
In Vitro Assessment of the Role of p53 on Chemotherapy Treatments in Neuroblastoma Cell Lines

 and
and
Abstract
:
1. Introduction
2. Results
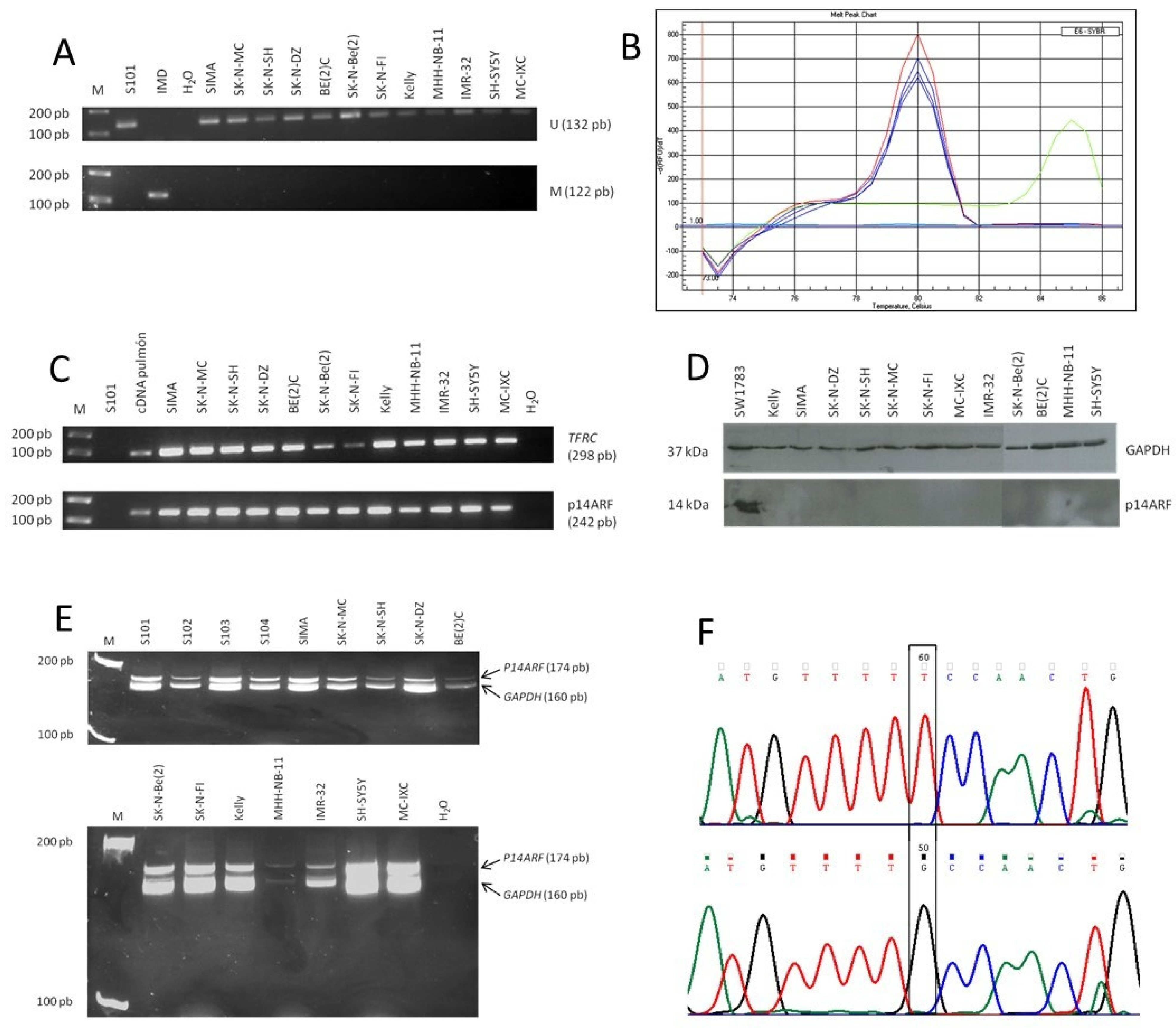
2.1. Methylation of the P14ARF Promoter
2.1.1. MSP (Methylation-Specific PCR)
2.1.2. MCA–Meth (Melting Curve Analysis–Methylation Assay)
2.2. p14ARF Expression
2.2.1. Detection of mRNA by Semi-Quantitative RT-PCR
2.2.2. Protein Detection by Western Blot
2.3. p14ARF Homozygous Deletions
2.4. TP53 Gene Mutations
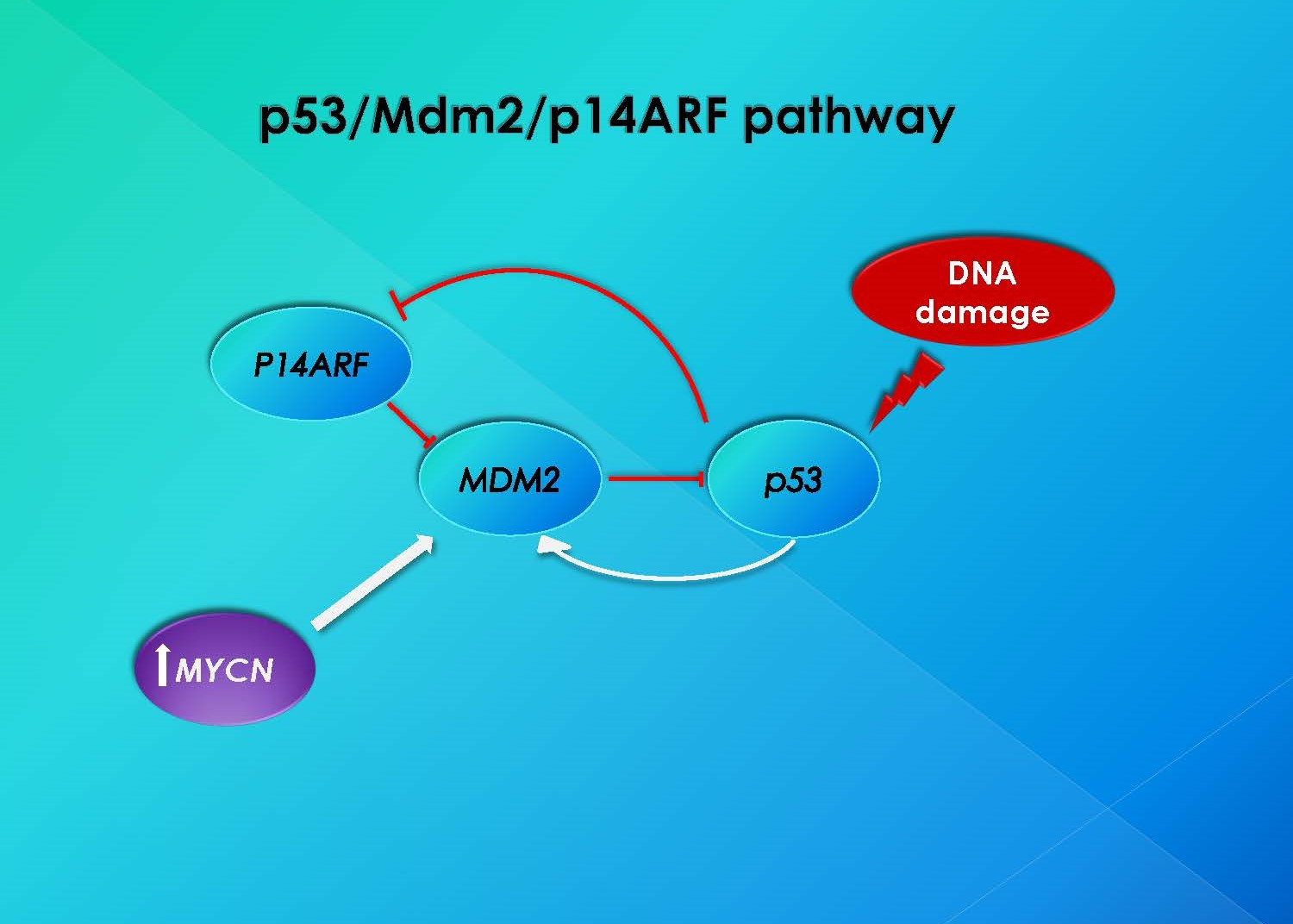
2.5. MYCN and MDM2 Gene Amplification by FISH
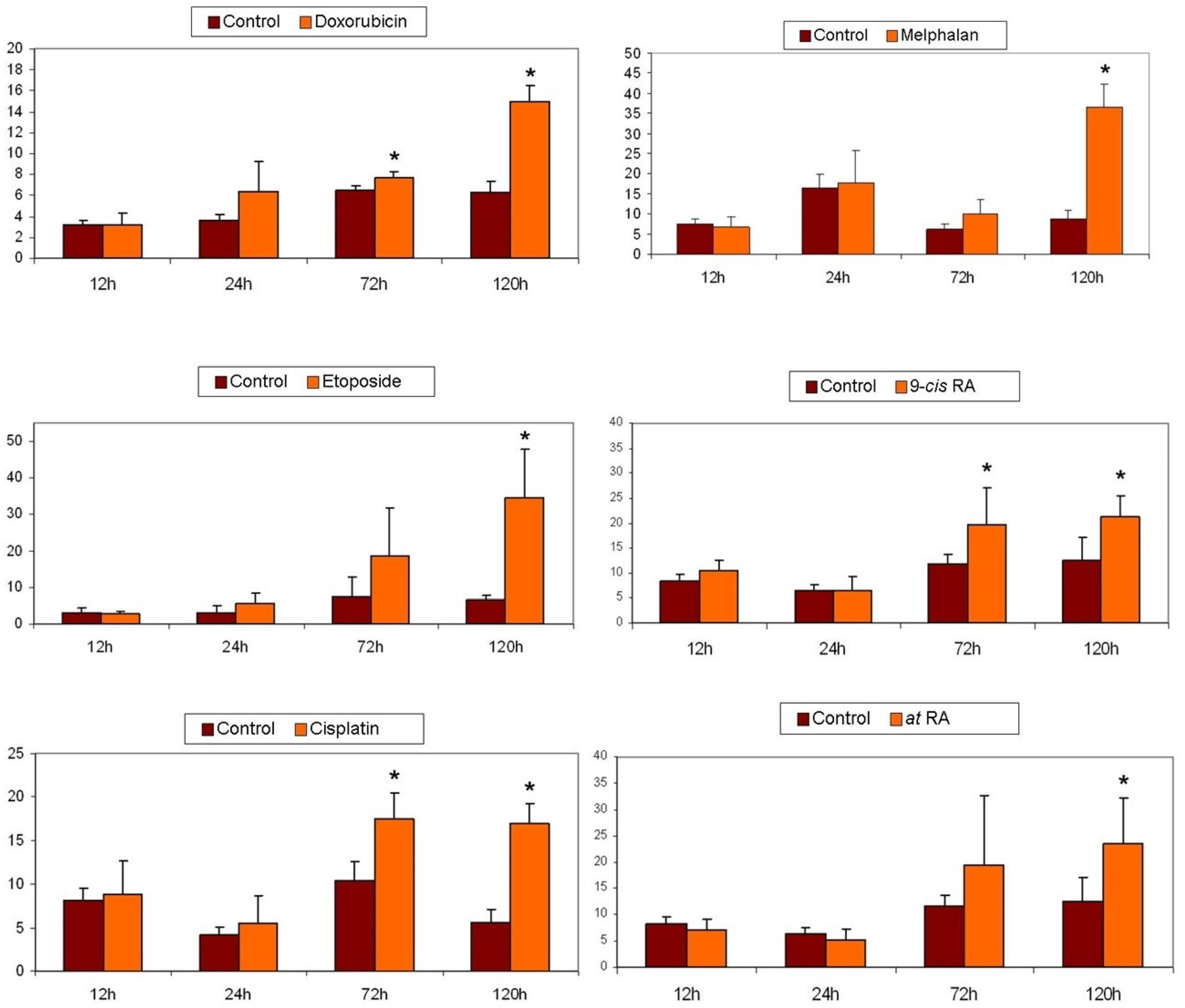
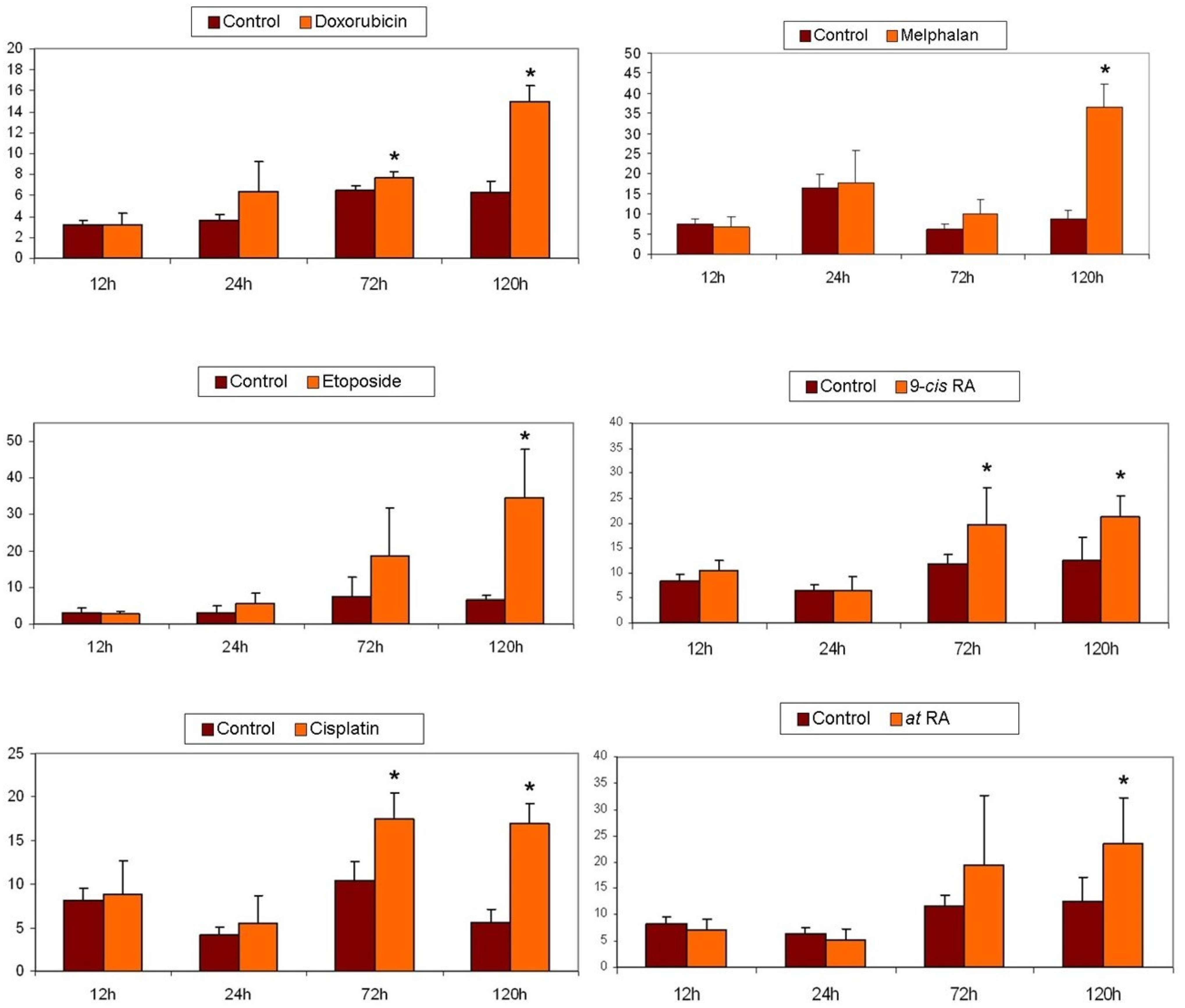
2.6. Cytotoxicity Assays
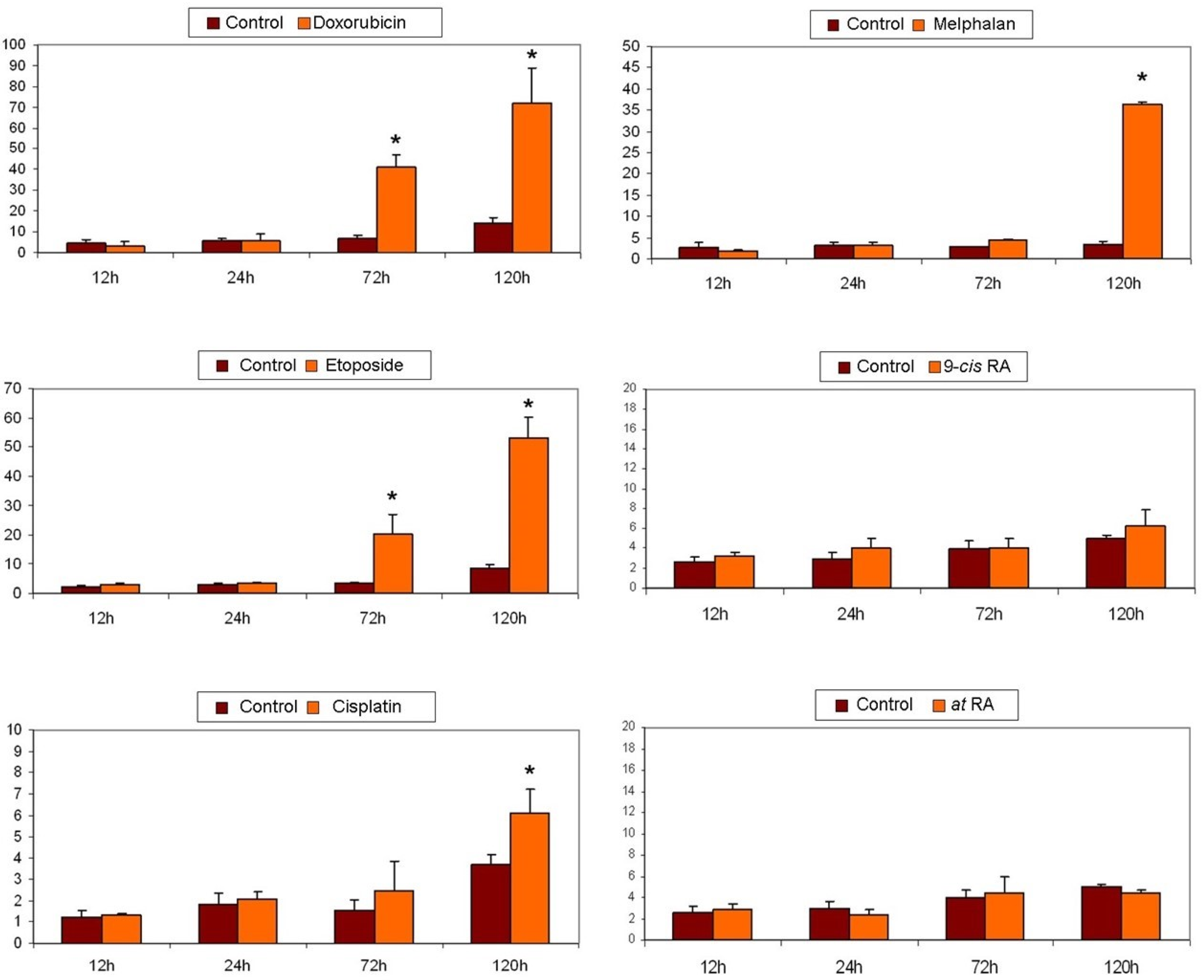
2.7. Apoptosis Detection
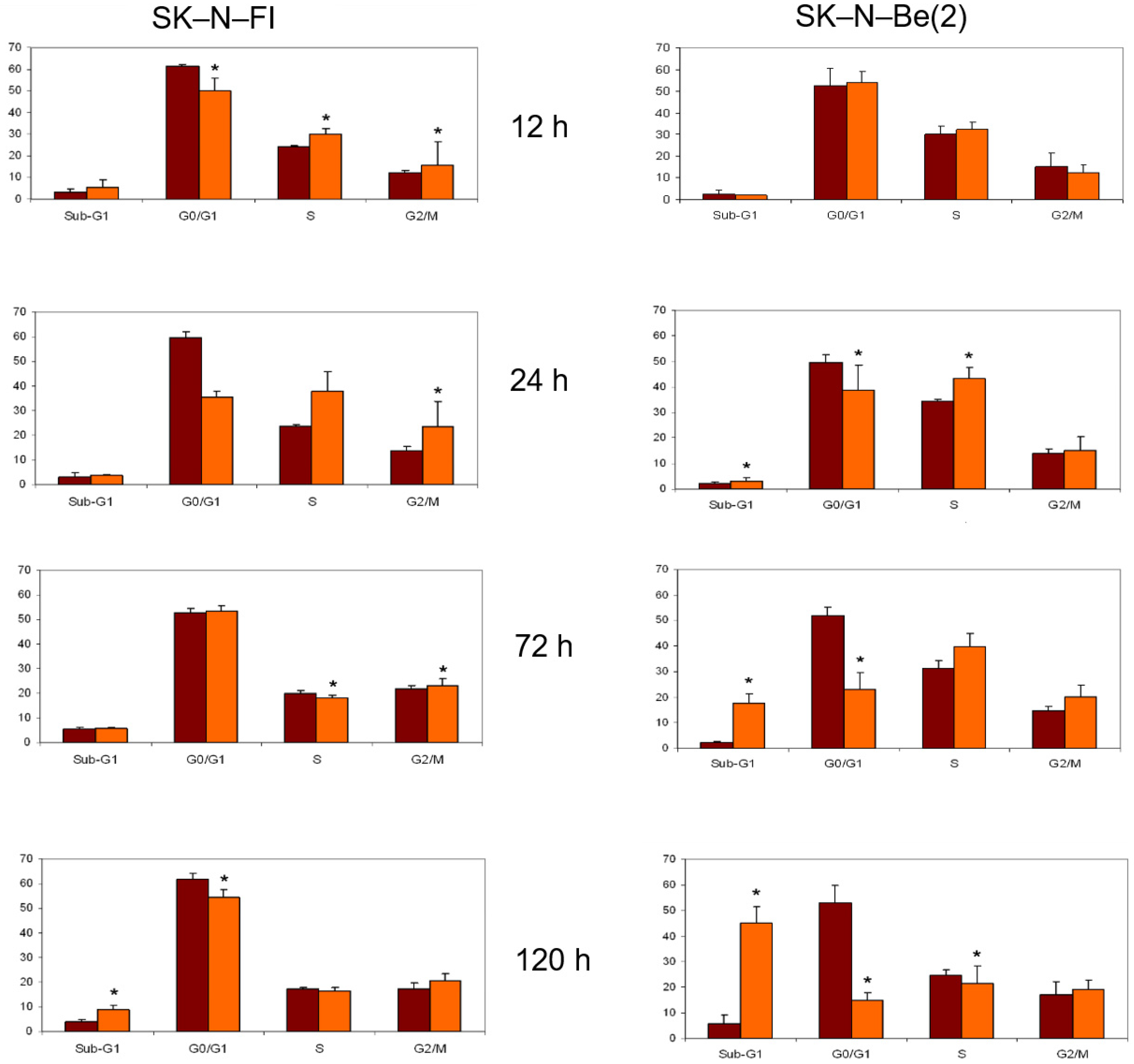
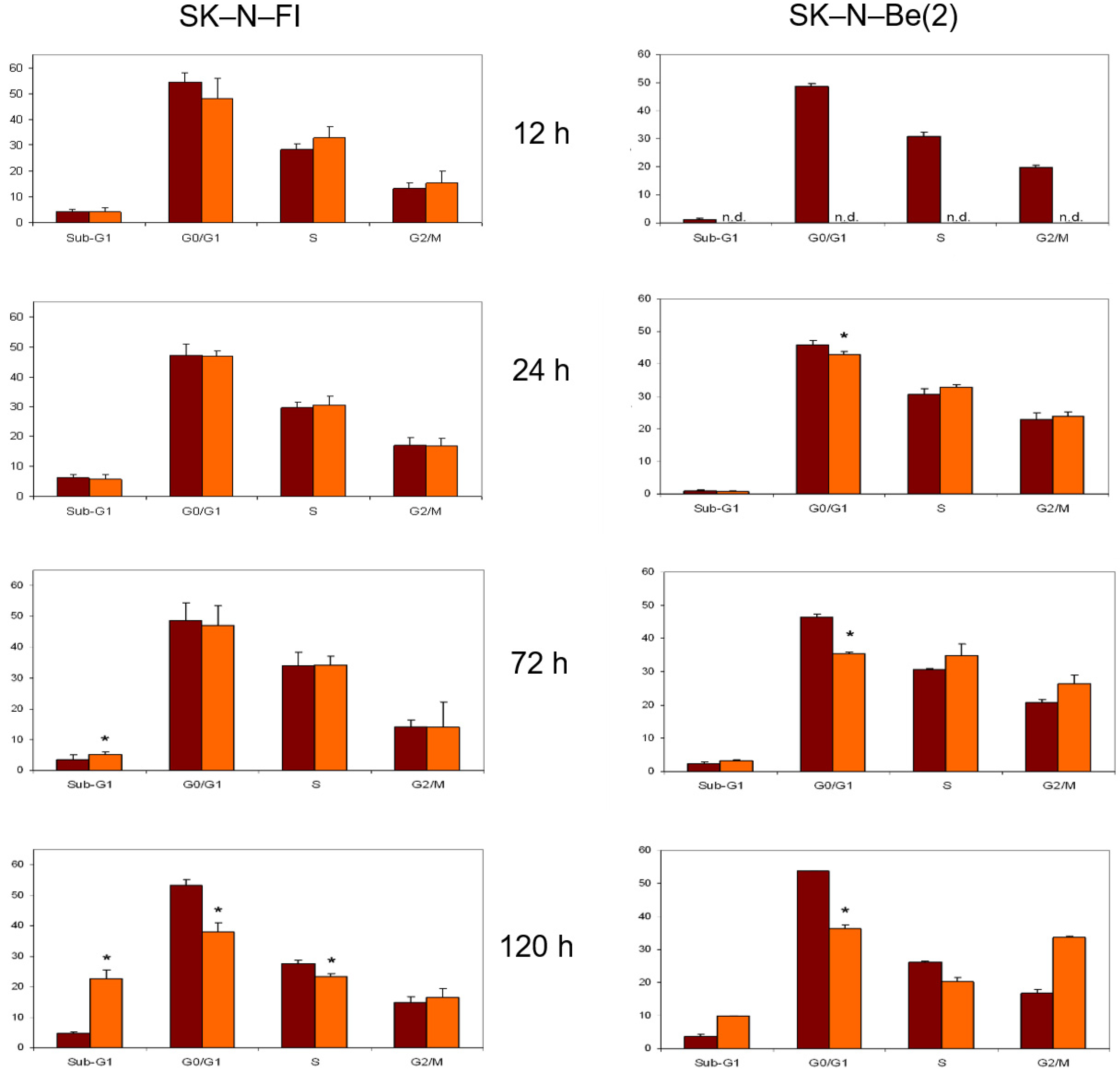
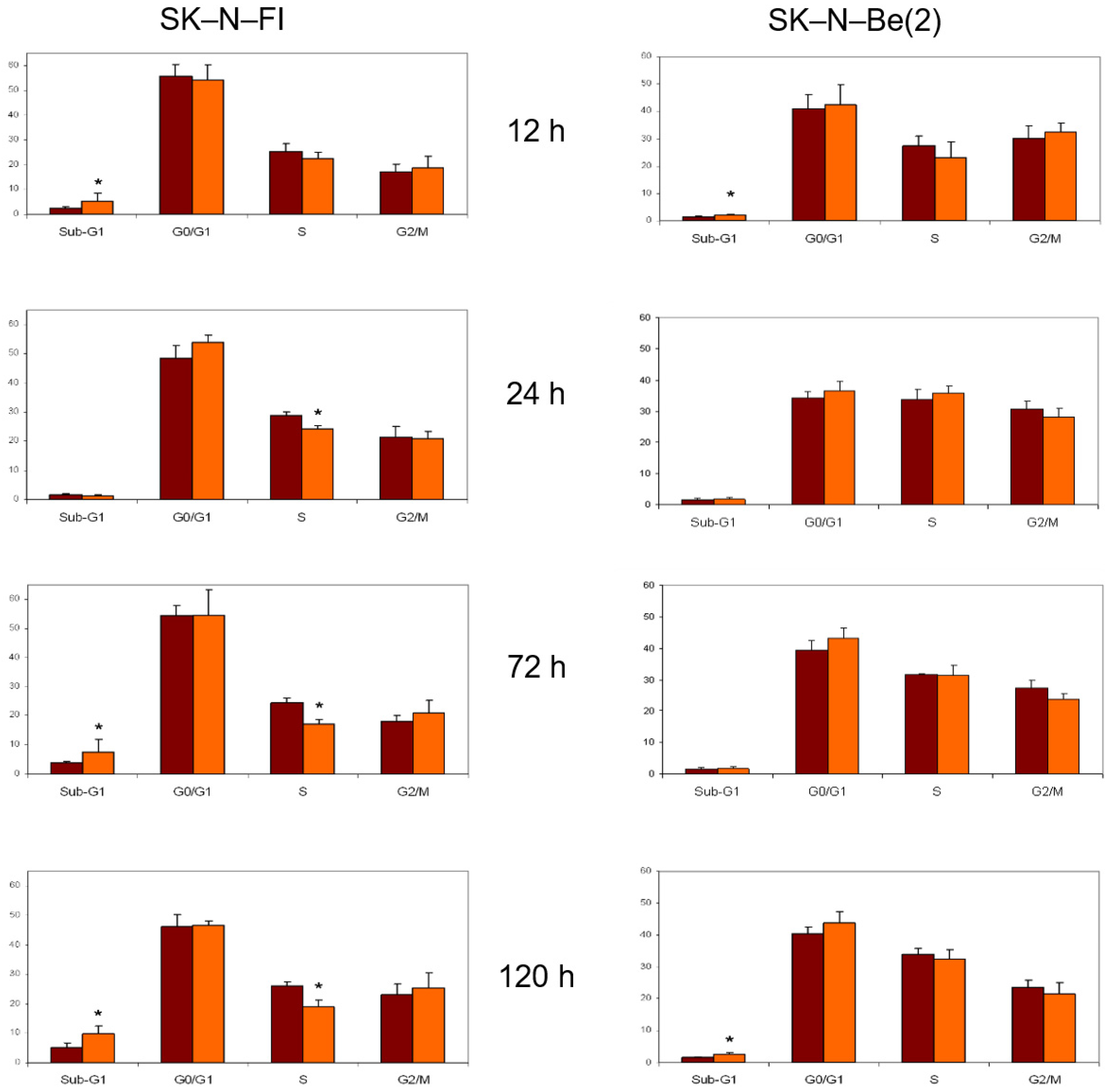
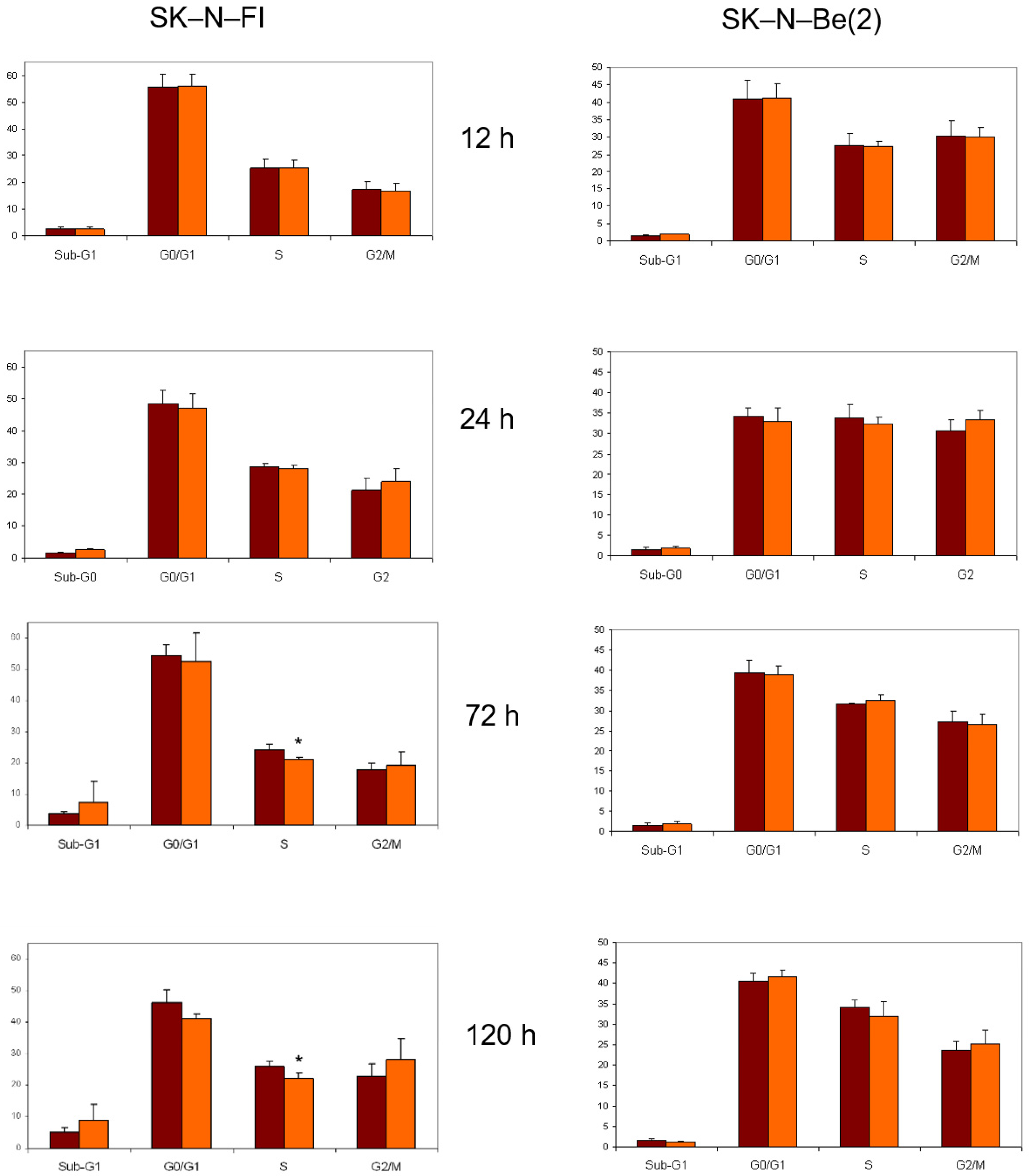
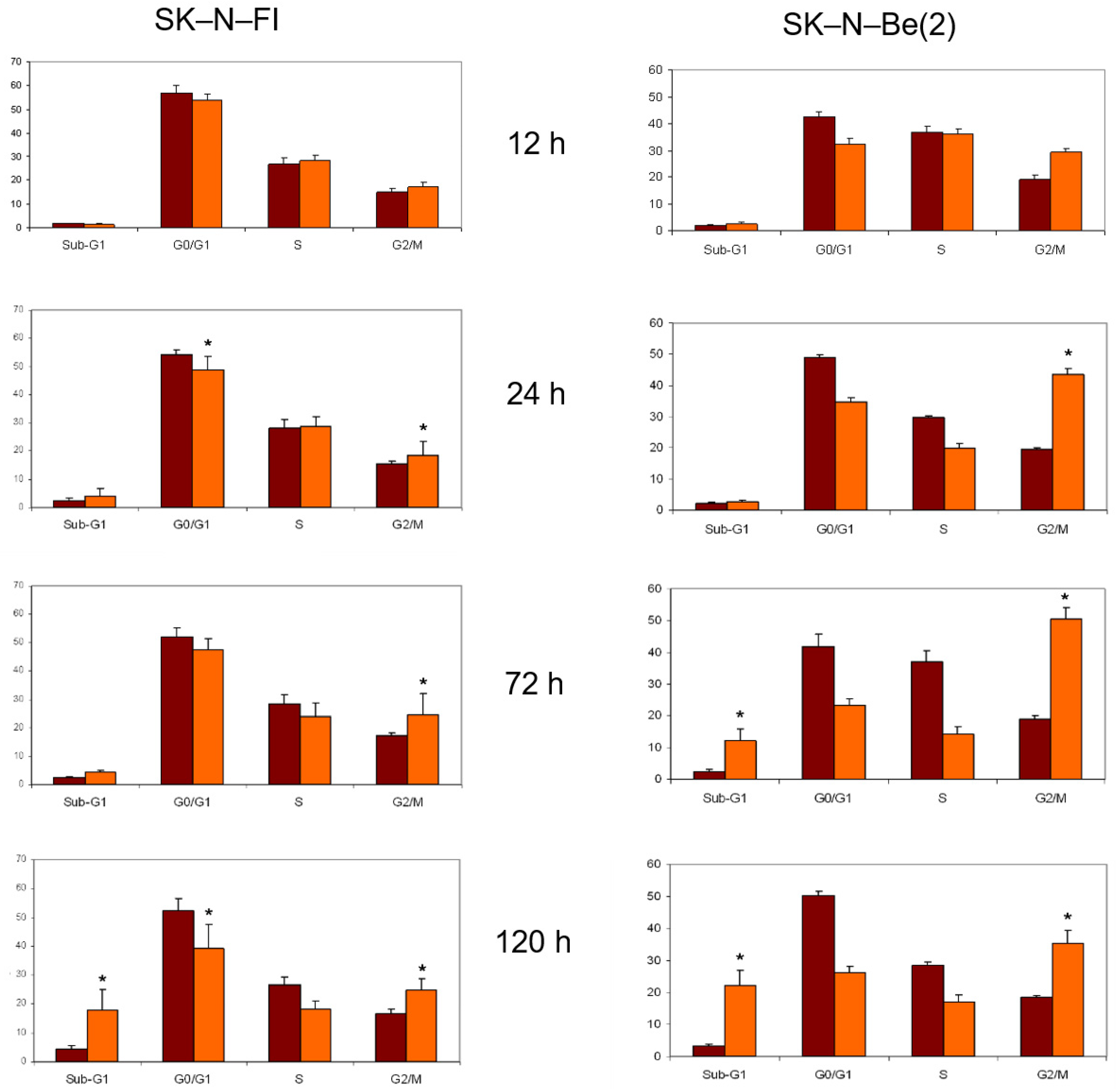
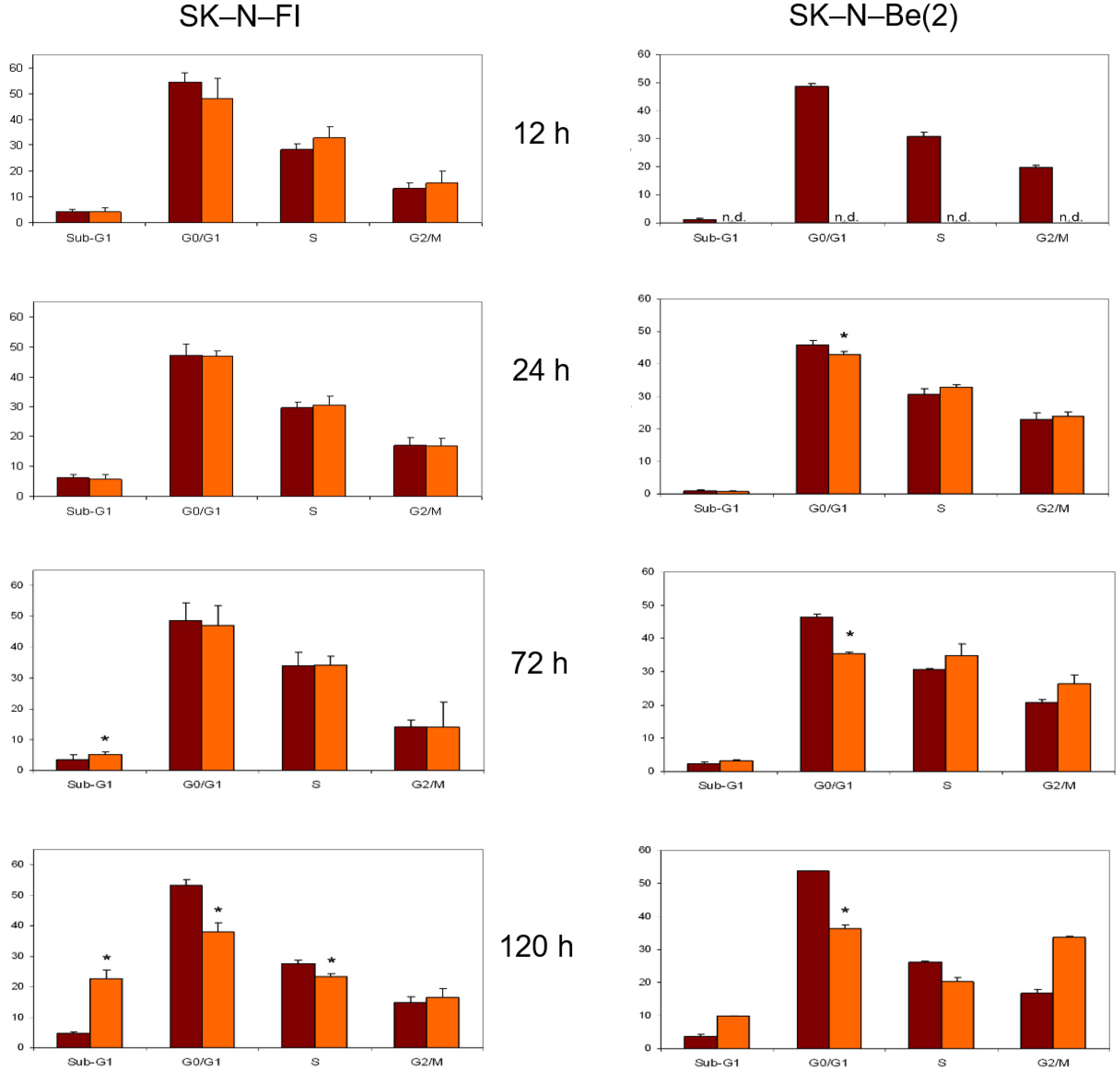
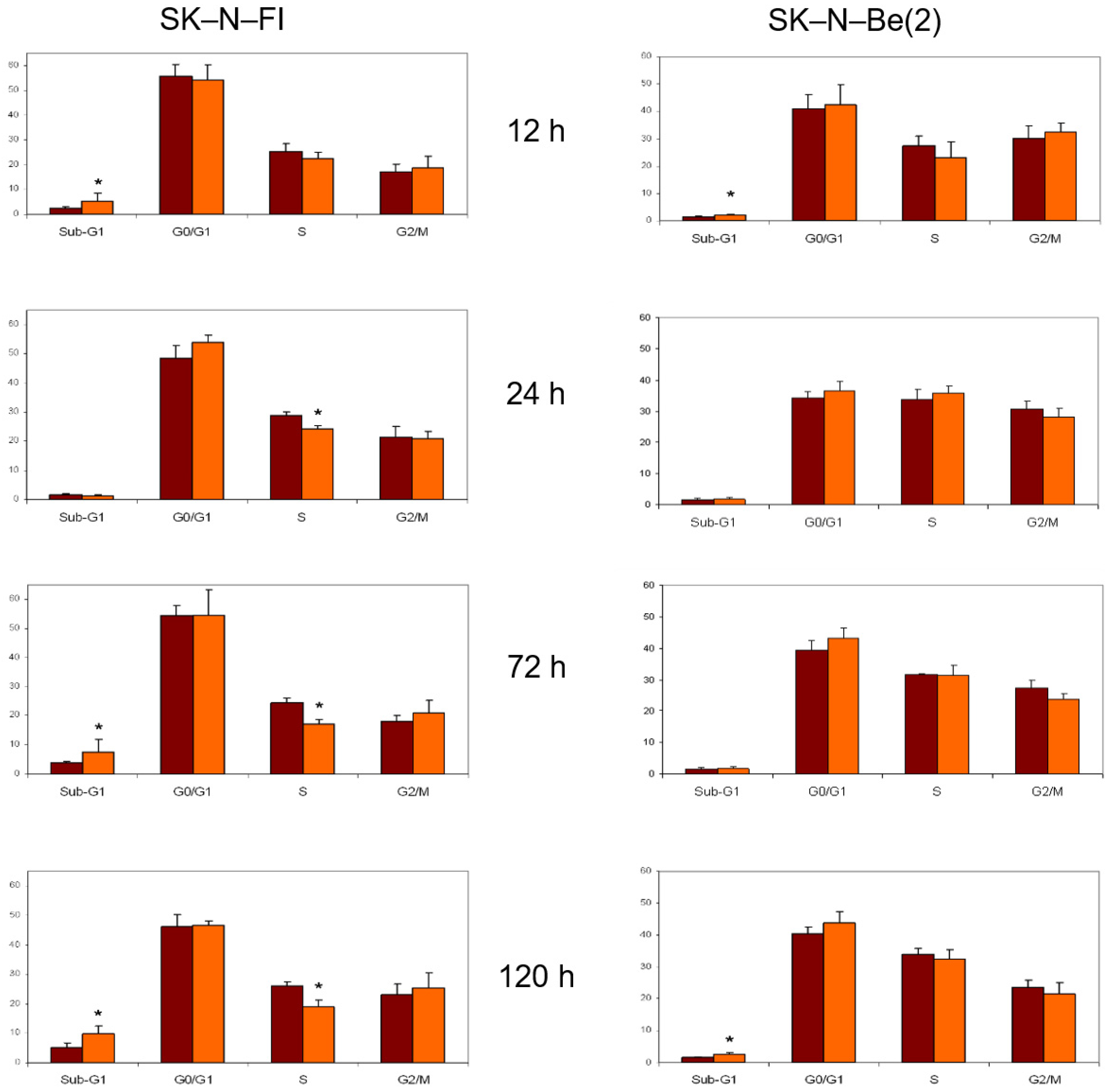
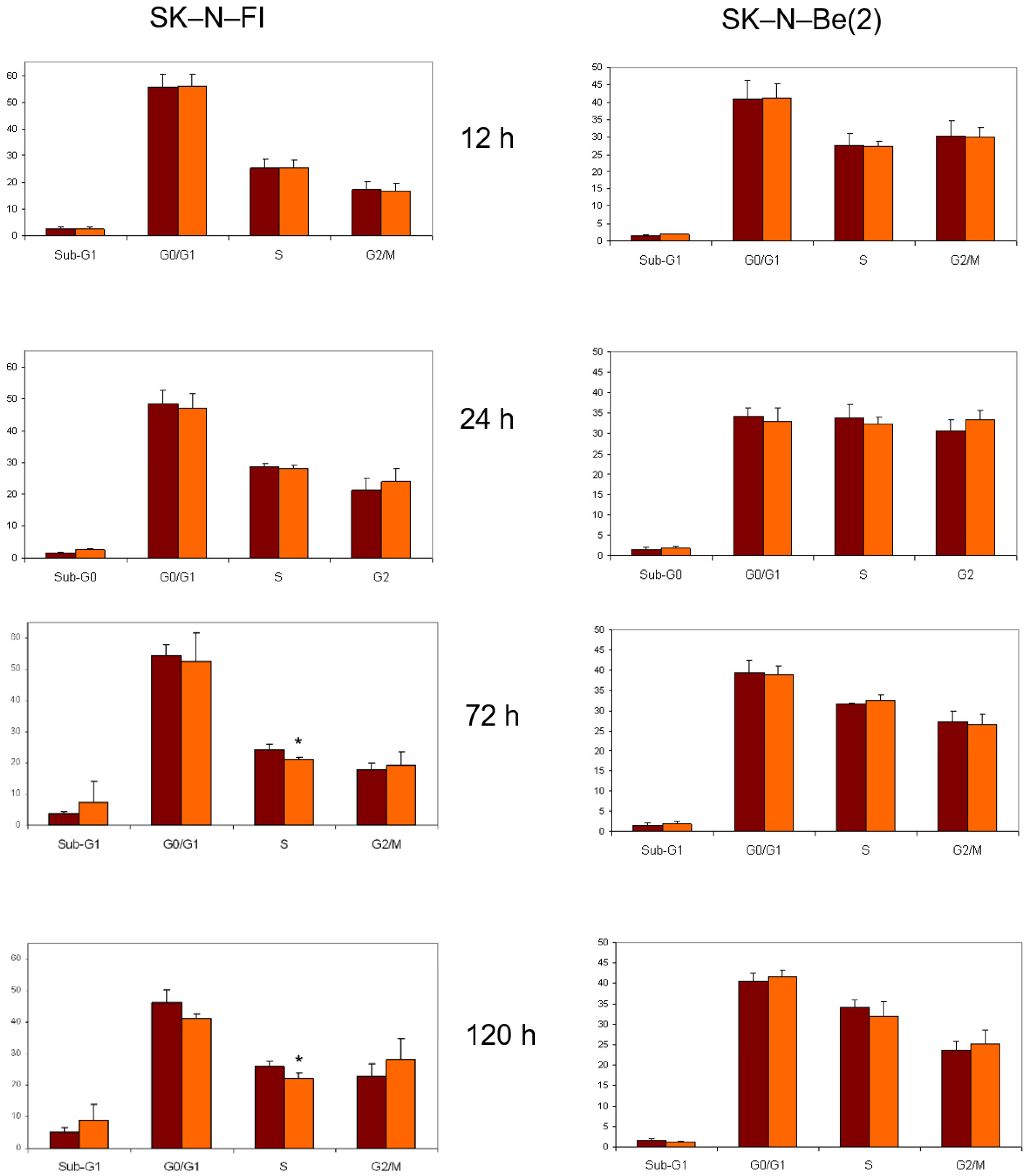
2.8. Cell Cycle Analysis
2.9. Protein Expression Analysis
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. p14ARF Gene Promoter Methylation
4.2.1. DNA Extraction and Bisulphite Treatment
4.2.2. Methylation-Specific PCR
4.2.3. Melting Curve Analysis–Methylation Assay
4.3. p14ARF Expression
4.3.1. RNA Extraction and Reverse Transcription
4.3.2. Semiquantitative RT-PCR
4.3.3. Protein Extraction
4.3.4. Western Blot Analysis
4.4. p14ARF Homozygous Deletions
4.5. TP53 Mutations
4.6. FISH (Fluorescence In Situ Hybridization) for MYCN and MDM2
4.6.1. Obtaining Cytogenetic Suspensions
4.6.2. Description of the FISH Probes Used
4.6.3. Locus-Specific FISH Probe Design
4.6.4. Culture of the FISH Probes
4.7. Detection of Apoptosis
4.8. Cell Cycle Analysis
4.9. Protein Expression
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tolbert, V.P.; Matthay, K.K. Neuroblastoma: Clinical and biological approach to risk stratification and treatment. Cell Tissue Res. 2018, 372, 195–209. [Google Scholar] [CrossRef]
- Van Arendonk, K.J.; Chung, D.H. Neuroblastoma: Tumor Biology and Its Implications for Staging and Treatment. Children 2019, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- George, R.E.; Diller, L.; Bernstein, M.L. Pharmacotherapy of neuroblastoma. Expert Opin. Pharmacother. 2010, 11, 1467–1478. [Google Scholar] [CrossRef]
- Paffhausen, T.; Schwab, M.; Westermann, F. Targeted MYCN expression affects cytotoxic potential of chemotherapeutic drugs in neuroblastoma cells. Cancer Lett. 2007, 250, 17–24. [Google Scholar] [CrossRef]
- Yanishevski, D.; McCarville, M.B.; Doubrovin, M.; Spiegl, H.R.; Zhao, X.; Lu, Z.; Federico, S.M.; Furman, W.L.; Murphy, A.J.; Davidoff, A.M. Impact of MYCN status on response of high-risk neuroblastoma to neoadjuvant chemotherapy. J. Pediatr. Surg. 2020, 55, 130–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthay, K.K.; Villablanca, J.G.; Seeger, R.C.; Stram, D.O.; Harris, R.E.; Ramsay, N.K.; Swift, P.; Shimada, H.; Black, C.T.; Brodeur, G.M.; et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N. Engl. J. Med. 1999, 341, 1165–1173. [Google Scholar] [CrossRef]
- Wagner, L.M.; Danks, M.K. New therapeutic targets for the treatment of high-risk neuroblastoma. J. Cell. Biochem. 2009, 107, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Zage, P.E. Novel Therapies for Relapsed and Refractory Neuroblastoma. Children 2018, 5, 148. [Google Scholar] [CrossRef] [Green Version]
- Bayeva, N.; Coll, E.; Piskareva, O. Differentiating Neuroblastoma: A Systematic Review of the Retinoic Acid, Its Derivatives, and Synergistic Interactions. J. Pers. Med. 2021, 11, 211. [Google Scholar] [CrossRef] [PubMed]
- Savory, J.G.; Edey, C.; Hess, B.; Mears, A.J.; Lohnes, D. Identification of novel retinoic acid target genes. Dev. Biol. 2014, 395, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef] [Green Version]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb. Perspect. Med. 2016, 6, a026179. [Google Scholar] [CrossRef] [Green Version]
- Wolter, J.; Angelini, P.; Irwin, M. p53 family: Therapeutic targets in neuroblastoma. Future Oncol. 2010, 6, 429–444. [Google Scholar] [CrossRef]
- Van Maerken, T.; Vandesompele, J.; Rihani, A.; De Paepe, A.; Speleman, F. Escape from p53-mediated tumor surveillance in neuroblastoma: Switching off the p14(ARF)-MDM2-p53 axis. Cell Death Differ. 2009, 16, 1563–1572. [Google Scholar] [CrossRef] [Green Version]
- Tweddle, D.A.; Pearson, A.D.; Haber, M.; Norris, M.D.; Xue, C.; Flemming, C.; Lunec, J. The p53 pathway and its inactivation in neuroblastoma. Cancer Lett. 2003, 197, 93–98. [Google Scholar] [CrossRef]
- Castresana, J.S.; Bello, M.J.; Rey, J.A.; Nebreda, P.; Queizán, A.; García-Miguel, P.; Pestaña, A. No TP53 mutations in neuroblastomas detected by PCR-SSCP analysis. Genes Chromosomes Cancer 1994, 10, 136–138. [Google Scholar] [CrossRef]
- Raj, N.; Attardi, L.D. The Transactivation Domains of the p53 Protein. Cold Spring Harb. Perspect. Med. 2017, 7, a026047. [Google Scholar] [CrossRef] [PubMed]
- Ronca, F.; Yee, K.S.; Yu, V.C. Retinoic acid confers resistance to p53-dependent apoptosis in SH-SY5Y neuroblastoma cells by modulating nuclear import of p53. J. Biol. Chem. 1999, 274, 18128–18134. [Google Scholar] [CrossRef] [Green Version]
- Shangary, S.; Wang, S. Targeting the MDM2-p53 interaction for cancer therapy. Clin. Cancer Res. 2008, 14, 5318–5324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, D.; Gu, W. Dual Roles of MDM2 in the Regulation of p53: Ubiquitination Dependent and Ubiquitination Independent Mechanisms of MDM2 Repression of p53 Activity. Genes Cancer 2012, 3, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Feng, Z.; Levine, A.J. The Regulation of Multiple p53 Stress Responses is Mediated through MDM2. Genes Cancer 2012, 3, 199–208. [Google Scholar] [CrossRef]
- Kim, E.; Shohet, J. Targeted molecular therapy for neuroblastoma: The ARF/MDM2/p53 axis. J. Natl. Cancer Inst. 2009, 101, 1527–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rössler, J.; Taylor, M.; Geoerger, B.; Farace, F.; Lagodny, J.; Peschka-Süss, R.; Niemeyer, C.M.; Vassal, G. Angiogenesis as a target in neuroblastoma. Eur. J. Cancer 2008, 44, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.N.; Singh, H.P.; Guo, W.; Cambier, L.; Riggan, L.; Shackleford, G.M.; Thornton, M.E.; Grubbs, B.H.; Erdreich-Epstein, A.; Qi, D.L.; et al. Reciprocal Induction of MDM2 and MYCN in Neural and Neuroendocrine Cancers. Front. Oncol. 2020, 10, 563156. [Google Scholar] [CrossRef]
- Gangopadhyay, S.; Jalali, F.; Reda, D.; Peacock, J.; Bristow, R.G.; Benchimol, S. Expression of different mutant p53 transgenes in neuroblastoma cells leads to different cellular responses to genotoxic agents. Exp. Cell Res. 2002, 275, 122–131. [Google Scholar] [CrossRef]
- McKenzie, P.P.; Guichard, S.M.; Middlemas, D.S.; Ashmun, R.A.; Danks, M.K.; Harris, L.C. Wild-type p53 can induce p21 and apoptosis in neuroblastoma cells but the DNA damage-induced G1 checkpoint function is attenuated. Clin. Cancer Res. 1999, 5, 4199–4207. [Google Scholar] [PubMed]
- Carr-Wilkinson, J.; O’Toole, K.; Wood, K.M.; Challen, C.C.; Baker, A.G.; Board, J.R.; Evans, L.; Cole, M.; Cheung, N.K.; Boos, J.; et al. High Frequency of p53/MDM2/p14ARF Pathway Abnormalities in Relapsed Neuroblastoma. Clin. Cancer Res. 2010, 16, 1108–1118. [Google Scholar] [CrossRef] [Green Version]
- Keshelava, N.; Zuo, J.J.; Chen, P.; Waidyaratne, S.N.; Luna, M.C.; Gomer, C.J.; Triche, T.J.; Reynolds, C.P. Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res. 2001, 61, 6185–6193. [Google Scholar]
- Gomes, S.; Raimundo, L.; Soares, J.; Loureiro, J.B.; Leão, M.; Ramos, H.; Monteiro, M.N.; Lemos, A.; Moreira, J.; Pinto, M.; et al. New inhibitor of the TAp73 interaction with MDM2 and mutant p53 with promising antitumor activity against neuroblastoma. Cancer Lett. 2019, 446, 90–102. [Google Scholar] [CrossRef]
- Monteleone, L.; Speciale, A.; Valenti, G.E.; Traverso, N.; Ravera, S.; Garbarino, O.; Leardi, R.; Farinini, E.; Roveri, A.; Ursini, F.; et al. PKCα Inhibition as a Strategy to Sensitize Neuroblastoma Stem Cells to Etoposide by Stimulating Ferroptosis. Antioxidants 2021, 10, 691. [Google Scholar] [CrossRef]
- Ödborn Jönsson, L.; Sahi, M.; Lopez-Lorenzo, X.; Keller, F.L.; Kostopoulou, O.N.; Herold, N.; Ährlund-Richter, L.; Shirazi Fard, S. Heterogeneities in Cell Cycle Checkpoint Activation Following Doxorubicin Treatment Reveal Targetable Vulnerabilities in TP53 Mutated Ultra High-Risk Neuroblastoma Cell Lines. Int. J. Mol. Sci. 2021, 22, 3664. [Google Scholar] [CrossRef]
- Rodrigo, M.A.M.; Michalkova, H.; Strmiska, V.; Casar, B.; Crespo, P.; de Los Rios, V.; Ignacio Casal, J.; Haddad, Y.; Guran, R.; Eckschlager, T.; et al. Metallothionein-3 promotes cisplatin chemoresistance remodelling in neuroblastoma. Sci. Rep. 2021, 11, 5496. [Google Scholar] [CrossRef]
- Granger, M.M.; Naranjo, A.; Bagatell, R.; DuBois, S.G.; McCune, J.S.; Tenney, S.C.; Weiss, B.D.; Mosse, Y.P.; Asgharzadeh, S.; Grupp, S.A.; et al. Myeloablative Busulfan/Melphalan Consolidation following Induction Chemotherapy for Patients with Newly Diagnosed High-Risk Neuroblastoma: Children’s Oncology Group Trial ANBL12P1. Transplant. Cell. Ther. 2021, 27, 490.e1–490.e8. [Google Scholar] [CrossRef]
- Tweddle, D.A.; Malcolm, A.J.; Bown, N.; Pearson, A.D.; Lunec, J. Evidence for the development of p53 mutations after cytotoxic therapy in a neuroblastoma cell line. Cancer Res. 2001, 61, 8–13. [Google Scholar]
- Keshelava, N.; Seeger, R.C.; Groshen, S.; Reynolds, C.P. Drug resistance patterns of human neuroblastoma cell lines derived from patients at different phases of therapy. Cancer Res. 1998, 58, 5396–5405. [Google Scholar] [PubMed]
- Xue, C.; Haber, M.; Flemming, C.; Marshall, G.M.; Lock, R.B.; MacKenzie, K.L.; Gurova, K.V.; Norris, M.D.; Gudkov, A.V. p53 determines multidrug sensitivity of childhood neuroblastoma. Cancer Res. 2007, 67, 10351–10360. [Google Scholar] [CrossRef] [Green Version]
- Keshelava, N.; Zuo, J.J.; Waidyaratne, N.S.; Triche, T.J.; Reynolds, C.P. p53 mutations and loss of p53 function confer multidrug resistance in neuroblastoma. Med. Pediatr. Oncol. 2000, 35, 563–568. [Google Scholar] [CrossRef]
- Carr, J.; Bell, E.; Pearson, A.D.; Kees, U.R.; Beris, H.; Lunec, J.; Tweddle, D.A. Increased frequency of aberrations in the p53/MDM2/p14(ARF) pathway in neuroblastoma cell lines established at relapse. Cancer Res. 2006, 66, 2138–2145. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.; Muralidharan, S.V.; Di Marco, M.; Juvvuna, P.K.; Kosalai, S.T.; Reischl, S.; Jachimowicz, D.; Subhash, S.; Raimondi, I.; Kurian, L.; et al. Subcellular Distribution of p53 by the p53-Responsive lncRNA NBAT1 Determines Chemotherapeutic Response in Neuroblastoma. Cancer Res. 2021, 81, 1457–1471. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Patel, N.H.; Gewirtz, D.A. Triangular Relationship between p53, Autophagy, and Chemotherapy Resistance. Int. J. Mol. Sci. 2020, 21, 8991. [Google Scholar] [CrossRef]
- Zhu, G.; Pan, C.; Bei, J.X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in Cancer Progression and Targeted Therapies. Front. Oncol. 2020, 10, 595187. [Google Scholar] [CrossRef] [PubMed]
- Waetzig, V.; Haeusgen, W.; Andres, C.; Frehse, S.; Reinecke, K.; Bruckmueller, H.; Boehm, R.; Herdegen, T.; Cascorbi, I. Retinoic acid-induced survival effects in SH-SY5Y neuroblastoma cells. J. Cell. Biochem. 2019, 120, 5974–5986. [Google Scholar] [CrossRef] [PubMed]
- Rettig, I.; Koeneke, E.; Trippel, F.; Mueller, W.C.; Burhenne, J.; Kopp-Schneider, A.; Fabian, J.; Schober, A.; Fernekorn, U.; von Deimling, A.; et al. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015, 6, e1657. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.; Chen, L.; Liu, T.; Marshall, G.M.; Lunec, J.; Tweddle, D.A. MYCN oncoprotein targets and their therapeutic potential. Cancer Lett. 2010, 293, 144–157. [Google Scholar] [CrossRef]
- Rashid, A.; Duan, X.; Gao, F.; Yang, M.; Yen, A. Roscovitine enhances all-trans retinoic acid (ATRA)-induced nuclear enrichment of an ensemble of activated signaling molecules and augments ATRA-induced myeloid cell differentiation. Oncotarget 2020, 11, 1017–1036. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Pearson, A.; Lunec, J. The MYCN oncoprotein as a drug development target. Cancer Lett. 2003, 197, 125–130. [Google Scholar] [CrossRef]
- Maeshima, R.; Moulding, D.; Stoker, A.W.; Hart, S.L. MYCN Silencing by RNAi Induces Neurogenesis and Suppresses Proliferation in Models of Neuroblastoma with Resistance to Retinoic Acid. Nucleic Acid Ther. 2020, 30, 237–248. [Google Scholar] [CrossRef]
- Gebauer, F.; Hentze, M.W. Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 2004, 5, 827–835. [Google Scholar] [CrossRef]
- Maugeri, M.; Barbagallo, D.; Barbagallo, C.; Banelli, B.; Di Mauro, S.; Purrello, F.; Magro, G.; Ragusa, M.; Di Pietro, C.; Romani, M.; et al. Altered expression of miRNAs and methylation of their promoters are correlated in neuroblastoma. Oncotarget 2016, 7, 83330–83341. [Google Scholar] [CrossRef] [Green Version]
- Nachtergaele, S.; He, C. The emerging biology of RNA post-transcriptional modifications. RNA Biol. 2017, 14, 156–163. [Google Scholar] [CrossRef]
- Mao, Y.; Dong, L.; Liu, X.M.; Guo, J.; Ma, H.; Shen, B.; Qian, S.B. m(6)A in mRNA coding regions promotes translation via the RNA helicase-containing YTHDC2. Nat. Commun. 2019, 10, 5332. [Google Scholar] [CrossRef]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef]
- Lin, W.; Wang, Z.; Wang, J.; Yan, H.; Han, Q.; Yao, W.; Li, K. circRNA-TBC1D4, circRNA-NAALAD2 and circRNA-TGFBR3: Selected Key circRNAs in Neuroblastoma and Their Associations with Clinical Features. Cancer Manag. Res. 2021, 13, 4271–4281. [Google Scholar] [CrossRef]
- Fang, Y.; Yao, Y.; Mao, K.; Zhong, Y.; Xu, Y. Circ_0132817 facilitates cell proliferation, migration, invasion and glycolysis by regulating the miR-432-5p/NOL4L axis in neuroblastoma. Exp. Brain Res. 2021, 239, 1841–1852. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, L.; Hu, X.; Li, Q.; Wu, M. Silencing of circular RNA circPDE5A suppresses neuroblastoma progression by targeting the miR-362-5p/NOL4L axis. Int. J. Neurosci. 2021, 1–11. [Google Scholar] [CrossRef]
- Zhang, L.; Zhou, H.; Li, J.; Wang, X.; Zhang, X.; Shi, T.; Feng, G. Comprehensive Characterization of Circular RNAs in Neuroblastoma Cell Lines. Technol. Cancer Res. Treat. 2020, 19, 1533033820957622. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Estlin, E.J.; Veal, G.J. Clinical and cellular pharmacology in relation to solid tumours of childhood. Cancer Treat. Rev. 2003, 29, 253–273. [Google Scholar] [CrossRef]
- Borriello, A.; Roberto, R.; Della Ragione, F.; Iolascon, A. Proliferate and survive: Cell division cycle and apoptosis in human neuroblastoma. Haematologica 2002, 87, 196–214. [Google Scholar] [PubMed]
- Lázcoz, P.; Muñoz, J.; Nistal, M.; Pestaña, A.; Encío, I.; Castresana, J.S. Frequent promoter hypermethylation of RASSF1A and CASP8 in neuroblastoma. BMC Cancer 2006, 6, 254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Cheung, I.Y.; Wei, X.X.; Tran, H.; Gao, X.; Cheung, N.K. Checkpoint kinase inhibitor synergizes with DNA-damaging agents in G1 checkpoint-defective neuroblastoma. Int. J. Cancer 2011, 129, 1953–1962. [Google Scholar] [CrossRef]
- Tweddle, D.A.; Malcolm, A.J.; Cole, M.; Pearson, A.D.; Lunec, J. p53 cellular localization and function in neuroblastoma: Evidence for defective G(1) arrest despite WAF1 induction in MYCN-amplified cells. Am. J. Pathol. 2001, 158, 2067–2077. [Google Scholar] [CrossRef]
- Van Maerken, T.; Speleman, F.; Vermeulen, J.; Lambertz, I.; De Clercq, S.; De Smet, E.; Yigit, N.; Coppens, V.; Philippé, J.; De Paepe, A.; et al. Small-molecule MDM2 antagonists as a new therapy concept for neuroblastoma. Cancer Res. 2006, 66, 9646–9655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Iraci, N.; Gherardi, S.; Gamble, L.D.; Wood, K.M.; Perini, G.; Lunec, J.; Tweddle, D.A. p53 is a direct transcriptional target of MYCN in neuroblastoma. Cancer Res. 2010, 70, 1377–1388. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Levine, A.J. The p53 functional circuit. J. Cell Sci. 2001, 114, 4139–4140. [Google Scholar] [CrossRef]
- Bell, E.; Premkumar, R.; Carr, J.; Lu, X.; Lovat, P.E.; Kees, U.R.; Lunec, J.; Tweddle, D.A. The role of MYCN in the failure of MYCN amplified neuroblastoma cell lines to G1 arrest after DNA damage. Cell Cycle 2006, 5, 2639–2647. [Google Scholar] [CrossRef]
- Bell, E.; Lunec, J.; Tweddle, D.A. Cell cycle regulation targets of MYCN identified by gene expression microarrays. Cell Cycle 2007, 6, 1249–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, J.A.; Cleveland, J.L. Myc pathways provoking cell suicide and cancer. Oncogene 2003, 22, 9007–9021. [Google Scholar] [CrossRef] [Green Version]
- Staege, M.S.; Hutter, C.; Neumann, I.; Foja, S.; Hattenhorst, U.E.; Hansen, G.; Afar, D.; Burdach, S.E. DNA microarrays reveal relationship of Ewing family tumors to both endothelial and fetal neural crest-derived cells and define novel targets. Cancer Res. 2004, 64, 8213–8221. [Google Scholar] [CrossRef] [Green Version]
- Li, L.C.; Dahiya, R. MethPrimer: Designing primers for methylation PCRs. Bioinformatics 2002, 18, 1427–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toguchida, J.; Yamaguchi, T.; Ritchie, B.; Beauchamp, R.L.; Dayton, S.H.; Herrera, G.E.; Yamamuro, T.; Kotoura, Y.; Sasaki, M.S.; Little, J.B.; et al. Mutation spectrum of the p53 gene in bone and soft tissue sarcomas. Cancer Res. 1992, 52, 6194–6199. [Google Scholar]
- Carr, J.; Bown, N.P.; Case, M.C.; Hall, A.G.; Lunec, J.; Tweddle, D.A. High-resolution analysis of allelic imbalance in neuroblastoma cell lines by single nucleotide polymorphism arrays. Cancer Genet. Cytogenet. 2007, 172, 127–138. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- De La Rosa, J.; Urdiciain, A.; Zelaya, M.V.; Zazpe, I.; Meléndez, B.; Rey, J.A.; Idoate, M.A.; Castresana, J.S. APR-246 combined with 3-deazaneplanocin A, panobinostat or temozolomide reduces clonogenicity and induces apoptosis in glioblastoma cells. Int. J. Oncol. 2021, 58, 312–330. [Google Scholar] [CrossRef]
- Menichini, P.; Monti, P.; Speciale, A.; Cutrona, G.; Matis, S.; Fais, F.; Taiana, E.; Neri, A.; Bomben, R.; Gentile, M.; et al. Antitumor Effects of PRIMA-1 and PRIMA-1(Met) (APR246) in Hematological Malignancies: Still a Mutant P53-Dependent Affair? Cells 2021, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bykov, V.J.N.; Wiman, K.G.; Zawacka-Pankau, J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018, 9, 439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maslah, N.; Diawara, Y.; Sebert, M.; Giraudier, S.; Fenaux, P.; Cassinat, B. In vitro assessment of the sensitivity to APR-246 + azacitidine combination predicts response to this combination in myelodysplastic/acute myeloid leukaemia patients. Br. J. Haematol. 2021, 194, e77–e79. [Google Scholar] [CrossRef]
- Li, X.L.; Zhou, J.; Xia, C.J.; Min, H.; Lu, Z.K.; Chen, Z.R. PRIMA-1(met) induces autophagy in colorectal cancer cells through upregulation of the mTOR/AMPK-ULK1-Vps34 signaling cascade. Oncol. Rep. 2021, 45, 86. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Rajaei, M.; Youn, J.Y.; Zafar, A.; Deokar, H.; Buolamwini, J.K.; Yang, J.; Foster, J.H.; Zhou, J.; et al. Targeting MDM2 for Neuroblastoma Therapy: In Vitro and In Vivo Anticancer Activity and Mechanism of Action. Cancers 2020, 12, 3651. [Google Scholar] [CrossRef]
- Zafar, A.; Wang, W.; Liu, G.; Xian, W.; McKeon, F.; Zhou, J.; Zhang, R. Targeting the p53-MDM2 pathway for neuroblastoma therapy: Rays of hope. Cancer Lett. 2021, 496, 16–29. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybridization Pattern | ||
|---|---|---|
| MYCN | MDM2 | |
| SH-SY5Y | 2G3R | normal |
| SK-N-SH | 2G3R | normal |
| BE(2)C | 2GampR | 3G3R |
| IMR-32 | 2GampR | normal |
| SK-N-Be(2) | 2GampR | normal |
| SK-N-FI | 1GampR | normal |
| Kelly | 1GampR | normal |
| SK-N-DZ | 4GampR | 5G4R |
| SIMA | 4GampR | 4G4R |
| MHH-NB-11 | 5GampR | 4G4R |
| SK-N-MC | normal | normal |
| MC-IXC | normal | 3G2R |
| MYCN amp | MDM2 amp | p14ARF meth | p14ARF exp | p14ARF hd | TP53 mut | |
|---|---|---|---|---|---|---|
| SH-SY5Y | − | − | − | − | − | − |
| SK-N-SH | − | − | − | − | − | − |
| BE(2)C | + | − | − | − | − | − |
| IMR-32 | + | − | − | − | − | − |
| SK-N-Be(2) | + | − | − | − | − | − |
| SK-N-FI | + | − | − | − | − | + |
| Kelly | + | − | − | − | − | − |
| SK-N-DZ | + | − | − | − | − | − |
| SIMA | + | − | − | − | − | − |
| MHH-NB-11 | + | − | − | − | − | − |
| SK-N-MC | − | − | − | − | − | − |
| MC-IXC | − | − | − | − | − | − |
| Study | Sequence (5′–3′) | Size (bp) | ||
|---|---|---|---|---|
| Methylation | MSP | P14ARF U–F: | 5′-TTTTTGGTGTTAAAGGGTGGTGTATAGT-3′ | 132 |
| P14ARF U–R: | 5′-CACAAAAACCCTCACTCACAACAA-3′ | |||
| P14ARF M–F: | 5′-GTGTTAAAGGGCGGCGTAGC-3′ | 122 | ||
| P14ARF M–R: | 5′-AAAACCCTCACTCGCGACGA-3′ | |||
| MCA–Meth | P14ARF–MET–F: | 5′-AAAAATGGGTTAGATATAAAG-3′ | 248 | |
| P14ARF–MET–R: | 5′-CCTCTTCTAAATTTAAAAAACA-3′ | |||
| Expression | RT–PCR | P14ARF–RT–F: | 5′-CCGCCGCGAGTGAGGGTTTT-3′ | 242 |
| P14ARF–RT–R: | 5′-GCACGGGTCGGGTGAGAGTGG-3′ | |||
| TFRC–F: | 5′-GTCAATGTCCCAAACGTCACCAGA-3′ | 298 | ||
| TFRC–R: | 5′-ATTTCGGGAATGCTGAGAAAACAGACAGA-3′ | |||
| Homozygous deletion | Differential PCR | P14ARF–DH–F: | 5′-TCCCAGTCTGCAGTTAAGGG-3′ | 174 |
| P14ARF–DH–R: | 5′-ACCACGAAAACCCTCACTCG-3′ | |||
| GAPDH–F: | 5′-AACGTGTCAGTGGTGGACCTG-3′ | 160 | ||
| GAPDH–R: | 5′-AGTGGGTGTCGCTGTTGAAGT-3′ | |||
| Exon | Sequence | Size (bp) | |
|---|---|---|---|
| Exon 5 (5′ end) | F: | 5′-TTATCTGTTCACTTGTGCCC-3′ | 189 |
| R: | 5′-TCATGTGCTGTGACTGCTTG-3′ | ||
| Exon 5 (3′ end) | F: | 5′-TTCCACACCCCCGCCCGGCA-3′ | 162 |
| R: | 5′-ACCCTGGGCAACCAGCCCTG-3′ | ||
| Exon 6 | F: | 5′-ACGACAGGGCTGGTTGCCCA-3′ | 201 |
| R: | 5′-CTCCCAGAGACCCCAGTTGC-3′ | ||
| Exon 7 | F: | 5′-GGCCTCATCTTGGGCCTGTG-3′ | 171 |
| R: | 5′-CAGTGTGCAGGGTGGCAAGT-3′ | ||
| Exon 8 | F: | 5′-CTGCCTCTTGCTTCTCTTTT-3′ | 204 |
| R: | 5′-TCTCCTCCACCGCTTCTTGT-3′ | ||
| Exon 9 | F: | 5′-GCAGTTATGCCTCAGATTCA-3′ | 185 |
| R: | 5′-GGCATTTTGAGTGTTAGACT-3′ | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco-Luquin, I.; Lázcoz, P.; Celay, J.; Castresana, J.S.; Encío, I.J. In Vitro Assessment of the Role of p53 on Chemotherapy Treatments in Neuroblastoma Cell Lines. Pharmaceuticals 2021, 14, 1184. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111184
Blanco-Luquin I, Lázcoz P, Celay J, Castresana JS, Encío IJ. In Vitro Assessment of the Role of p53 on Chemotherapy Treatments in Neuroblastoma Cell Lines. Pharmaceuticals. 2021; 14(11):1184. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111184
Chicago/Turabian StyleBlanco-Luquin, Idoia, Paula Lázcoz, Jon Celay, Javier S. Castresana, and Ignacio J. Encío. 2021. "In Vitro Assessment of the Role of p53 on Chemotherapy Treatments in Neuroblastoma Cell Lines" Pharmaceuticals 14, no. 11: 1184. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14111184