
Reversing an Oncogenic Epithelial-to-Mesenchymal Transition Program in Breast Cancer Reveals Actionable Immune Suppressive Pathways

 ,
,  ,
,
Abstract
:1. Introduction
2. Experimental Models of Breast Cancer Metastasis and Epithelial-to-Mesenchymal Transition (EMT)
2.1. Modeling Breast Cancer Metastasis
2.1.1. Immune-Competent Models of Breast Cancer Metastasis
2.1.2. Patient Derived Xenograft Models of Breast Cancer Metastasis
2.2. Manipulating EMT in Breast Cancer Models
2.2.1. Introduction to EMT
2.2.2. Methods to Manipulate EMT in Breast Cancer Models
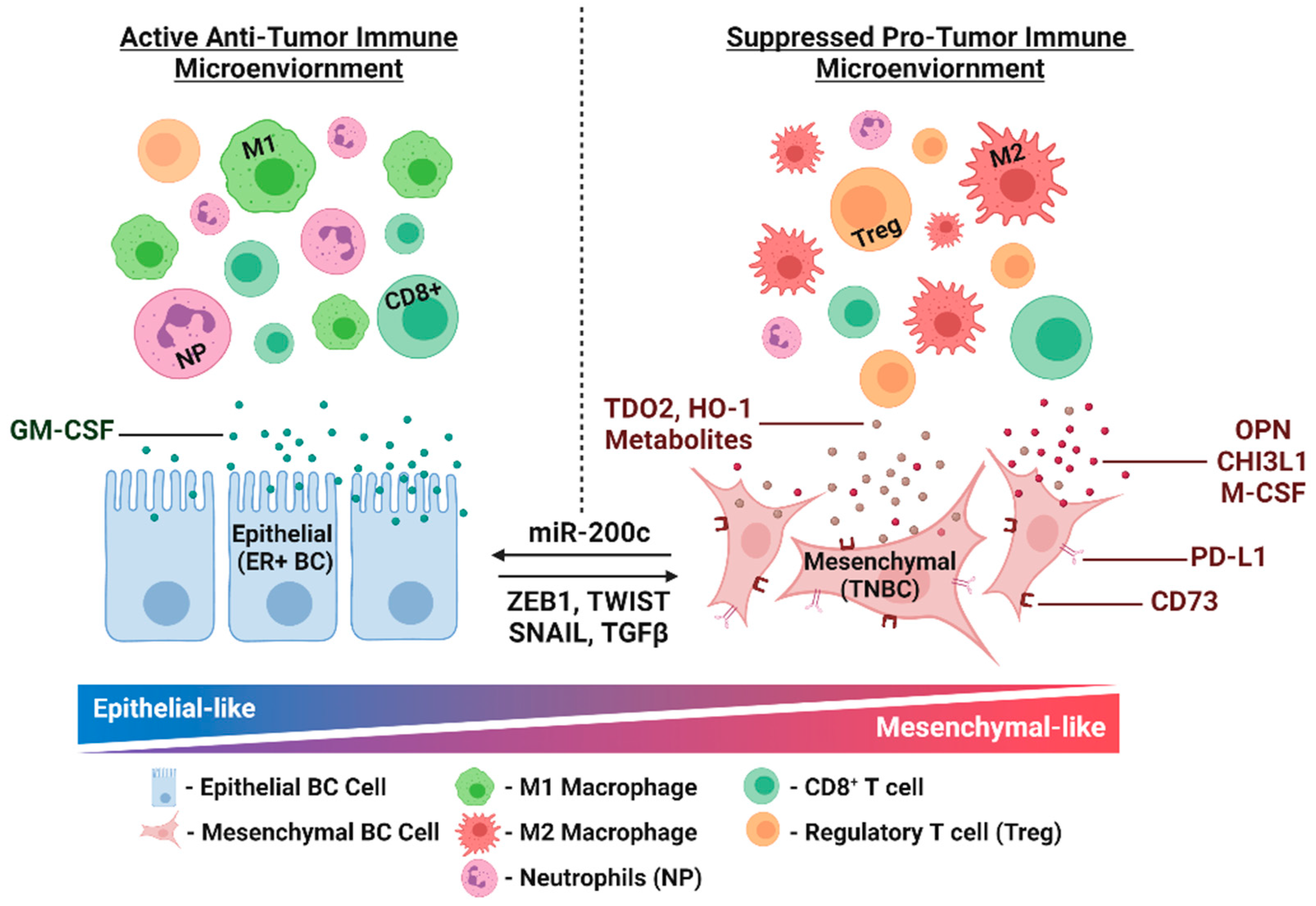
3. EMT and Immune Modulation
3.1. Preclinical and Clinical Evidence for Immune Modulation via EMT in Breast Cancer
3.2. Manipulation of EMT in Breast Cancer Models Reveals Additional Immune Modulators
4. Modulation of EMT Reveals Immune Suppressive Enzymes
4.1. 5′-Nucleotidase (NT5E)/CD73
4.1.1. CD73 and Breast Cancer Metastasis
4.1.2. Adenosine and the Breast Cancer Microenvironment
4.1.3. Clinical Targeting of CD73 in Breast Cancer
4.2. Heme Oxgenase-1 (HO-1)
4.2.1. HO-1 and Breast Cancer Metastasis
4.2.2. HO-1 and Immune Suppression in Breast Cancer
4.3. Tryptophan 2,3-Dioxygenase (TDO2)
4.3.1. TDO2 and Breast Cancer Metastasis
4.3.2. Tryptophan Catabolism and the Breast Cancer Microenvironment
4.3.3. Clinical Targeting of Tryptophan Catabolism
5. EMT Regulated Immune Modulatory Cytokines
5.1. Granulocyte-Macrophage Colony-Stimulatory Factor (GM-CSF)
5.1.1. GM-CSF and Breast Cancer Metastasis
5.1.2. Clinical Testing of GM-CSF Therapy
5.2. Macrophage Colony-Stimulating Factor (M-CSF)/Colony Stimulating Factor-1 (CSF-1)
5.2.1. M-CSF and Breast Cancer Progression
5.2.2. Preclinical and Clinical Targeting of M-CSF in Breast Cancer
6. Other Secreted Immune Suppressive Factors Identified via EMT Modulation
6.1. Chitinase-3 Like-1 (CHI3L1)
6.1.1. CHI3L1 and Breast Cancer Metastasis
6.1.2. Impact of CHI3L1 on the Tumor Microenvironment
6.2. Secreted Phosphoprotein 1 (SPP1)/Osteopontin (OPN)
6.2.1. OPN and Breast Cancer Metastasis
6.2.2. OPN and the Breast Cancer Microenvironment
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Morante, Z.; De la Cruz Ku, G.A.; Enriquez, D.; Saavedra, A.; Luján, M.; Luque, R.; Eyzaguirre, E.; Guardamino, D.; Valcárcel, B.; Araujo, J.M.; et al. Post-recurrence survival in triple negative breast cancer. J. Clin. Oncol. 2018, 36, e13120. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Zhu, L.; Narloch, J.L.; Onkar, S.; Joy, M.; Broadwater, G.; Luedke, C.; Hall, A.; Kim, R.; Pogue-Geile, K.; Sammons, S.; et al. Metastatic breast cancers have reduced immune cell recruitment but harbor increased macrophages relative to their matched primary tumors. J. Immunother Cancer 2019, 7, 265. [Google Scholar] [CrossRef] [Green Version]
- Hutchinson, K.E.; Yost, S.E.; Chang, C.W.; Johnson, R.M.; Carr, A.R.; McAdam, P.R.; Halligan, D.L.; Chang, C.C.; Schmolze, D.; Liang, J.; et al. Comprehensive Profiling of Poor-Risk Paired Primary and Recurrent Triple-Negative Breast Cancers Reveals Immune Phenotype Shifts. Clin. Cancer Res. 2020, 26, 657–668. [Google Scholar] [CrossRef] [Green Version]
- Roarty, K.; Echeverria, G.V. Laboratory Models for Investigating Breast Cancer Therapy Resistance and Metastasis. Front. Oncol. 2021, 11, 645698. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, P.; Zhang, A.; Mao, X. Advances in Rodent Models for Breast Cancer Formation, Progression, and Therapeutic Testing. Front. Oncol. 2021, 11, 593337. [Google Scholar] [CrossRef] [PubMed]
- Attalla, S.; Taifour, T.; Bui, T.; Muller, W. Insights from transgenic mouse models of PyMT-induced breast cancer: Recapitulating human breast cancer progression in vivo. Oncogene 2021, 40, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Jones, J.G.; Li, P.; Zhu, L.; Whitney, K.D.; Muller, W.J.; Pollard, J.W. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 2003, 163, 2113–2126. [Google Scholar] [CrossRef] [Green Version]
- Aslakson, C.J.; Miller, F.R. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992, 52, 1399–1405. [Google Scholar]
- Borowsky, A.D.; Namba, R.; Young, L.J.; Hunter, K.W.; Hodgson, J.G.; Tepper, C.G.; McGoldrick, E.T.; Muller, W.J.; Cardiff, R.D.; Gregg, J.P. Syngeneic mouse mammary carcinoma cell lines: Two closely related cell lines with divergent metastatic behavior. Clin. Exp. Metastasis 2005, 22, 47–59. [Google Scholar] [CrossRef]
- Lelekakis, M.; Moseley, J.M.; Martin, T.J.; Hards, D.; Williams, E.; Ho, P.; Lowen, D.; Javni, J.; Miller, F.R.; Slavin, J.; et al. A novel orthotopic model of breast cancer metastasis to bone. Clin. Exp. Metastasis 1999, 17, 163–170. [Google Scholar] [CrossRef]
- Rashid, O.M.; Nagahashi, M.; Ramachandran, S.; Dumur, C.I.; Schaum, J.C.; Yamada, A.; Aoyagi, T.; Milstien, S.; Spiegel, S.; Takabe, K. Is tail vein injection a relevant breast cancer lung metastasis model? J. Thorac. Dis. 2013, 5, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Goddard, E.T.; Fischer, J.; Schedin, P. A Portal Vein Injection Model to Study Liver Metastasis of Breast Cancer. J. Vis. Exp. 2016, 118, e54903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, J.K.; Hildreth, B.E., 3rd; Supsavhad, W.; Elshafae, S.M.; Hassan, B.B.; Dirksen, W.P.; Toribio, R.E.; Rosol, T.J. Animal Models of Bone Metastasis. Vet. Pathol. 2015, 52, 827–841. [Google Scholar] [CrossRef]
- Zhang, X.; Claerhout, S.; Prat, A.; Dobrolecki, L.E.; Petrovic, I.; Lai, Q.; Landis, M.D.; Wiechmann, L.; Schiff, R.; Giuliano, M.; et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013, 73, 4885–4897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeRose, Y.S.; Wang, G.; Lin, Y.C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajra, K.M.; Chen, D.Y.; Fearon, E.R. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002, 62, 1613–1618. [Google Scholar]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongre, A.; Rashidian, M.; Eaton, E.N.; Reinhardt, F.; Thiru, P.; Zagorulya, M.; Nepal, S.; Banaz, T.; Martner, A.; Spranger, S.; et al. Direct and Indirect Regulators of Epithelial-Mesenchymal Transition-Mediated Immunosuppression in Breast Carcinomas. Cancer Discov. 2021, 11, 1286–1305. [Google Scholar] [CrossRef] [PubMed]
- Cesi, V.; Casciati, A.; Sesti, F.; Tanno, B.; Calabretta, B.; Raschella, G. TGFbeta-induced c-Myb affects the expression of EMT-associated genes and promotes invasion of ER+ breast cancer cells. Cell Cycle 2011, 10, 4149–4161. [Google Scholar] [CrossRef] [Green Version]
- Cochrane, D.R.; Cittelly, D.M.; Howe, E.N.; Spoelstra, N.S.; McKinsey, E.L.; LaPara, K.; Elias, A.; Yee, D.; Richer, J.K. MicroRNAs link estrogen receptor alpha status and Dicer levels in breast cancer. Horm. Cancer 2010, 1, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Knezevic, J.; Pfefferle, A.D.; Petrovic, I.; Greene, S.B.; Perou, C.M.; Rosen, J.M. Expression of miR-200c in claudin-low breast cancer alters stem cell functionality, enhances chemosensitivity and reduces metastatic potential. Oncogene 2015, 34, 5997–6006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cochrane, D.R.; Spoelstra, N.S.; Howe, E.N.; Nordeen, S.K.; Richer, J.K. MicroRNA-200c mitigates invasiveness and restores sensitivity to microtubule-targeting chemotherapeutic agents. Mol. Cancer 2009, 8, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Howe, E.N.; Cochrane, D.R.; Richer, J.K. Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res. 2011, 13, R45. [Google Scholar] [CrossRef] [Green Version]
- Mak, M.P.; Tong, P.; Diao, L.; Cardnell, R.J.; Gibbons, D.L.; William, W.N.; Skoulidis, F.; Parra, E.R.; Rodriguez-Canales, J.; Wistuba, I.I.; et al. A Patient-Derived, Pan-Cancer EMT Signature Identifies Global Molecular Alterations and Immune Target Enrichment Following Epithelial-to-Mesenchymal Transition. Clin. Cancer Res. 2016, 22, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Rogers, T.J.; Christenson, J.L.; Greene, L.I.; O’Neill, K.I.; Williams, M.M.; Gordon, M.A.; Nemkov, T.; D’Alessandro, A.; Degala, G.D.; Shin, J.; et al. Reversal of Triple-Negative Breast Cancer EMT by miR-200c Decreases Tryptophan Catabolism and a Program of Immunosuppression. Mol. Cancer Res. 2019, 17, 30–41. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.M.; Christenson, J.L.; O’Neill, K.I.; Hafeez, S.A.; Ihle, C.L.; Spoelstra, N.S.; Slansky, J.E.; Richer, J.K. MicroRNA-200c restoration reveals a cytokine profile to enhance M1 macrophage polarization in breast cancer. NPJ Breast Cancer 2021, 7, 64. [Google Scholar] [CrossRef]
- Cancer Genome Atlas, N. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Tsang, J.Y.S.; Hlaing, T.; Hu, J.; Ni, Y.B.; Chan, S.K.; Cheung, S.Y.; Tse, G.M. Distinct Tertiary Lymphoid Structure Associations and Their Prognostic Relevance in HER2 Positive and Negative Breast Cancers. Oncologist 2017, 22, 1316–1324. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 2017, 6, e1263412. [Google Scholar] [CrossRef]
- Kim, I.S.; Gao, Y.; Welte, T.; Wang, H.; Liu, J.; Janghorban, M.; Sheng, K.; Niu, Y.; Goldstein, A.; Zhao, N.; et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat. Cell Biol. 2019, 21, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Halldorsson, S.; Rohatgi, N.; Magnusdottir, M.; Choudhary, K.S.; Gudjonsson, T.; Knutsen, E.; Barkovskaya, A.; Hilmarsdottir, B.; Perander, M.; Maelandsmo, G.M.; et al. Metabolic re-wiring of isogenic breast epithelial cell lines following epithelial to mesenchymal transition. Cancer Lett. 2017, 396, 117–129. [Google Scholar] [CrossRef] [Green Version]
- Hua, W.; Kostidis, S.; Mayboroda, O.; Giera, M.; Hornsveld, M.; Ten Dijke, P. Metabolic Reprogramming of Mammary Epithelial Cells during TGF-beta-Induced Epithelial-to-Mesenchymal Transition. Metabolites 2021, 11, 626. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Qian, Y.; Yu, J.; Wong, C.C. Metabolic rewiring in the promotion of cancer metastasis: Mechanisms and therapeutic implications. Oncogene 2020, 39, 6139–6156. [Google Scholar] [CrossRef]
- Jennings, M.R.; Munn, D.; Blazeck, J. Immunosuppressive metabolites in tumoral immune evasion: Redundancies, clinical efforts, and pathways forward. J. ImmunoTherapy Cancer 2021, 9, e003013. [Google Scholar] [CrossRef]
- Blay, J.; White, T.D.; Hoskin, D.W. The Extracellular Fluid of Solid Carcinomas Contains Immunosuppressive Concentrations of Adenosine. Cancer Res. 1997, 57, 2602. [Google Scholar] [PubMed]
- Leth-Larsen, R.; Lund, R.; Hansen, H.V.; Laenkholm, A.V.; Tarin, D.; Jensen, O.N.; Ditzel, H.J. Metastasis-related plasma membrane proteins of human breast cancer cells identified by comparative quantitative mass spectrometry. Mol. Cell Proteom. 2009, 8, 1436–1449. [Google Scholar] [CrossRef] [Green Version]
- Buisseret, L.; Pommey, S.; Allard, B.; Garaud, S.; Bergeron, M.; Cousineau, I.; Ameye, L.; Bareche, Y.; Paesmans, M.; Crown, J.P.A.; et al. Clinical significance of CD73 in triple-negative breast cancer: Multiplex analysis of a phase III clinical trial. Ann. Oncol. 2018, 29, 1056–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, G.; Cheng, P.; Pan, J.; Wang, S.; Ding, Q.; Jiang, Z.; Cheng, L.; Shao, X.; Huang, L.; Huang, J. An IL6-Adenosine Positive Feedback Loop between CD73(+) gammadeltaTregs and CAFs Promotes Tumor Progression in Human Breast Cancer. Cancer Immunol. Res. 2020, 8, 1273–1286. [Google Scholar] [CrossRef]
- Samanta, D.; Park, Y.; Ni, X.; Li, H.; Zahnow, C.A.; Gabrielson, E.; Pan, F.; Semenza, G.L. Chemotherapy induces enrichment of CD47(+)/CD73(+)/PDL1(+) immune evasive triple-negative breast cancer cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1239–E1248. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Gu, C.; Yao, X.; Guo, W.; Wang, H.; Lin, T.; Li, F.; Chen, D.; Wu, J.; Ye, G.; et al. CD73 promotes tumor metastasis by modulating RICS/RhoA signaling and EMT in gastric cancer. Cell Death Dis. 2020, 11, 202. [Google Scholar] [CrossRef]
- Petruk, N.; Tuominen, S.; Akerfelt, M.; Mattsson, J.; Sandholm, J.; Nees, M.; Yegutkin, G.G.; Jukkola, A.; Tuomela, J.; Selander, K.S. CD73 facilitates EMT progression and promotes lung metastases in triple-negative breast cancer. Sci. Rep. 2021, 11, 6035. [Google Scholar] [CrossRef]
- Etique, N.; Grillier-Vuissoz, I.; Lecomte, J.; Flament, S. Crosstalk between adenosine receptor (A2A isoform) and ERα mediates ethanol action in MCF-7 breast cancer cells. Oncol. Rep. 2009, 21, 977–981. [Google Scholar] [CrossRef] [Green Version]
- Lan, J.; Lu, H.; Samanta, D.; Salman, S.; Lu, Y.; Semenza, G.L. Hypoxia-inducible factor 1-dependent expression of adenosine receptor 2B promotes breast cancer stem cell enrichment. Proc. Natl. Acad. Sci. USA 2018, 115, E9640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmet, C.J.; Gallenne, T.; Prieur, A.; Reyal, F.; Visser, N.L.; Wittner, B.S.; Smit, M.A.; Geiger, T.R.; Laoukili, J.; Iskit, S.; et al. Identification of a pharmacologically tractable Fra-1/ADORA2B axis promoting breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 5139–5144. [Google Scholar] [CrossRef] [Green Version]
- Mittal, D.; Sinha, D.; Barkauskas, D.; Young, A.; Kalimutho, M.; Stannard, K.; Caramia, F.; Haibe-Kains, B.; Stagg, J.; Khanna, K.K.; et al. Adenosine 2B Receptor Expression on Cancer Cells Promotes Metastasis. Cancer Res. 2016, 76, 4372–4382. [Google Scholar] [CrossRef] [Green Version]
- Stagg, J.; Divisekera, U.; McLaughlin, N.; Sharkey, J.; Pommey, S.; Denoyer, D.; Dwyer, K.M.; Smyth, M.J. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1547–1552. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, G.; Vuerich, M.; Pellegatti, P.; Marimpietri, D.; Emionite, L.; Marigo, I.; Bronte, V.; Di Virgilio, F.; Pistoia, V.; Raffaghello, L. ATP/P2X7 axis modulates myeloid-derived suppressor cell functions in neuroblastoma microenvironment. Cell Death Dis. 2014, 5, e1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjaergaard, J.; Hatfield, S.; Jones, G.; Ohta, A.; Sitkovsky, M. A2A Adenosine Receptor Gene Deletion or Synthetic A2A Antagonist Liberate Tumor-Reactive CD8(+) T Cells from Tumor-Induced Immunosuppression. J. Immunol. 2018, 201, 782–791. [Google Scholar] [CrossRef] [Green Version]
- Vasiukov, G.; Novitskaya, T.; Zijlstra, A.; Owens, P.; Ye, F.; Zhao, Z.; Moses, H.L.; Blackwell, T.; Feoktistov, I.; Novitskiy, S.V. Myeloid Cell–Derived TGFβ Signaling Regulates ECM Deposition in Mammary Carcinoma via Adenosine-Dependent Mechanisms. Cancer Res. 2020, 80, 2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, M.; Guo, G.; Huang, L.; Deng, L.; Chang, C.-S.; Achyut, B.R.; Canning, M.; Xu, N.; Arbab, A.S.; Bollag, R.J.; et al. CD73 on cancer-associated fibroblasts enhanced by the A2B-mediated feedforward circuit enforces an immune checkpoint. Nat. Commun. 2020, 11, 515. [Google Scholar] [CrossRef]
- Cekic, C.; Sag, D.; Li, Y.; Theodorescu, D.; Strieter, R.M.; Linden, J. Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J. Immunol. 2012, 188, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Loi, S.; Pommey, S.; Haibe-Kains, B.; Beavis, P.A.; Darcy, P.K.; Smyth, M.J.; Stagg, J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 11091–11096. [Google Scholar] [CrossRef] [Green Version]
- Allard, B.; Pommey, S.; Smyth, M.J.; Stagg, J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin. Cancer Res. 2013, 19, 5626–5635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagg, J.; Smyth, M.J. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 2010, 29, 5346–5358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidders, B.; Zhang, P.; Goodwin, K.; O’Connor, G.; Russell, D.L.; Borodovsky, A.; Armenia, J.; McEwen, R.; Linghu, B.; Bendell, J.C.; et al. Adenosine Signaling Is Prognostic for Cancer Outcome and Has Predictive Utility for Immunotherapeutic Response. Clin. Cancer Res. 2020, 26, 2176–2187. [Google Scholar] [CrossRef]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Hay, C.M.; Sult, E.; Huang, Q.; Mulgrew, K.; Fuhrmann, S.R.; McGlinchey, K.A.; Hammond, S.A.; Rothstein, R.; Rios-Doria, J.; Poon, E.; et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology 2016, 5, e1208875. [Google Scholar] [CrossRef]
- Bendell, J.C.; LoRusso, P.; Overman, M.J.; Noonan, A.M.; Kim, D.-W.; Strickler, J.; Kim, S.-W.; Clarke, S.J.; George, T.J.; Grimison, P.S.; et al. Safety and efficacy of the anti-CD73 monoclonal antibody (mAb) oleclumab ± durvalumab in patients (pts) with advanced colorectal cancer (CRC), pancreatic ductal adenocarcinoma (PDAC), or EGFR-mutant non-small cell lung cancer (EGFRm NSCLC). J. Clin. Oncol. 2021, 39, 9047. [Google Scholar] [CrossRef]
- De Caluwe, A.; Buisseret, L.; Poortmans, P.; Van Gestel, D.; Salgado, R.; Sotiriou, C.; Larsimont, D.; Paesmans, M.; Craciun, L.; Stylianos, D.; et al. Neo-CheckRay: Radiation therapy and adenosine pathway blockade to increase benefit of immuno-chemotherapy in early stage luminal B breast cancer, a randomized phase II trial. BMC Cancer 2021, 21, 899. [Google Scholar] [CrossRef]
- Luke, J.J.; Powderly, J.D.; Merchan, J.R.; Barve, M.A.; Hotson, A.N.; Mobasher, M.; Kwei, L.; Luciano, G.; Buggy, J.J.; Piccione, E.; et al. Immunobiology, preliminary safety, and efficacy of CPI-006, an anti-CD73 antibody with immune modulating activity, in a phase 1 trial in advanced cancers. J. Clin. Oncol. 2019, 37, 2505. [Google Scholar] [CrossRef]
- Powderly, J.D.; Souza, P.L.d.; Gutierrez, R.; Horvath, L.; Seitz, L.; Ashok, D.; Park, A.; Walters, M.J.; Karakunnel, J.J.; Berry, W.; et al. AB928, a novel dual adenosine receptor antagonist, combined with chemotherapy or AB122 (anti-PD-1) in patients (pts) with advanced tumors: Preliminary results from ongoing phase I studies. J. Clin. Oncol. 2019, 37, 2604. [Google Scholar] [CrossRef]
- Zhong, Y.; Zhang, F.; Sun, Z.; Zhou, W.; Li, Z.Y.; You, Q.D.; Guo, Q.L.; Hu, R. Drug resistance associates with activation of Nrf2 in MCF-7/DOX cells, and wogonin reverses it by down-regulating Nrf2-mediated cellular defense response. Mol. Carcinog 2013, 52, 824–834. [Google Scholar] [CrossRef]
- Tan, Q.; Wang, H.; Hu, Y.; Hu, M.; Li, X.; Aodengqimuge; Ma, Y.; Wei, C.; Song, L. Src/STAT3-dependent heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to doxorubicin by promoting autophagy. Cancer Sci. 2015, 106, 1023–1032. [Google Scholar] [CrossRef]
- Pei, L.; Kong, Y.; Shao, C.; Yue, X.; Wang, Z.; Zhang, N. Heme oxygenase-1 induction mediates chemoresistance of breast cancer cells to pharmorubicin by promoting autophagy via PI3K/Akt pathway. J. Cell Mol. Med. 2018, 22, 5311–5321. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.F.; Li, W.; Ma, J.Y.; Shao, N.; Zhang, Y.J.; Liu, R.M.; Wu, W.B.; Lin, Y.; Wang, S.M. Knockdown of heme oxygenase-1 promotes apoptosis and autophagy and enhances the cytotoxicity of doxorubicin in breast cancer cells. Oncol. Lett. 2015, 10, 2974–2980. [Google Scholar] [CrossRef] [Green Version]
- Deng, R.; Wang, S.M.; Yin, T.; Ye, T.H.; Shen, G.B.; Li, L.; Zhao, J.Y.; Sang, Y.X.; Duan, X.G.; Wei, Y.Q. Inhibition of tumor growth and alteration of associated macrophage cell type by an HO-1 inhibitor in breast carcinoma-bearing mice. Oncol. Res. 2013, 20, 473–482. [Google Scholar] [CrossRef]
- Muliaditan, T.; Opzoomer, J.W.; Caron, J.; Okesola, M.; Kosti, P.; Lall, S.; Van Hemelrijck, M.; Dazzi, F.; Tutt, A.; Grigoriadis, A.; et al. Repurposing Tin Mesoporphyrin as an Immune Checkpoint Inhibitor Shows Therapeutic Efficacy in Preclinical Models of Cancer. Clin. Cancer Res. 2018, 24, 1617–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.; Fuchs, S.Y.; et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Investig. 2015, 125, 2592–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [Google Scholar] [CrossRef]
- Noh, S.J.; Bae, J.S.; Jamiyandorj, U.; Park, H.S.; Kwon, K.S.; Jung, S.H.; Youn, H.J.; Lee, H.; Park, B.H.; Chung, M.J.; et al. Expression of nerve growth factor and heme oxygenase-1 predict poor survival of breast carcinoma patients. BMC Cancer 2013, 13, 516. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Peng, F.H.; Peng, L.K. MiR-200c sensitizes clear-cell renal cell carcinoma cells to sorafenib and imatinib by targeting heme oxygenase-1. Neoplasma 2014, 61, 680–689. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Liu, Q.; Cheng, W.; Wei, H.; Jiang, W.; Fang, E.; Yu, Y.; Jin, J.; Zou, C. Heme Oxygenase-1 Inhibits Tumor Metastasis Mediated by Notch1 Pathway in Murine Mammary Carcinoma. Oncol. Res. 2019, 27, 643–651. [Google Scholar] [CrossRef]
- Zhu, X.; Huang, S.; Zeng, L.; Ma, J.; Sun, S.; Zeng, F.; Kong, F.; Cheng, X. HMOX-1 inhibits TGF-beta-induced epithelial-mesenchymal transition in the MCF-7 breast cancer cell line. Int J. Mol. Med. 2017, 40, 411–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khojandi, N.; Kuehm, L.M.; Piening, A.; Donlin, M.J.; Hsueh, E.C.; Schwartz, T.L.; Farrell, K.; Richart, J.M.; Geerling, E.; Pinto, A.K.; et al. Oxidized Lipoproteins Promote Resistance to Cancer Immunotherapy Independent of Patient Obesity. Cancer Immunol. Res. 2021, 9, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Alaluf, E.; Vokaer, B.; Detavernier, A.; Azouz, A.; Splittgerber, M.; Carrette, A.; Boon, L.; Libert, F.; Soares, M.; Le Moine, A.; et al. Heme oxygenase-1 orchestrates the immunosuppressive program of tumor-associated macrophages. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Muliaditan, T.; Caron, J.; Okesola, M.; Opzoomer, J.W.; Kosti, P.; Georgouli, M.; Gordon, P.; Lall, S.; Kuzeva, D.M.; Pedro, L.; et al. Macrophages are exploited from an innate wound healing response to facilitate cancer metastasis. Nat. Commun. 2018, 9, 2951. [Google Scholar] [CrossRef] [Green Version]
- Kirkby, K.A.; Adin, C.A. Products of heme oxygenase and their potential therapeutic applications. Am. J. Physiol. Ren. Physiol. 2006, 290, F563–F571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, K.; McDaid, J.; Ollinger, R.; Tsui, T.Y.; Berberat, P.O.; Usheva, A.; Csizmadia, E.; Smith, R.N.; Soares, M.P.; Bach, F.H. Biliverdin, a natural product of heme catabolism, induces tolerance to cardiac allografts. FASEB J. 2004, 18, 765–767. [Google Scholar] [CrossRef] [PubMed]
- Fondevila, C.; Shen, X.D.; Tsuchiyashi, S.; Yamashita, K.; Csizmadia, E.; Lassman, C.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Bach, F.H. Biliverdin therapy protects rat livers from ischemia and reperfusion injury. Hepatology 2004, 40, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Li, P.; Lu, J.; Xiong, W.; Oger, J.; Tetzlaff, W.; Cynader, M. Bilirubin possesses powerful immunomodulatory activity and suppresses experimental autoimmune encephalomyelitis. J. Immunol. 2008, 181, 1887–1897. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Ma, J.; Fan, S.T.; Schlitt, H.J.; Tsui, T.Y. Bilirubin derived from heme degradation suppresses MHC class II expression in endothelial cells. Biochem. Biophys. Res. Commun. 2005, 338, 890–896. [Google Scholar] [CrossRef]
- Vetvicka, V.; Miler, I.; Sima, P.; Taborsky, L.; Fornusek, L. The effect of bilirubin on the Fc receptor expression and phagocytic activity of mouse peritoneal macrophages. Folia Microbiol. 1985, 30, 373–380. [Google Scholar] [CrossRef]
- Rocuts, F.; Zhang, X.; Yan, J.; Yue, Y.; Thomas, M.; Bach, F.H.; Czismadia, E.; Wang, H. Bilirubin promotes de novo generation of T regulatory cells. Cell Transpl. 2010, 19, 443–451. [Google Scholar] [CrossRef]
- Haga, Y.; Tempero, M.A.; Kay, D.; Zetterman, R.K. Intracellular accumulation of unconjugated bilirubin inhibits phytohemagglutin-induced proliferation and interleukin-2 production of human lymphocytes. Dig. Dis. Sci. 1996, 41, 1468–1474. [Google Scholar] [CrossRef]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef] [PubMed]
- Valaes, T.; Petmezaki, S.; Henschke, C.; Drummond, G.S.; Kappas, A. Control of jaundice in preterm newborns by an inhibitor of bilirubin production: Studies with tin-mesoporphyrin. Pediatrics 1994, 93, 1–11. [Google Scholar] [PubMed]
- Fernandez-Fierro, A.; Funes, S.C.; Rios, M.; Covian, C.; Gonzalez, J.; Kalergis, A.M. Immune Modulation by Inhibitors of the HO System. Int. J. Mol. Sci. 2020, 22, 294. [Google Scholar] [CrossRef]
- Rafice, S.A.; Chauhan, N.; Efimov, I.; Basran, J.; Raven, E.L. Oxidation of L-tryptophan in biology: A comparison between tryptophan 2,3-dioxygenase and indoleamine 2,3-dioxygenase. Biochem. Soc. Trans. 2009, 37, 408–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan. Res. 2017, 10, 1178646917691938. [Google Scholar] [CrossRef] [Green Version]
- Opitz, C.A.; Somarribas Patterson, L.F.; Mohapatra, S.R.; Dewi, D.L.; Sadik, A.; Platten, M.; Trump, S. The therapeutic potential of targeting tryptophan catabolism in cancer. Br. J. Cancer 2020, 122, 30–44. [Google Scholar] [CrossRef]
- Muller, A.J.; Manfredi, M.G.; Zakharia, Y.; Prendergast, G.C. Inhibiting IDO pathways to treat cancer: Lessons from the ECHO-301 trial and beyond. Semin Immunopathol. 2019, 41, 41–48. [Google Scholar] [CrossRef]
- D’Amato, N.C.; Rogers, T.J.; Gordon, M.A.; Greene, L.I.; Cochrane, D.R.; Spoelstra, N.S.; Nemkov, T.G.; D’Alessandro, A.; Hansen, K.C.; Richer, J.K. A TDO2-AhR signaling axis facilitates anoikis resistance and metastasis in triple-negative breast cancer. Cancer Res. 2015, 75, 4651–4664. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhai, J.; Kong, X.; Wang, X.; Wang, Z.; Fang, Y.; Wang, J. Comprehensive Analysis of the Expression and Prognosis for TDO2 in Breast Cancer. Mol. Oncolytics 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Novikov, O.; Wang, Z.; Stanford, E.A.; Parks, A.J.; Ramirez-Cardenas, A.; Landesman, E.; Laklouk, I.; Sarita-Reyes, C.; Gusenleitner, D.; Li, A.; et al. An Aryl Hydrocarbon Receptor-Mediated Amplification Loop That Enforces Cell Migration in ER-/PR-/Her2- Human Breast Cancer Cells. Mol. Pharm. 2016, 90, 674–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, L.; Bell, A.; Ladomersky, E.; Lauing, K.L.; Bollu, L.; Sosman, J.A.; Zhang, B.; Wu, J.D.; Miller, S.D.; Meeks, J.J.; et al. Immunosuppressive IDO in Cancer: Mechanisms of Action, Animal Models, and Targeting Strategies. Front. Immunol. 2020, 11, 1185. [Google Scholar] [CrossRef]
- Terness, P.; Bauer, T.M.; Rose, L.; Dufter, C.; Watzlik, A.; Simon, H.; Opelz, G. Inhibition of allogeneic T cell proliferation by indoleamine 2,3-dioxygenase-expressing dendritic cells: Mediation of suppression by tryptophan metabolites. J. Exp. Med. 2002, 196, 447–457. [Google Scholar] [CrossRef] [Green Version]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Mezrich, J.D.; Fechner, J.H.; Zhang, X.; Johnson, B.P.; Burlingham, W.J.; Bradfield, C.A. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010, 185, 3190–3198. [Google Scholar] [CrossRef] [Green Version]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef]
- Marshall, N.B.; Kerkvliet, N.I. Dioxin and immune regulation: Emerging role of aryl hydrocarbon receptor in the generation of regulatory T cells. Ann. N. Y. Acad. Sci. 2010, 1183, 25–37. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Liu, L.; Liu, K.; Bizargity, P.; Hancock, W.W.; Visner, G.A. Reduced cytotoxic function of effector CD8+ T cells is responsible for indoleamine 2,3-dioxygenase-dependent immune suppression. J. Immunol. 2009, 183, 1022–1031. [Google Scholar] [CrossRef] [Green Version]
- Greene, L.I.; Bruno, T.C.; Christenson, J.L.; D’Alessandro, A.; Culp-Hill, R.; Torkko, K.; Borges, V.F.; Slansky, J.E.; Richer, J.K. A Role for Tryptophan-2,3-dioxygenase in CD8 T-cell Suppression and Evidence of Tryptophan Catabolism in Breast Cancer Patient Plasma. Mol. Cancer Res. 2019, 17, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlova, A.; Frederick, R. Current state on tryptophan 2,3-dioxygenase inhibitors: A patent review. Expert Opin. Pat. 2019, 29, 11–23. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Salter, M.; Hazelwood, R.; Pogson, C.I.; Iyer, R.; Madge, D.J. The effects of a novel and selective inhibitor of tryptophan 2,3-dioxygenase on tryptophan and serotonin metabolism in the rat. Biochem. Pharm. 1995, 49, 1435–1442. [Google Scholar] [CrossRef]
- Dolusic, E.; Larrieu, P.; Moineaux, L.; Stroobant, V.; Pilotte, L.; Colau, D.; Pochet, L.; Van den Eynde, B.; Masereel, B.; Wouters, J.; et al. Tryptophan 2,3-dioxygenase (TDO) inhibitors. 3-(2-(pyridyl)ethenyl)indoles as potential anticancer immunomodulators. J. Med. Chem. 2011, 54, 5320–5334. [Google Scholar] [CrossRef]
- Sari, S.; Tomek, P.; Leung, E.; Reynisson, J. Discovery and Characterisation of Dual Inhibitors of Tryptophan 2,3-Dioxygenase (TDO2) and Indoleamine 2,3-Dioxygenase 1 (IDO1) Using Virtual Screening. Molecules 2019, 24, 4346. [Google Scholar] [CrossRef] [Green Version]
- Gyulveszi, G.; Fischer, C.; Mirolo, M.; Stern, M.; Green, L.; Ceppi, M.; Wang, H.; Bürgi, B.; Staempfli, A.; Muster, W.; et al. Abstract LB-085: RG70099: A novel, highly potent dual IDO1/TDO inhibitor to reverse metabolic suppression of immune cells in the tumor micro-environment. Cancer Res. 2016, 76, LB-085. [Google Scholar] [CrossRef]
- Gullapalli, S.; Roychowdhury, A.; Khaladkar, T.; Sawargave, S.; Janrao, R.; Kalhapure, V.; Urunkar, G.; Kulathingal, J.; Lekkala, R.R.; Bhadra, S.; et al. Abstract 1701: EPL-1410, a novel fused heterocycle based orally active dual inhibitor of IDO1/TDO2, as a potential immune-oncology therapeutic. Cancer Res. 2018, 78, 1701. [Google Scholar] [CrossRef]
- Kim, C.; Kim, J.H.; Kim, J.S.; Chon, H.J.; Kim, J.-H. A novel dual inhibitor of IDO and TDO, CMG017, potently suppresses the kynurenine pathway and overcomes resistance to immune checkpoint inhibitors. J. Clin. Oncol. 2019, 37, e14228. [Google Scholar] [CrossRef]
- Zhang, M.; Stone, E.; Triplett, T.A.; Triplett, K.; Lamb, C.; Karamitros, C.S.; Blazek, J.; Georgiou, G.; Manfredi, M.G. Abstract 5570: A novel approach to targeting the IDO/TDO pathway through degradation of the immunosuppressive metabolite kynurenine. Cancer Res. 2017, 77, 5570. [Google Scholar] [CrossRef]
- Coma, S.; Cavanaugh, J.; Nolan, J.; Tchaicha, J.; McGovern, K.; Stone, E.; Lamb, C.; Karamitros, C.; Blazek, J.; Garrison, K.; et al. Abstract 3757: Targeting the IDO/TDO pathway through degradation of the immunosuppressive metabolite kynurenine. Cancer Res. 2018, 78, 3757. [Google Scholar] [CrossRef]
- Brenot, A.; Knolhoff, B.L.; DeNardo, D.G.; Longmore, G.D. SNAIL1 action in tumor cells influences macrophage polarization and metastasis in breast cancer through altered GM-CSF secretion. Oncogenesis 2018, 7, 32. [Google Scholar] [CrossRef]
- Eubank, T.D.; Roberts, R.D.; Khan, M.; Curry, J.M.; Nuovo, G.J.; Kuppusamy, P.; Marsh, C.B. Granulocyte macrophage colony-stimulating factor inhibits breast cancer growth and metastasis by invoking an anti-angiogenic program in tumor-educated macrophages. Cancer Res. 2009, 69, 2133–2140. [Google Scholar] [CrossRef] [Green Version]
- Su, X.; Xu, Y.; Fox, G.C.; Xiang, J.; Kwakwa, K.A.; Davis, J.L.; Belle, J.I.; Lee, W.C.; Wong, W.H.; Fontana, F.; et al. Breast cancer derived GM-CSF regulates arginase 1 in myeloid cells to promote an immunosuppressive microenvironment. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Parmiani, G.; Castelli, C.; Pilla, L.; Santinami, M.; Colombo, M.P.; Rivoltini, L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann. Oncol. 2007, 18, 226–232. [Google Scholar] [CrossRef]
- Morales, J.K.; Kmieciak, M.; Knutson, K.L.; Bear, H.D.; Manjili, M.H. GM-CSF is one of the main breast tumor-derived soluble factors involved in the differentiation of CD11b-Gr1- bone marrow progenitor cells into myeloid-derived suppressor cells. Breast Cancer Res. Treat. 2010, 123, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, B.K.; Zhang, H.; Zeng, Q.; Dai, J.; Keller, E.T.; Giordano, T.; Gu, K.; Shah, V.; Pei, L.; Zarbo, R.J.; et al. NF-kappaB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nat. Med. 2007, 13, 62–69. [Google Scholar] [CrossRef]
- Ogawa, T.; Kusumoto, M.; Mizumoto, K.; Sato, N.; Tanaka, M. Adenoviral GM-CSF Gene Transduction into Breast Cancer Cells Induced Long-Lasting Antitumor Immunity in Mice. Breast Cancer 1999, 6, 301–304. [Google Scholar] [CrossRef] [PubMed]
- Ghirelli, C.; Reyal, F.; Jeanmougin, M.; Zollinger, R.; Sirven, P.; Michea, P.; Caux, C.; Bendriss-Vermare, N.; Donnadieu, M.H.; Caly, M.; et al. Breast Cancer Cell-Derived GM-CSF Licenses Regulatory Th2 Induction by Plasmacytoid Predendritic Cells in Aggressive Disease Subtypes. Cancer Res. 2015, 75, 2775–2787. [Google Scholar] [CrossRef] [Green Version]
- Machiels, J.P.; Reilly, R.T.; Emens, L.A.; Ercolini, A.M.; Lei, R.Y.; Weintraub, D.; Okoye, F.I.; Jaffee, E.M. Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice. Cancer Res. 2001, 61, 3689–3697. [Google Scholar]
- Cheng, Y.C.; Valero, V.; Davis, M.L.; Green, M.C.; Gonzalez-Angulo, A.M.; Theriault, R.L.; Murray, J.L.; Hortobagyi, G.N.; Ueno, N.T. Addition of GM-CSF to trastuzumab stabilises disease in trastuzumab-resistant HER2+ metastatic breast cancer patients. Br. J. Cancer 2010, 103, 1331–1334. [Google Scholar] [CrossRef]
- Clifton, G.T.; Peace, K.M.; Holmes, J.P.; Vreeland, T.J.; Hale, D.F.; Herbert, G.S.; Litton, J.K.; Murthy, R.K.; Lukas, J.; Peoples, G.E.; et al. Initial safety analysis of a randomized phase II trial of nelipepimut-S+GM-CSF and trastuzumab compared to trastuzumab alone to prevent recurrence in breast cancer patients with HER2 low-expressing tumors. Clin. Immunol. 2019, 201, 48–54. [Google Scholar] [CrossRef]
- Chick, R.C.; Clifton, G.T.; Hale, D.F.; Vreeland, T.J.; Hickerson, A.T.; Kemp Bohan, P.M.; McCarthy, P.M.; Litton, J.K.; Alatrash, G.; Murthy, R.K.; et al. Subgroup analysis of nelipepimut-S plus GM-CSF combined with trastuzumab versus trastuzumab alone to prevent recurrences in patients with high-risk, HER2 low-expressing breast cancer. Clin. Immunol. 2021, 225, 108679. [Google Scholar] [CrossRef]
- Wiseman, C.L.; Kharazi, A. Objective clinical regression of metastatic breast cancer in disparate sites after use of whole-cell vaccine genetically modified to release sargramostim. Breast J. 2006, 12, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Lacher, M.D.; Bauer, G.; Fury, B.; Graeve, S.; Fledderman, E.L.; Petrie, T.D.; Coleal-Bergum, D.P.; Hackett, T.; Perotti, N.H.; Kong, Y.Y.; et al. SV-BR-1-GM, a Clinically Effective GM-CSF-Secreting Breast Cancer Cell Line, Expresses an Immune Signature and Directly Activates CD4(+) T Lymphocytes. Front. Immunol. 2018, 9, 776. [Google Scholar] [CrossRef]
- Emens, L.A.; Asquith, J.M.; Leatherman, J.M.; Kobrin, B.J.; Petrik, S.; Laiko, M.; Levi, J.; Daphtary, M.M.; Biedrzycki, B.; Wolff, A.C.; et al. Timed sequential treatment with cyclophosphamide, doxorubicin, and an allogeneic granulocyte-macrophage colony-stimulating factor-secreting breast tumor vaccine: A chemotherapy dose-ranging factorial study of safety and immune activation. J. Clin. Oncol. 2009, 27, 5911–5918. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Gupta, R.; Petrik, S.; Laiko, M.; Leatherman, J.M.; Asquith, J.M.; Daphtary, M.M.; Garrett-Mayer, E.; Davidson, N.E.; Hirt, K.; et al. A feasibility study of cyclophosphamide, trastuzumab, and an allogeneic GM-CSF-secreting breast tumor vaccine for HER2+ metastatic breast cancer. Cancer Immunol. Res. 2014, 2, 949–961. [Google Scholar] [CrossRef] [Green Version]
- Honkoop, A.H.; Luykx-de Bakker, S.A.; Hoekman, K.; Meyer, S.; Meyer, O.W.; van Groeningen, C.J.; van Diest, P.J.; Boven, E.; van der Wall, E.; Giaccone, G.; et al. Prolonged neoadjuvant chemotherapy with GM-CSF in locally advanced breast cancer. Oncologist 1999, 4, 106–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clifton, G.T.; Hale, D.; Vreeland, T.J.; Hickerson, A.T.; Litton, J.K.; Alatrash, G.; Murthy, R.K.; Qiao, N.; Philips, A.V.; Lukas, J.J.; et al. Results of a Randomized Phase IIb Trial of Nelipepimut-S + Trastuzumab versus Trastuzumab to Prevent Recurrences in Patients with High-Risk HER2 Low-Expressing Breast Cancer. Clin. Cancer Res. 2020, 26, 2515–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lum, L.G.; Thakur, A.; Al-Kadhimi, Z.; Colvin, G.A.; Cummings, F.J.; Legare, R.D.; Dizon, D.S.; Kouttab, N.; Maizel, A.; Colaiace, W.; et al. Targeted T-cell Therapy in Stage IV Breast Cancer: A Phase I Clinical Trial. Clin. Cancer Res. 2015, 21, 2305–2314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peace, K.M.; Mittendorf, E.A.; Perez, S.A.; Tzonis, P.; Pistamaltzian, N.F.; Anastasopoulou, E.A.; Vreeland, T.J.; Hale, D.F.; Clifton, G.T.; Litton, J.K.; et al. Subgroup efficacy evaluation of the AE37 HER2 vaccine in breast cancer patients in the adjuvant setting. J. Clin. Oncol. 2017, 35, 3088. [Google Scholar] [CrossRef]
- Disis, M.L.; Dang, Y.; Coveler, A.L.; Higgins, D.; Childs, J.; Salazar, L.G. Final report and long-term outcomes: Phase I trial of a HER2 intracellular plasmid-based vaccine in HER2+ advanced stage breast cancer. J. Clin. Oncol. 2021, 39, 2619. [Google Scholar] [CrossRef]
- Wesolowski, R.; Sharma, N.; Reebel, L.; Rodal, M.B.; Peck, A.; West, B.L.; Marimuthu, A.; Severson, P.; Karlin, D.A.; Dowlati, A.; et al. Phase Ib study of the combination of pexidartinib (PLX3397), a CSF-1R inhibitor, and paclitaxel in patients with advanced solid tumors. Adv. Med. Oncol. 2019, 11, 1758835919854238. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Roca, C.A.; Italiano, A.; Le Tourneau, C.; Cassier, P.A.; Toulmonde, M.; D’Angelo, S.P.; Campone, M.; Weber, K.L.; Loirat, D.; Cannarile, M.A.; et al. Phase I study of emactuzumab single agent or in combination with paclitaxel in patients with advanced/metastatic solid tumors reveals depletion of immunosuppressive M2-like macrophages. Ann. Oncol. 2019, 30, 1381–1392. [Google Scholar] [CrossRef]
- Dowlati, A.; Harvey, R.D.; Carvajal, R.D.; Hamid, O.; Klempner, S.J.; Kauh, J.S.W.; Peterson, D.A.; Yu, D.; Chapman, S.C.; Szpurka, A.M.; et al. LY3022855, an anti-colony stimulating factor-1 receptor (CSF-1R) monoclonal antibody, in patients with advanced solid tumors refractory to standard therapy: Phase 1 dose-escalation trial. Investig. New Drugs 2021, 39, 1057–1071. [Google Scholar] [CrossRef]
- Kuemmel, S.; Campone, M.; Loirat, D.; Lopez Lopez, R.; Beck, J.T.; De Laurentiis, M.; Im, S.A.; Kim, S.B.; Kwong, A.; Steger, G.G.; et al. A Randomized Phase II Study of Anti-CSF-1 Monoclonal Antibody Lacnotuzumab (MCS110) Combined with Gemcitabine and Carboplatin in Advanced Triple Negative Breast Cancer. Clin. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Calvo, A.; Joensuu, H.; Sebastian, M.; Naing, A.; Bang, Y.-J.; Martin, M.; Roda, D.; Hodi, F.S.; Veloso, A.; Mataraza, J.; et al. Phase Ib/II study of lacnotuzumab (MCS110) combined with spartalizumab (PDR001) in patients (pts) with advanced tumors. J. Clin. Oncol. 2018, 36, 3014. [Google Scholar] [CrossRef]
- Kai, K.; Iwamoto, T.; Zhang, D.; Shen, L.; Takahashi, Y.; Rao, A.; Thompson, A.; Sen, S.; Ueno, N.T. CSF-1/CSF-1R axis is associated with epithelial/mesenchymal hybrid phenotype in epithelial-like inflammatory breast cancer. Sci. Rep. 2018, 8, 9427. [Google Scholar] [CrossRef]
- Kluger, H.M.; Dolled-Filhart, M.; Rodov, S.; Kacinski, B.M.; Camp, R.L.; Rimm, D.L. Macrophage colony-stimulating factor-1 receptor expression is associated with poor outcome in breast cancer by large cohort tissue microarray analysis. Clin. Cancer Res. 2004, 10, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Richardsen, E.; Uglehus, R.D.; Johnsen, S.H.; Busund, L.T. Macrophage-colony stimulating factor (CSF1) predicts breast cancer progression and mortality. Anticancer Res. 2015, 35, 865–874. [Google Scholar]
- Aharinejad, S.; Salama, M.; Paulus, P.; Zins, K.; Berger, A.; Singer, C.F. Elevated CSF1 serum concentration predicts poor overall survival in women with early breast cancer. Endocr. Relat. Cancer 2013, 20, 777–783. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Beck, A.H.; Webster, J.A.; Espinosa, I.; Montgomery, K.; Varma, S.; van de Rijn, M.; Jensen, K.C.; West, R.B. Analysis of stromal signatures in the tumor microenvironment of ductal carcinoma in situ. Breast Cancer Res. Treat. 2010, 123, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.H.; Espinosa, I.; Edris, B.; Li, R.; Montgomery, K.; Zhu, S.; Varma, S.; Marinelli, R.J.; van de Rijn, M.; West, R.B. The macrophage colony-stimulating factor 1 response signature in breast carcinoma. Clin. Cancer Res. 2009, 15, 778–787. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–740. [Google Scholar] [CrossRef] [Green Version]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [Green Version]
- Wyckoff, J.B.; Wang, Y.; Lin, E.Y.; Li, J.F.; Goswami, S.; Stanley, E.R.; Segall, J.E.; Pollard, J.W.; Condeelis, J. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007, 67, 2649–2656. [Google Scholar] [CrossRef] [Green Version]
- Linde, N.; Casanova-Acebes, M.; Sosa, M.S.; Mortha, A.; Rahman, A.; Farias, E.; Harper, K.; Tardio, E.; Reyes Torres, I.; Jones, J.; et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swierczak, A.; Cook, A.D.; Lenzo, J.C.; Restall, C.M.; Doherty, J.P.; Anderson, R.L.; Hamilton, J.A. The promotion of breast cancer metastasis caused by inhibition of CSF-1R/CSF-1 signaling is blocked by targeting the G-CSF receptor. Cancer Immunol. Res. 2014, 2, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renkema, G.H.; Boot, R.G.; Au, F.L.; Donker-Koopman, W.E.; Strijland, A.; Muijsers, A.O.; Hrebicek, M.; Aerts, J.M. Chitotriosidase, a chitinase, and the 39-kDa human cartilage glycoprotein, a chitin-binding lectin, are homologues of family 18 glycosyl hydrolases secreted by human macrophages. Eur. J. Biochem. 1998, 251, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Krause, S.W.; Rehli, M.; Kreutz, M.; Schwarzfischer, L.; Paulauskis, J.D.; Andreesen, R. Differential screening identifies genetic markers of monocyte to macrophage maturation. J. Leukoc. Biol. 1996, 60, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, R.B.; Matico, R.E.; McNulty, D.E.; Strickler, J.E.; Rosenberg, M. An abundantly secreted glycoprotein from Drosophila melanogaster is related to mammalian secretory proteins produced in rheumatoid tissues and by activated macrophages. Gene 1995, 153, 147–154. [Google Scholar] [CrossRef]
- Volck, B.; Price, P.A.; Johansen, J.S.; Sorensen, O.; Benfield, T.L.; Nielsen, H.J.; Calafat, J.; Borregaard, N. YKL-40, a mammalian member of the chitinase family, is a matrix protein of specific granules in human neutrophils. Proc. Assoc. Am. Physicians 1998, 110, 351–360. [Google Scholar] [PubMed]
- Hakala, B.E.; White, C.; Recklies, A.D. Human cartilage gp-39, a major secretory product of articular chondrocytes and synovial cells, is a mammalian member of a chitinase protein family. J. Biol. Chem. 1993, 268, 25803–25810. [Google Scholar] [CrossRef]
- Santana, J.O.; Haft, J.I.; LaMarche, N.S.; Goldstein, J.E. Coronary angioplasty in patients eighty years of age or older. Am. Heart J. 1992, 124, 13–18. [Google Scholar] [CrossRef]
- Kim, D.H.; Park, H.J.; Lim, S.; Koo, J.H.; Lee, H.G.; Choi, J.O.; Oh, J.H.; Ha, S.J.; Kang, M.J.; Lee, C.M.; et al. Regulation of chitinase-3-like-1 in T cell elicits Th1 and cytotoxic responses to inhibit lung metastasis. Nat. Commun. 2018, 9, 503. [Google Scholar] [CrossRef]
- Rusak, A.; Jablonska, K.; Piotrowska, A.; Grzegrzolka, J.; Nowak, A.; Wojnar, A.; Dziegiel, P. The Role of CHI3L1 Expression in Angiogenesis in Invasive Ductal Breast Carcinoma. Anticancer Res. 2018, 38, 3357–3366. [Google Scholar] [CrossRef]
- James, A.J.; Reinius, L.E.; Verhoek, M.; Gomes, A.; Kupczyk, M.; Hammar, U.; Ono, J.; Ohta, S.; Izuhara, K.; Bel, E.; et al. Increased YKL-40 and Chitotriosidase in Asthma and Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2016, 193, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Libreros, S.; Garcia-Areas, R.; Iragavarapu-Charyulu, V. CHI3L1 plays a role in cancer through enhanced production of pro-inflammatory/pro-tumorigenic and angiogenic factors. Immunol. Res. 2013, 57, 99–105. [Google Scholar] [CrossRef] [Green Version]
- Hughes, K.; Wickenden, J.A.; Allen, J.E.; Watson, C.J. Conditional deletion of Stat3 in mammary epithelium impairs the acute phase response and modulates immune cell numbers during post-lactational regression. J. Pathol. 2012, 227, 106–117. [Google Scholar] [CrossRef]
- Shao, R.; Cao, Q.J.; Arenas, R.B.; Bigelow, C.; Bentley, B.; Yan, W. Breast cancer expression of YKL-40 correlates with tumour grade, poor differentiation, and other cancer markers. Br. J. Cancer 2011, 105, 1203–1209. [Google Scholar] [CrossRef] [Green Version]
- Johansen, J.S.; Christensen, I.J.; Riisbro, R.; Greenall, M.; Han, C.; Price, P.A.; Smith, K.; Brunner, N.; Harris, A.L. High serum YKL-40 levels in patients with primary breast cancer is related to short recurrence free survival. Breast Cancer Res. Treat. 2003, 80, 15–21. [Google Scholar] [CrossRef]
- Kang, E.J.; Jung, H.; Woo, O.H.; Park, K.H.; Woo, S.U.; Yang, D.S.; Kim, A.R.; Lee, J.B.; Kim, Y.H.; Kim, J.S.; et al. YKL-40 expression could be a poor prognostic marker in the breast cancer tissue. Tumour Biol. 2014, 35, 277–286. [Google Scholar] [CrossRef]
- Jensen, B.V.; Johansen, J.S.; Price, P.A. High levels of serum HER-2/neu and YKL-40 independently reflect aggressiveness of metastatic breast cancer. Clin. Cancer Res. 2003, 9, 4423–4434. [Google Scholar] [PubMed]
- Williams, M.M.; Spoelstra, N.S.; Arnesen, S.; O’Neill, K.I.; Christenson, J.L.; Reese, J.; Torkko, K.C.; Goodspeed, A.; Rosas, E.; Hanamura, T.; et al. Steroid Hormone Receptor and Infiltrating Immune Cell Status Reveals Therapeutic Vulnerabilities of ESR1-Mutant Breast Cancer. Cancer Res. 2021, 81, 732–746. [Google Scholar] [CrossRef]
- Morera, E.; Steinhauser, S.S.; Budkova, Z.; Ingthorsson, S.; Kricker, J.; Krueger, A.; Traustadottir, G.A.; Gudjonsson, T. YKL-40/CHI3L1 facilitates migration and invasion in HER2 overexpressing breast epithelial progenitor cells and generates a niche for capillary-like network formation. Vitr. Cell Dev. Biol. Anim. 2019, 55, 838–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, K.I.; Bhan, A.; Liu, X.; Chen, M.Y.; Jandial, R. Astrocytic IGFBP2 and CHI3L1 in cerebrospinal fluid drive cortical metastasis of HER2+breast cancer. Clin. Exp. Metastasis 2020, 37, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Wang, L.; Chen, H.; Xie, L.; Bai, T.; Liu, H.; Wang, D. YKL-40 promotes the migration and invasion of prostate cancer cells by regulating epithelial mesenchymal transition. Am. J. Transl. Res. 2017, 9, 3749–3757. [Google Scholar] [PubMed]
- Jefri, M.; Huang, Y.N.; Huang, W.C.; Tai, C.S.; Chen, W.L. YKL-40 regulated epithelial-mesenchymal transition and migration/invasion enhancement in non-small cell lung cancer. BMC Cancer 2015, 15, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, N.; Shani, O.; Raz, Y.; Sharon, Y.; Hoffman, D.; Abramovitz, L.; Erez, N. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene 2017, 36, 4457–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libreros, S.; Garcia-Areas, R.; Shibata, Y.; Carrio, R.; Torroella-Kouri, M.; Iragavarapu-Charyulu, V. Induction of proinflammatory mediators by CHI3L1 is reduced by chitin treatment: Decreased tumor metastasis in a breast cancer model. Int. J. Cancer 2012, 131, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Herzog, E.L.; Lee, C.G.; Peng, X.; Lee, C.M.; Chen, X.; Rockwell, S.; Koo, J.S.; Kluger, H.; Herbst, R.S.; et al. Role of chitinase 3-like-1 and semaphorin 7a in pulmonary melanoma metastasis. Cancer Res. 2015, 75, 487–496. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Herzog, E.L.; Moore, M.; Lee, C.M.; Na, S.H.; Lee, C.G.; Elias, J.A. RIG-like Helicase Regulation of Chitinase 3-like 1 Axis and Pulmonary Metastasis. Sci. Rep. 2016, 6, 26299. [Google Scholar] [CrossRef]
- Pang, H.; Lu, H.; Song, H.; Meng, Q.; Zhao, Y.; Liu, N.; Lan, F.; Liu, Y.; Yan, S.; Dong, X.; et al. Prognostic values of osteopontin-c, E-cadherin and beta-catenin in breast cancer. Cancer Epidemiol. 2013, 37, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Zduniak, K.; Ziolkowski, P.; Ahlin, C.; Agrawal, A.; Agrawal, S.; Blomqvist, C.; Fjallskog, M.L.; Weber, G.F. Nuclear osteopontin-c is a prognostic breast cancer marker. Br. J. Cancer 2015, 112, 729–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, M.; Zheng, X. Osteopontin and vasculogenic mimicry formation are associated with response to neoadjuvant chemotherapy in advanced breast cancer. Onco Targets 2017, 10, 4121–4127. [Google Scholar] [CrossRef] [Green Version]
- Gothlin Eremo, A.; Lagergren, K.; Othman, L.; Montgomery, S.; Andersson, G.; Tina, E. Evaluation of SPP1/osteopontin expression as predictor of recurrence in tamoxifen treated breast cancer. Sci. Rep. 2020, 10, 1451. [Google Scholar] [CrossRef] [Green Version]
- Macur, K.; Hagen, L.; Ciesielski, T.M.; Konieczna, L.; Skokowski, J.; Jenssen, B.M.; Slupphaug, G.; Baczek, T. A targeted mass spectrometry immunoassay to quantify osteopontin in fresh-frozen breast tumors and adjacent normal breast tissues. J. Proteom. 2019, 208, 103469. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Chen, C.; Fan, G. Association between the expression of secreted phosphoprotein—Related genes and prognosis of human cancer. BMC Cancer 2019, 19, 1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patani, N.; Jouhra, F.; Jiang, W.; Mokbel, K. Osteopontin expression profiles predict pathological and clinical outcome in breast cancer. Anticancer Res. 2008, 28, 4105–4110. [Google Scholar]
- Anborgh, P.H.; Caria, L.B.; Chambers, A.F.; Tuck, A.B.; Stitt, L.W.; Brackstone, M. Role of plasma osteopontin as a biomarker in locally advanced breast cancer. Am. J. Transl. Res. 2015, 7, 723–732. [Google Scholar]
- Wei, T.; Bi, G.; Bian, Y.; Ruan, S.; Yuan, G.; Xie, H.; Zhao, M.; Shen, R.; Zhu, Y.; Wang, Q.; et al. The Significance of Secreted Phosphoprotein 1 in Multiple Human Cancers. Front. Mol. Biosci. 2020, 7, 565383. [Google Scholar] [CrossRef]
- Sangaletti, S.; Tripodo, C.; Sandri, S.; Torselli, I.; Vitali, C.; Ratti, C.; Botti, L.; Burocchi, A.; Porcasi, R.; Tomirotti, A.; et al. Osteopontin shapes immunosuppression in the metastatic niche. Cancer Res. 2014, 74, 4706–4719. [Google Scholar] [CrossRef] [Green Version]
- Sharon, Y.; Raz, Y.; Cohen, N.; Ben-Shmuel, A.; Schwartz, H.; Geiger, T.; Erez, N. Tumor-derived osteopontin reprograms normal mammary fibroblasts to promote inflammation and tumor growth in breast cancer. Cancer Res. 2015, 75, 963–973. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.S.; Chan, C.; Vilaire, G.; Mousa, S.A.; DeGrado, W.F. Agonist-activated alphavbeta3 on platelets and lymphocytes binds to the matrix protein osteopontin. J. Biol. Chem. 1997, 272, 8137–8140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leblanc, R.; Lee, S.C.; David, M.; Bordet, J.C.; Norman, D.D.; Patil, R.; Miller, D.; Sahay, D.; Ribeiro, J.; Clezardin, P.; et al. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood 2014, 124, 3141–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chellaiah, M.A.; Hruska, K.A. The integrin alpha(v)beta(3) and CD44 regulate the actions of osteopontin on osteoclast motility. Calcif. Tissue Int. 2003, 72, 197–205. [Google Scholar] [CrossRef]
- Sodek, J.; Ganss, B.; McKee, M.D. Osteopontin. Crit. Rev. Oral Biol. Med. 2000, 11, 279–303. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M.; Schwartz, S.M.; Liaw, L. Molecular and cellular biology of osteopontin Potential role in cardiovascular disease. Trends Cardiovasc. Med. 1995, 5, 88–95. [Google Scholar] [CrossRef]
- Lund, S.A.; Giachelli, C.M.; Scatena, M. The role of osteopontin in inflammatory processes. J. Cell Commun. Signal. 2009, 3, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Denhardt, D.T.; Giachelli, C.M.; Rittling, S.R. Role of osteopontin in cellular signaling and toxicant injury. Annu. Rev. Pharm. Toxicol. 2001, 41, 723–749. [Google Scholar] [CrossRef] [PubMed]
- Buback, F.; Renkl, A.C.; Schulz, G.; Weiss, J.M. Osteopontin and the skin: Multiple emerging roles in cutaneous biology and pathology. Exp. Derm. 2009, 18, 750–759. [Google Scholar] [CrossRef]
- Park, Y.H.; Lal, S.; Lee, J.E.; Choi, Y.L.; Wen, J.; Ram, S.; Ding, Y.; Lee, S.H.; Powell, E.; Lee, S.K.; et al. Chemotherapy induces dynamic immune responses in breast cancers that impact treatment outcome. Nat. Commun. 2020, 11, 6175. [Google Scholar] [CrossRef] [PubMed]
- Roller, A.; Ferreira, C.; Jarassier, L.; Heller, A.; Dietmann, G.; Korski, K.; Gomes, B.; Cannarile, M.A. Abstract 2051: Precision immune phenotyping from primary tumor and metastatic lesions reveals novel insights into therapeutic intervention in cancer immunotherapy. Cancer Res. 2020, 80, 2051. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Top Pathways Downregulated by miR-200c | |||||
|---|---|---|---|---|---|
| Term | Count | ES | NES | NOM p-Val. | FDR q-Val. |
| HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION | 198 | −0.75 | −3.39 | <0.00001 | <0.00001 |
| HALLMARK_MYOGENESIS | 198 | −0.54 | −2.47 | <0.00001 | <0.00001 |
| HALLMARK_KRAS_SIGNALING_UP | 196 | −0.54 | −2.45 | <0.00001 | <0.00001 |
| HALLMARK_UV_RESPONSE_DN | 142 | −0.56 | −2.44 | <0.00001 | <0.00001 |
| HALLMARK_COAGULATION | 134 | −0.56 | −2.43 | <0.00001 | <0.00001 |
| HALLMARK_TNFA_SIGNALING_VIA_NFKB | 198 | −0.53 | −2.40 | <0.00001 | <0.00001 |
| HALLMARK_ANGIOGENESIS | 36 | −0.68 | −2.39 | <0.00001 | <0.00001 |
| HALLMARK_APICAL_JUNCTION | 195 | −0.51 | −2.31 | <0.00001 | <0.00001 |
| HALLMARK_TGF_BETA_SIGNALING | 54 | −0.59 | −2.21 | <0.00001 | <0.00001 |
| HALLMARK_ALLOGRAFT_REJECTION | 193 | −0.48 | −2.20 | <0.00001 | <0.00001 |
| HALLMARK_INFLAMMATORY_RESPONSE | 198 | −0.48 | −2.20 | <0.00001 | <0.00001 |
| HALLMARK_COMPLEMENT | 197 | −0.47 | −2.16 | <0.00001 | <0.00001 |
| HALLMARK_APOPTOSIS | 160 | −0.48 | −2.14 | <0.00001 | <0.00001 |
| HALLMARK_IL2_STAT5_SIGNALING | 195 | −0.42 | −1.93 | <0.00001 | 0.000068 |
| HALLMARK_INTERFERON_GAMMA_RESPONSE | 192 | −0.42 | −1.93 | <0.00001 | 0.000063 |
| HALLMARK_HYPOXIA | 199 | −0.41 | −1.90 | <0.00001 | 0.000059 |
| HALLMARK_IL6_JAK_STAT3_SIGNALING | 83 | −0.43 | −1.76 | <0.00001 | 0.000908 |
| HALLMARK_ANDROGEN_RESPONSE | 96 | −0.41 | −1.68 | 0.001431 | 0.002189 |
| HALLMARK_P53_PATHWAY | 194 | −0.37 | −1.67 | <0.00001 | 0.002450 |
| HALLMARK_BILE_ACID_METABOLISM | 112 | −0.39 | −1.63 | <0.00001 | 0.003382 |
| Target | Drug | Clinical Trial Number | Study Phase | Cancer Type | Combination Therapy | Results; Publications |
|---|---|---|---|---|---|---|
| CD73 | Oleclumab (MEDI9447) | NCT02503774 | I | Solid tumors | PD-1 | Ongoing; [64,65] |
| NCT03616886 | I/II | Inoperable or mTNBC | Paclitaxel, Carboplatin, PD-1 | Recruiting | ||
| NCT03875573 | II | ER+ breast cancer | Doxorubicin-cyclophosphamide, pre-operative radiation | Ongoing; [66] | ||
| LY3475070 | NCT04148937 | I | Advanced solid tumors | PD-1 | Ongoing | |
| CD73/A2AR | NZV930 | NCT03549000 | I/Ib | Advanced cancers | PD-1, A2AR | Recruiting |
| CPI-006 | NCT03454451 | I/Ib | Select advanced cancers | PD-1 or A2AR | Recruiting; well-tolerated, some anti-tumor activity [67] | |
| A2AR | NIR178 | NCT03207867 | II | Solid tumors | PD-1 | Recruiting |
| A2AR+A2BR | Etrumadenant (AB928) | NCT03719326 | I | mTNBC or ovarian | Doxorubicin and PI3Kγ or paclitaxel | Ongoing; favorable safety profile [68] |
| NCT03629756 | I | Advanced cancers | PD-1 |
| Target | Drug | Clinical Trial Number | Study Phase | Cancer Type | Combination Therapy | Results; Publications |
|---|---|---|---|---|---|---|
| GM-CSF | NeuVax vaccine | NCT01479244 | III | HER2-low breast | - | Well-tolerated, no change DFS [138] |
| NCT01570036 | II | HER2-low breast | Trastuzumab | Well-tolerated, increased DFS in TNBC patients [139] | ||
| Sargramostim | NCT00027807 | I | Stage IV breast | IL-2 and autologous T cells | Increased cytotoxic T cells [140] | |
| NCT00524277 | II | Breast | HER2 vaccine | Increased DFS for TNBC [141] | ||
| NCT00436254 | I | HER2+ breast and ovarian | HER2 vaccine | Immunogenic out to 60 weeks [142] | ||
| NCT02636582 | II | DCIS | HER2 vaccine | Ongoing | ||
| CSF1R | Pexidartinib (PLX-3397) | NCT01525602 | I | Advanced solid tumors | Paclitaxel | Well-tolerated, promising decrease in monocyte recruitment [143] |
| NCT01596751 | I/II | Metastatic breast | Eribulin | Completed, results not published | ||
| NCT01042379 | II | Breast | - | Ongoing | ||
| Emactuzumab (RG7155) | NCT01494688 | I | Advanced or metastatic tumors | Paclitaxel | Decreased immune suppressive TAMs, no anti-tumor activity [144] | |
| LY3022855 | NCT01346358 | I | Advanced solid tumors | - | Dose dependent pharmacokinetics, no clinical activity [145] | |
| M-CSF | Lacnotuzumab (MCS110) | NCT02435680 | II | Advanced TNBC | Carboplatin and gemcitabine | On-target, no change in PFS [146] |
| NCT02807844 | Ib/II | Advanced tumors | PD-1 | Anti-tumor response in pancreatic tumors [147] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, M.M.; Hafeez, S.A.; Christenson, J.L.; O’Neill, K.I.; Hammond, N.G.; Richer, J.K. Reversing an Oncogenic Epithelial-to-Mesenchymal Transition Program in Breast Cancer Reveals Actionable Immune Suppressive Pathways. Pharmaceuticals 2021, 14, 1122. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111122
Williams MM, Hafeez SA, Christenson JL, O’Neill KI, Hammond NG, Richer JK. Reversing an Oncogenic Epithelial-to-Mesenchymal Transition Program in Breast Cancer Reveals Actionable Immune Suppressive Pathways. Pharmaceuticals. 2021; 14(11):1122. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111122
Chicago/Turabian StyleWilliams, Michelle M., Sabrina A. Hafeez, Jessica L. Christenson, Kathleen I. O’Neill, Nia G. Hammond, and Jennifer K. Richer. 2021. "Reversing an Oncogenic Epithelial-to-Mesenchymal Transition Program in Breast Cancer Reveals Actionable Immune Suppressive Pathways" Pharmaceuticals 14, no. 11: 1122. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111122