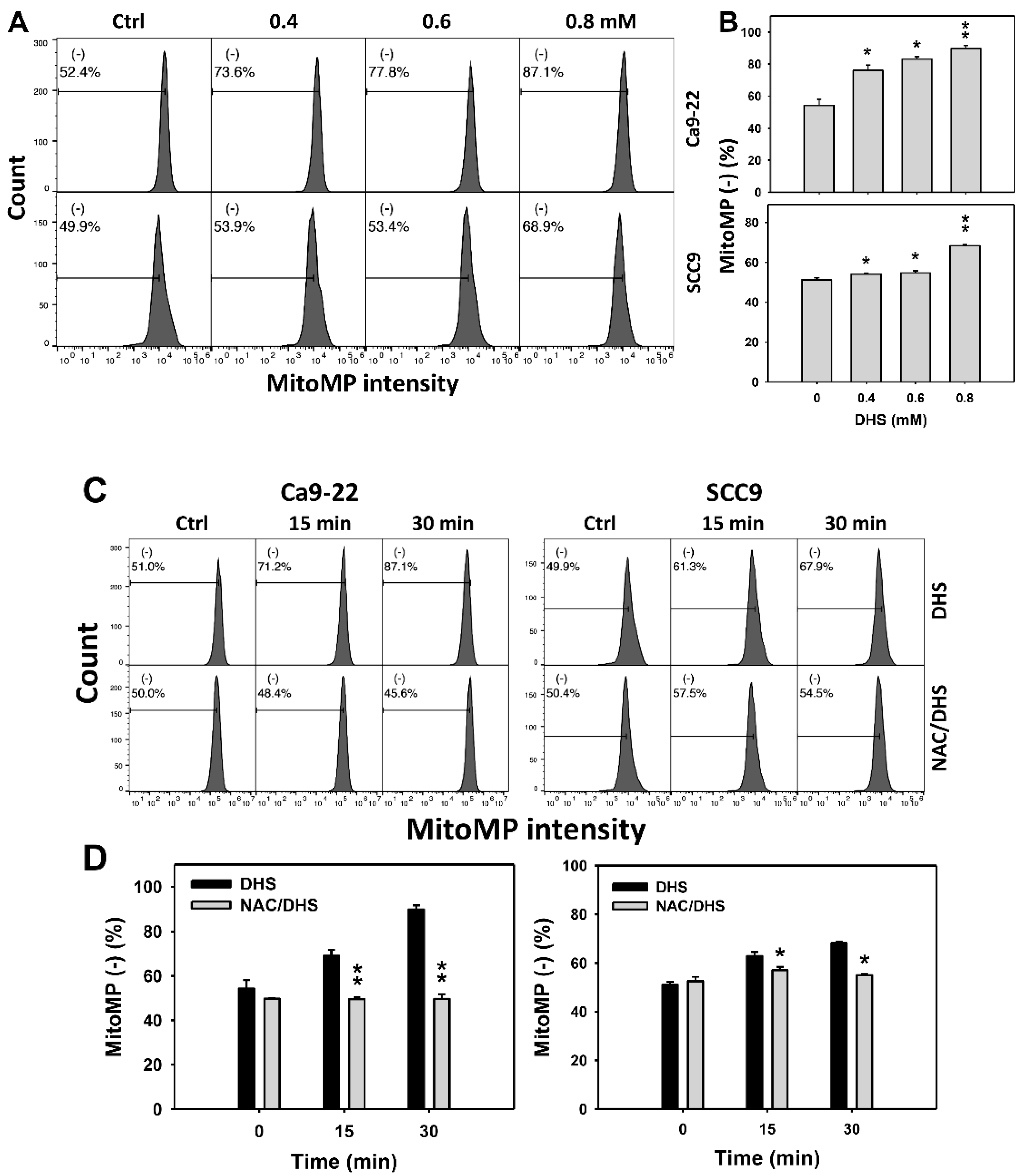
2.7. DHS Induces MitoMP Depletion Stress of Oral Cancer Cells Alleviated by NAC
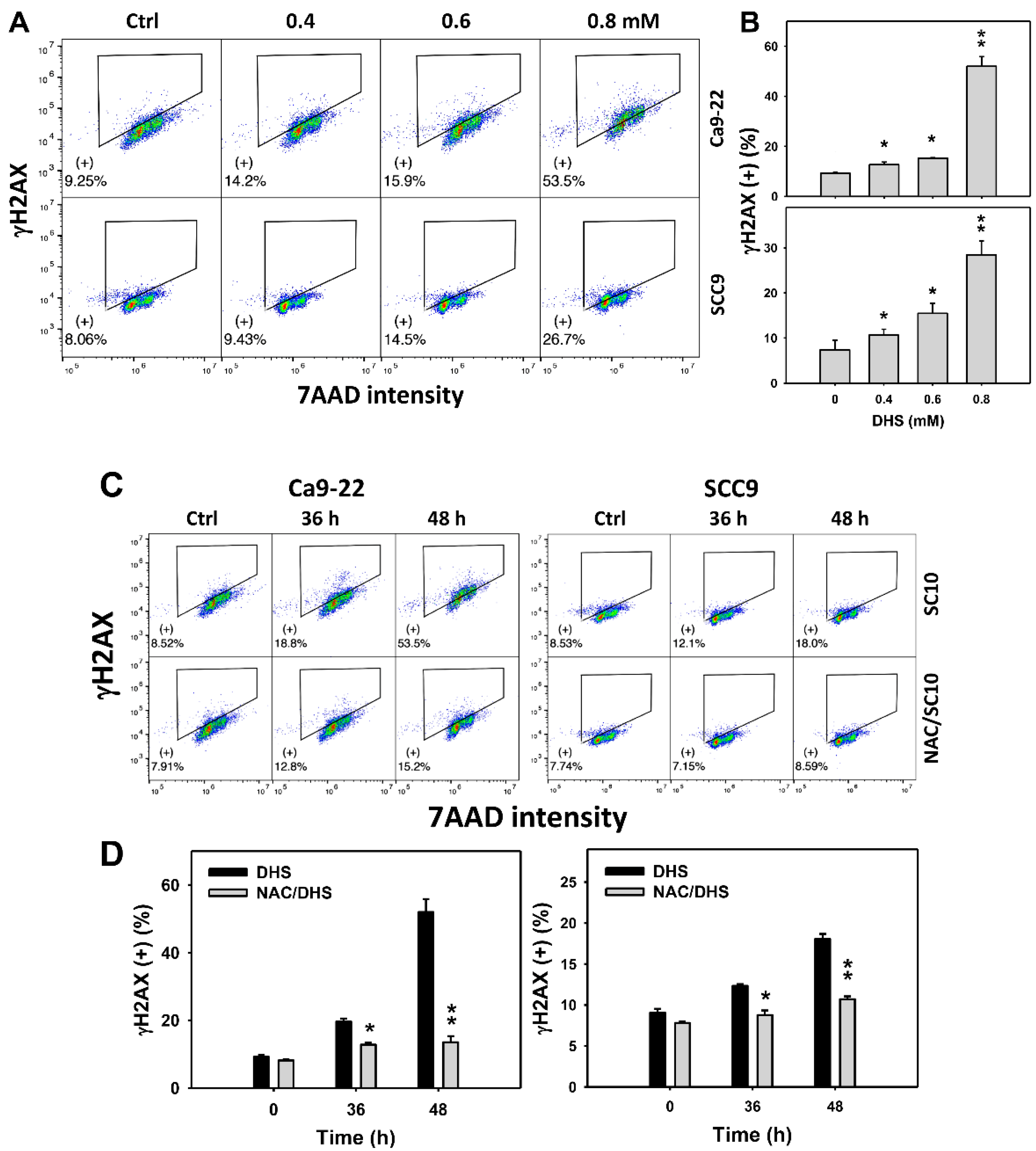
2.8. DHS Induces γH2AX Phosphorylation of Oral Cancer Cells Alleviated by NAC
3. Discussion
3.1. Oxidative Stress Effect of DHS
3.2. Antiproliferation Effect of DHS against Oral Cancer Cells
3.3. DHS Induced Apoptosis and Causes DNA Damage Effects on Oral Cancer Cells
3.4. Oxidative Stress Is Important in DHS-Induced Antiproliferation Mechanisms on Oral Cancer Cells
3.5. The Influence of Toxicity on Apoptosis, Oxidative Stress, and DNA Damage in Oral Cancer Cells
4. Materials and Methods
4.1. Preparation of DHS and Inhibitors
4.2. Cell Culture and Viability
4.3. Cell Cycle
4.4. Apoptosis Assays by Annexin V/7AAD, Pancaspase, and Caspase 3
4.5. Oxidative Stress Assays for ROS, MitoSOX, and MitoMP
4.6. DNA Damage Assays by γH2AX Phosphorylation
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warnakulasuriya, S.; Kerr, A.R. Oral cancer screening: Past, present, and future. J. Dent. Res. 2021, 220345211014795. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Sarode, G.; Maniyar, N.; Sarode, S.C.; Choudhary, N.; Mehta, V.; Gopalakrishnan, D.; Yerwadekar, S.; Joshi, S.; Pendyala, G.; Patil, S. Oral cancer in young vs old individuals: A systematic review. J. Contemp. Dent. Pract. 2021, 22, 435–451. [Google Scholar] [PubMed]
- Inchingolo, F.; Santacroce, L.; Ballini, A.; Topi, S.; Dipalma, G.; Haxhirexha, K.; Bottalico, L.; Charitos, I.A. Oral cancer: A historical review. Int. J. Environ. Res. Public Health 2020, 17, 3168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Wang, F.X.; Jia, K.K.; Kong, L.D. Natural product interventions for chemotherapy and radiotherapy-induced side effects. Front. Pharmacol. 2018, 9, 1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, A.M.S.; Rodriguez, A.D.; Taglialatela-Scafati, O.; Fusetani, N. Marine pharmacology in 2012–2013: Marine compounds with antibacterial, antidiabetic, antifungal, anti-inflammatory, antiprotozoal, antituberculosis, and antiviral activities; affecting the immune and nervous systems, and other miscellaneous mechanisms of action. Mar. Drugs 2017, 15, 273. [Google Scholar]
- Lichota, A.; Gwozdzinski, K. Anticancer activity of natural compounds from plant and marine environment. Int. J. Mol. Sci. 2018, 19, 3533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matulja, D.; Wittine, K.; Malatesti, N.; Laclef, S.; Turks, M.; Markovic, M.K.; Ambrozic, G.; Markovic, D. Marine natural products with high anticancer activities. Curr. Med. Chem. 2020, 27, 1243–1307. [Google Scholar] [CrossRef]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine natural products: A source of novel anticancer drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef] [Green Version]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Zhou, F.; Al-Kareef, A.M.; Wang, H. Anticancer agents from marine sponges. J. Asian Nat. Prod. Res. 2015, 17, 64–88. [Google Scholar] [CrossRef]
- Yun, C.W.; Kim, H.J.; Lee, S.H. Therapeutic application of diverse marine-derived natural products in cancer therapy. Anticancer Res. 2019, 39, 5261–5284. [Google Scholar] [CrossRef] [Green Version]
- Abdelaleem, E.R.; Samy, M.N.; Desoukey, S.Y.; Liu, M.; Quinn, R.J.; Abdelmohsen, U.R. Marine natural products from sponges (Porifera) of the order Dictyoceratida (2013 to 2019); a promising source for drug discovery. RSC Adv. 2020, 10, 34959–34976. [Google Scholar] [CrossRef]
- Saeed, A.; Su, J.; Ouyang, S. Marine-derived drugs: Recent advances in cancer therapy and immune signaling. Biomed. Pharmacother. 2021, 134, 111091. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Huang, T.Y.; Chang, W.J.; Pan, Y.S.; Chu, H.R.; Li, Z.L.; Unson, S.; Chin, Y.T.; Lin, C.Y.; Huang, H.M.; et al. Combined treatment of heteronemin and tetrac induces antiproliferation in oral cancer cells. Mar. Drugs 2020, 18, 348. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.R.; Lu, M.C.; El-Shazly, M.; Wu, S.L.; Lai, K.H.; Su, J.H. Aquaculture soft coral Lobophytum crassum as a producer of anti-proliferative cembranoids. Mar. Drugs 2018, 16, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, M.; Nath, A.K.; Tang, Y.; Choi, Y.J.; Debnath, T.; Choi, E.J.; Kim, E.K. Investigation of the anti-prostate cancer properties of marine-derived compounds. Mar. Drugs 2018, 16, 160. [Google Scholar] [CrossRef] [Green Version]
- Su, J.H.; Ahmed, A.F.; Sung, P.J.; Chao, C.H.; Kuo, Y.H.; Sheu, J.H. Manaarenolides A-I, diterpenoids from the soft coral Sinularia manaarensis. J. Nat. Prod. 2006, 69, 1134–1139. [Google Scholar] [CrossRef]
- Tsai, T.C.; Chen, H.Y.; Sheu, J.H.; Chiang, M.Y.; Wen, Z.H.; Dai, C.F.; Su, J.H. Structural elucidation and structure-anti-inflammatory activity relationships of cembranoids from cultured soft corals Sinularia sandensis and Sinularia flexibilis. J. Agric. Food Chem. 2015, 63, 7211–7218. [Google Scholar] [CrossRef]
- Weinheimer, A.J.; Matson, J.A.; Hossain, M.B.; van der Helm, D. Marine anticancer agents: Sinularin and dihydrosinularin, new cembranolides from the soft coral, Sinularia flexibilis. Tetrahedron. Lett. 1977, 18, 2923–2926. [Google Scholar] [CrossRef]
- Ma, Q.; Meng, X.Y.; Wu, K.R.; Cao, J.Z.; Yu, R.; Yan, Z.J. Sinularin exerts anti-tumor effects against human renal cancer cells relies on the generation of ROS. J. Cancer 2019, 10, 5114–5123. [Google Scholar] [CrossRef]
- Chung, T.W.; Lin, S.C.; Su, J.H.; Chen, Y.K.; Lin, C.C.; Chan, H.L. Sinularin induces DNA damage, G2/M phase arrest, and apoptosis in human hepatocellular carcinoma cells. BMC Complement. Altern. Med. 2017, 17, 62. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.T.; Wu, C.Y.; Tang, J.Y.; Huang, C.Y.; Liaw, C.C.; Wu, S.H.; Sheu, J.H.; Chang, H.W. Sinularin induces oxidative stress-mediated G2/M arrest and apoptosis in oral cancer cells. Environ. Toxicol. 2017, 32, 2124–2132. [Google Scholar] [CrossRef]
- Wu, Y.J.; Wong, B.S.; Yea, S.H.; Lu, C.I.; Weng, S.H. Sinularin induces apoptosis through mitochondria dysfunction and inactivation of the PI3K/Akt/mTOR Pathway in gastric carcinoma cells. Mar. Drugs 2016, 14, 142. [Google Scholar] [CrossRef] [Green Version]
- Su, T.R.; Lin, J.J.; Chiu, C.C.; Chen, J.Y.; Su, J.H.; Cheng, Z.J.; Hwang, W.I.; Huang, H.H.; Wu, Y.J. Proteomic investigation of anti-tumor activities exerted by sinularin against A2058 melanoma cells. Electrophoresis 2012, 33, 1139–1152. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.C.; Li, R.N.; Lin, L.C.; Tang, J.Y.; Su, J.H.; Sheu, J.H.; Chang, H.W. Comparison of antioxidant and anticancer properties of soft coral-derived sinularin and dihydrosinularin. Molecules 2021, 26, 3853. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.W.; Chang, F.R.; McPhail, A.T.; Lee, K.H.; Wu, Y.C. New cembranolide analogues from the formosan soft coral Sinularia flexibilis and their cytotoxicity. Nat. Prod. Res. 2003, 17, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Peng, B.R.; Hsu, K.C.; El-Shazly, M.; Shih, S.P.; Lin, T.E.; Kuo, F.W.; Chou, Y.C.; Lin, H.Y.; Lu, M.C. 13-Acetoxysarcocrassolide exhibits cytotoxic activity against oral cancer cells through the interruption of the Keap1/Nrf2/p62/SQSTM1 pathway: The need to move beyond classical concepts. Mar. Drugs 2020, 18, 382. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Cai, J.; Jones, D.P. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J. Biol. Chem. 1998, 273, 11401–11404. [Google Scholar] [CrossRef] [Green Version]
- Ly, J.D.; Grubb, D.R.; Lawen, A. The mitochondrial membrane potential (deltapsi(m)) in apoptosis; an update. Apoptosis 2003, 8, 115–128. [Google Scholar] [CrossRef]
- Ricci, C.; Pastukh, V.; Leonard, J.; Turrens, J.; Wilson, G.; Schaffer, D.; Schaffer, S.W. Mitochondrial DNA damage triggers mitochondrial-superoxide generation and apoptosis. Am. J. Physiol. Cell Physiol. 2008, 294, C413–C422. [Google Scholar] [CrossRef] [Green Version]
- Su, J.H.; Wen, Z.H. Bioactive cembrane-based diterpenoids from the soft coral Sinularia triangular. Mar. Drugs 2011, 9, 944–951. [Google Scholar] [CrossRef] [Green Version]
- Chandra, J.; Samali, A.; Orrenius, S. Triggering and modulation of apoptosis by oxidative stress. Free Radic. Biol. Med. 2000, 29, 323–333. [Google Scholar] [CrossRef]
- Bouayed, J.; Bohn, T. Exogenous antioxidants--Double-edged swords in cellular redox state: Health beneficial effects at physiologic doses versus deleterious effects at high doses. Oxid. Med. Cell. Longev. 2010, 3, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Widodo, N.; Priyandoko, D.; Shah, N.; Wadhwa, R.; Kaul, S.C. Selective killing of cancer cells by Ashwagandha leaf extract and its component Withanone involves ROS signaling. PLoS ONE 2010, 5, e13536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.C.; Hou, M.F.; Huang, H.W.; Chang, F.R.; Yeh, C.C.; Tang, J.Y.; Chang, H.W. Marine algal natural products with anti-oxidative, anti-inflammatory, and anti-cancer properties. Cancer Cell Int. 2013, 13, 55. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.Y.; Ou-Yang, F.; Hou, M.F.; Huang, H.W.; Wang, H.R.; Li, K.T.; Fayyaz, S.; Shu, C.W.; Chang, H.W. Oxidative stress-modulating drugs have preferential anticancer effects—Involving the regulation of apoptosis, DNA damage, endoplasmic reticulum stress, autophagy, metabolism, and migration. Semin. Cancer Biol. 2019, 58, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Sanduja, R.; Sanduja, S.K.; Weinheimer, A.J.; Alam, M.; Martin, G.E. Isolation of the cembranolide diterpenes dihydrosinularin and 11-epi-sinulariolide from the marine mollusk Planaxis sulcatus. J. Nat. Prod. 1986, 49, 718–719. [Google Scholar] [CrossRef]
- Longton, E.; Schmit, K.; Fransolet, M.; Clement, F.; Michiels, C. Appropriate sequence for afatinib and cisplatin combination improves anticancer activity in head and neck squamous cell carcinoma. Front. Oncol. 2018, 8, 432. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, E.J. Hormesis: Why it is important to toxicology and toxicologists. Environ. Toxicol. Chem. 2008, 27, 1451–1474. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Segura, J.A.; Alonso, F.J.; Marquez, J. Oxidative stress in apoptosis and cancer: An update. Arch. Toxicol. 2012, 86, 1649–1665. [Google Scholar] [CrossRef] [PubMed]
- Guerra, J.R.; Cárdenas, A.B.; Ochoa-Zarzosa, A.; Meza, J.L.; Pérez, A.U.; Fierro-Medina, R.; Monroy, Z.J.R.; Castañeda, J.E.G. The tetrameric peptide LfcinB (20–25) 4 derived from bovine lactoferricin induces apoptosis in the MCF-7 breast cancer cell line. RSC Adv. 2019, 9, 20497–20504. [Google Scholar] [CrossRef] [Green Version]
- Barzilai, A.; Yamamoto, K. DNA damage responses to oxidative stress. DNA Repair 2004, 3, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. gammaH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef] [Green Version]
- Firsanov, D.V.; Solovjeva, L.V.; Svetlova, M.P. H2AX phosphorylation at the sites of DNA double-strand breaks in cultivated mammalian cells and tissues. Clin. Epigenetics 2011, 2, 283–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAdam, E.; Brem, R.; Karran, P. Oxidative stress-induced protein damage inhibits DNA repair and determines mutation risk and therapeutic efficacy. Mol. Cancer Res. 2016, 14, 612–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tam, L.M.; Price, N.E.; Wang, Y. Molecular mechanisms of arsenic-induced disruption of DNA repair. Chem. Res. Toxicol. 2020, 33, 709–726. [Google Scholar] [CrossRef]
- Bravard, A.; Vacher, M.; Gouget, B.; Coutant, A.; de Boisferon, F.H.; Marsin, S.; Chevillard, S.; Radicella, J.P. Redox regulation of human OGG1 activity in response to cellular oxidative stress. Mol. Cell. Biol. 2006, 26, 7430–7436. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Li, X.; Xu, R.; Ye, L.; Kong, H.; Zeng, X.; Wang, H.; Xie, W. The synergistic effect of resveratrol in combination with cisplatin on apoptosis via modulating autophagy in A549 cells. Acta Biochim. Biophys. Sin. 2016, 48, 528–535. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Fan, J.; Ai, G.; Liu, J.; Luo, N.; Li, C.; Cheng, Z. Berberine in combination with cisplatin induces necroptosis and apoptosis in ovarian cancer cells. Biol. Res. 2019, 52, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.Z.; Liu, W.Z.; Hsieh, W.T.; Tang, F.Y.; Chung, J.G.; Leung, H.W. Oxidative stress involvement in Physalis angulata-induced apoptosis in human oral cancer cells. Food Chem. Toxicol. 2009, 47, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.S.; Lin, C.P.; Chen, Y.P.; Chao, M.R.; Li, C.C.; Liu, K.L. CYP450-mediated mitochondrial ROS production involved in arecoline N-oxide-induced oxidative damage in liver cell lines. Environ. Toxicol. 2018, 33, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.; Kuan, C.P.; Lin, J.Y.; Lai, J.S.; Ho, T.F. Tanshinone IIA facilitates TRAIL sensitization by up-regulating DR5 through the ROS-JNK-CHOP signaling axis in human ovarian carcinoma cell lines. Chem. Res. Toxicol. 2015, 28, 1574–1583. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Meng, C.L. Regulation of PG synthase by EGF and PDGF in human oral, breast, stomach, and fibrosarcoma cancer cell lines. J. Dent. Res. 1994, 73, 1407–1415. [Google Scholar] [CrossRef]
- Huang, H.W.; Tang, J.Y.; Ou-Yang, F.; Wang, H.R.; Guan, P.Y.; Huang, C.Y.; Chen, C.Y.; Hou, M.F.; Sheu, J.H.; Chang, H.W. Sinularin selectively kills breast cancer cells showing G2/M arrest, apoptosis, and oxidative DNA damage. Molecules 2018, 23, 849. [Google Scholar] [CrossRef] [Green Version]
- Sas, A.; Bento, L.; Muncunill, J.; Martínez-Serra, J.; Ros, T.; Asensio, V.; Vögler, O.; Salar, A.; Sampol, A.; Alemany, R. In vitro validation of micro-RNAs (miRNAs) associated to treatment failure in diffuse large B-cell lymphoma (DLBCL). Blood 2020, 136, 21. [Google Scholar] [CrossRef]
- Vignon, C.; Debeissat, C.; Georget, M.T.; Bouscary, D.; Gyan, E.; Rosset, P.; Herault, O. Flow cytometric quantification of all phases of the cell cycle and apoptosis in a two-color fluorescence plot. PLoS ONE 2013, 8, e68425. [Google Scholar] [CrossRef] [Green Version]
- Carbonari, M. New use for an old reagent: Cell cycle analysis of DNA content using flow cytometry in formamide treated cells. Cytom. Part. A 2016, 89, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wang, J.; Liu, M.; Chen, G. Fluvastatin protects neuronal cells from hydrogen peroxide-induced toxicity with decreasing oxidative damage and increasing PI3K/Akt/mTOR signalling. J. Pharm. Pharmacol. 2021, 73, 515–521. [Google Scholar] [CrossRef]
- Lee, C.H.; Shih, Y.L.; Lee, M.H.; Au, M.K.; Chen, Y.L.; Lu, H.F.; Chung, J.G. Bufalin induces apoptosis of human osteosarcoma U-2 OS cells through endoplasmic reticulum stress, caspase- and mitochondria-dependent signaling pathways. Molecules 2017, 22, 437. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Halicka, D.; Traganos, F.; Darzynkiewicz, Z. Cytometric analysis of DNA damage: Phosphorylation of histone H2AX as a marker of DNA double-strand breaks (DSBs). Methods Mol. Biol 2009, 523, 161–168. [Google Scholar]
- Abdi, H.; Williams, L.J. Tukey’s honestly significant difference (HSD) test. Encycl. Res. Des. 2010, 3, 1–5. [Google Scholar]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, K.-H.; Lin, Y.-S.; Wang, S.-C.; Lee, M.-Y.; Tang, J.-Y.; Chang, F.-R.; Chuang, Y.-T.; Sheu, J.-H.; Chang, H.-W. Soft Coral-Derived Dihydrosinularin Exhibits Antiproliferative Effects Associated with Apoptosis and DNA Damage in Oral Cancer Cells. Pharmaceuticals 2021, 14, 994. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14100994
Yang K-H, Lin Y-S, Wang S-C, Lee M-Y, Tang J-Y, Chang F-R, Chuang Y-T, Sheu J-H, Chang H-W. Soft Coral-Derived Dihydrosinularin Exhibits Antiproliferative Effects Associated with Apoptosis and DNA Damage in Oral Cancer Cells. Pharmaceuticals. 2021; 14(10):994. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14100994
Chicago/Turabian StyleYang, Kun-Han, Yu-Sheng Lin, Sheng-Chieh Wang, Min-Yu Lee, Jen-Yang Tang, Fang-Rong Chang, Ya-Ting Chuang, Jyh-Horng Sheu, and Hsueh-Wei Chang. 2021. "Soft Coral-Derived Dihydrosinularin Exhibits Antiproliferative Effects Associated with Apoptosis and DNA Damage in Oral Cancer Cells" Pharmaceuticals 14, no. 10: 994. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14100994