
De Novo Molecular Design of Caspase-6 Inhibitors by a GRU-Based Recurrent Neural Network Combined with a Transfer Learning Approach

 ,
,
Abstract
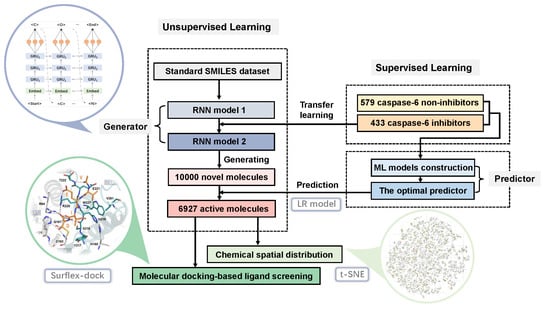
:
1. Introduction
2. Methods
2.1. Datasets
2.2. Machine Learning Based Classification Models of Caspase-6 Inhibitors
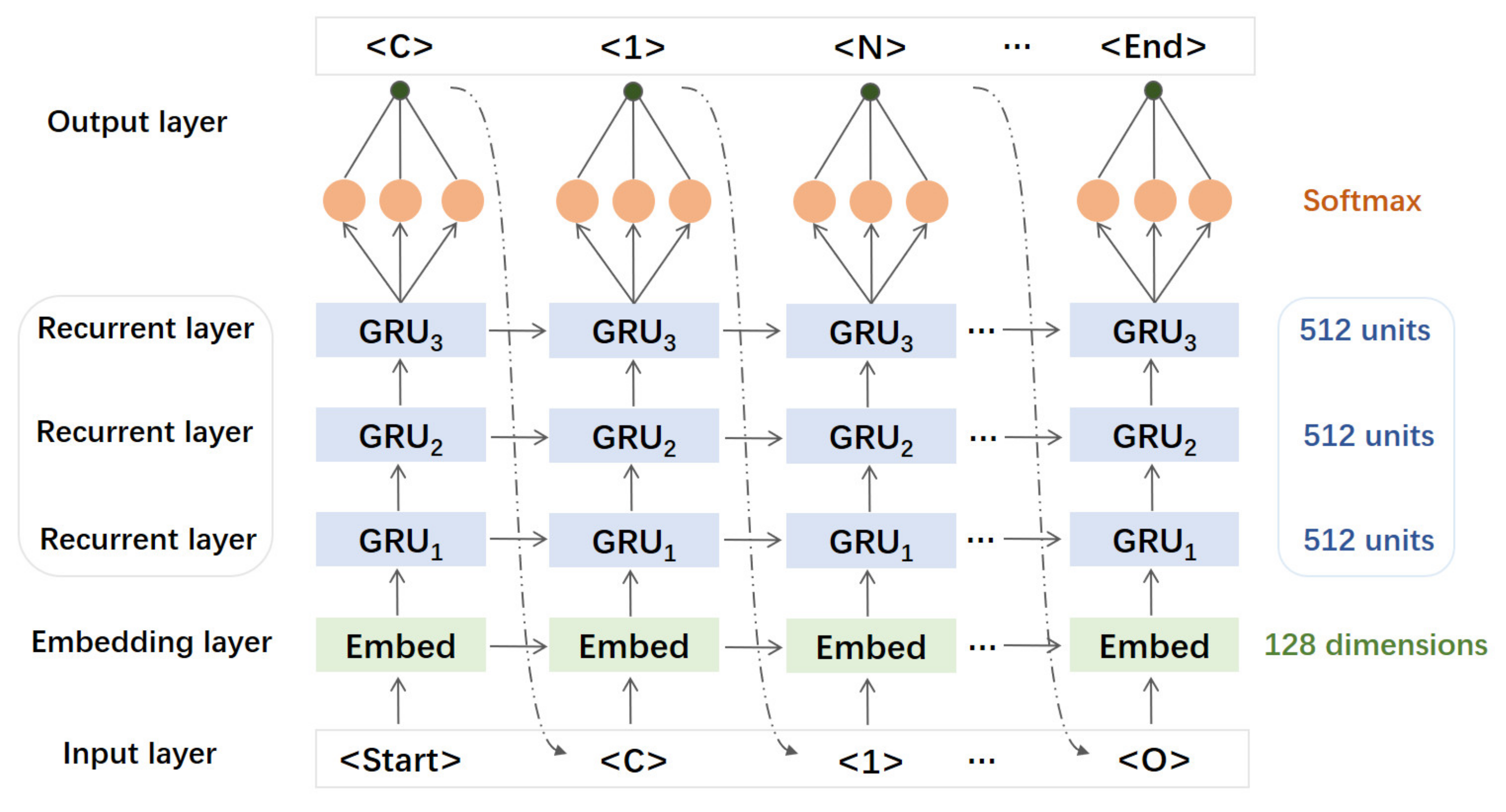
2.3. Generative RNN Modeling and Transfer Learning
2.4. Molecular Docking
3. Results and Discussion
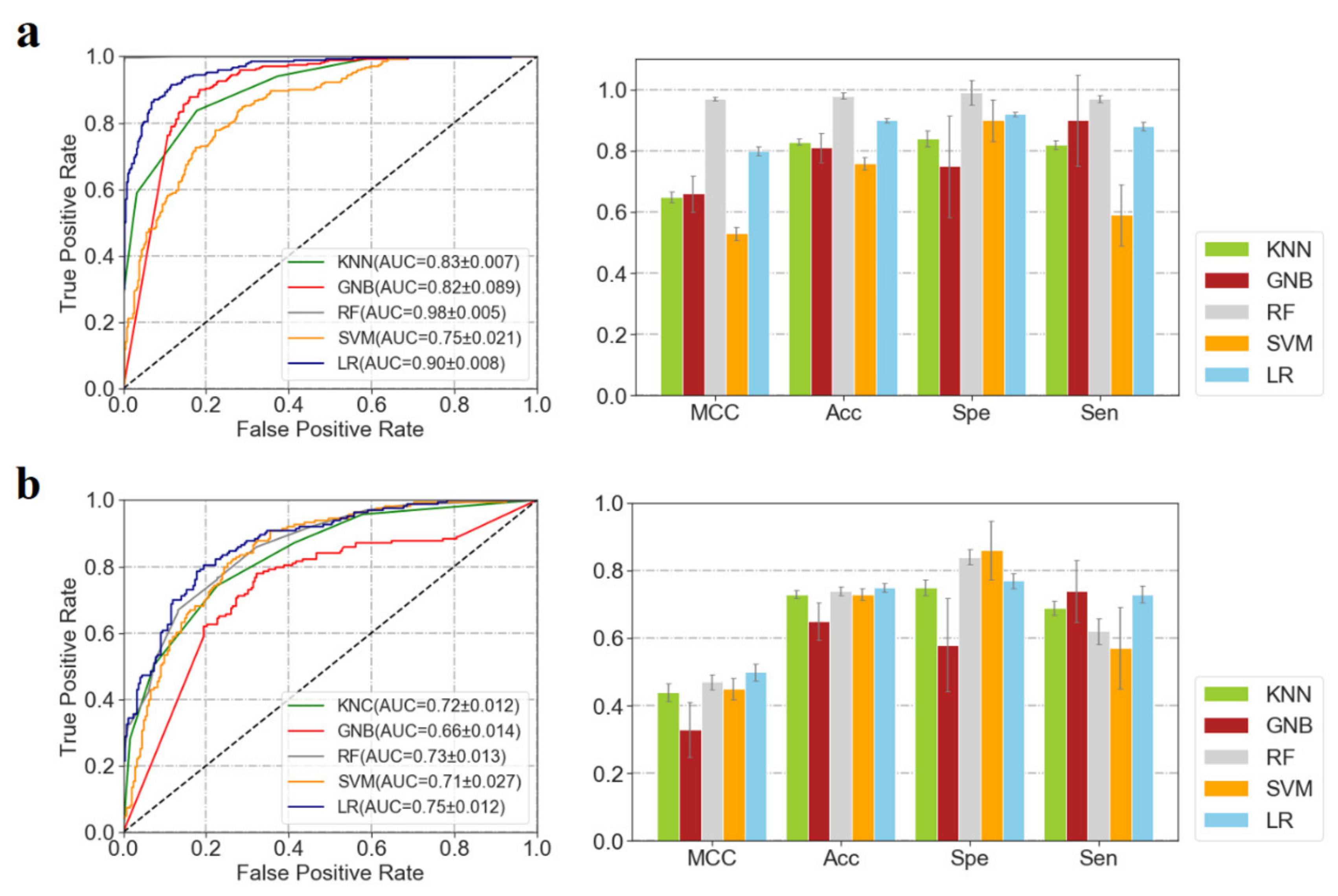
3.1. Performances of ML Predictors
3.2. The Generative RNN Modeling
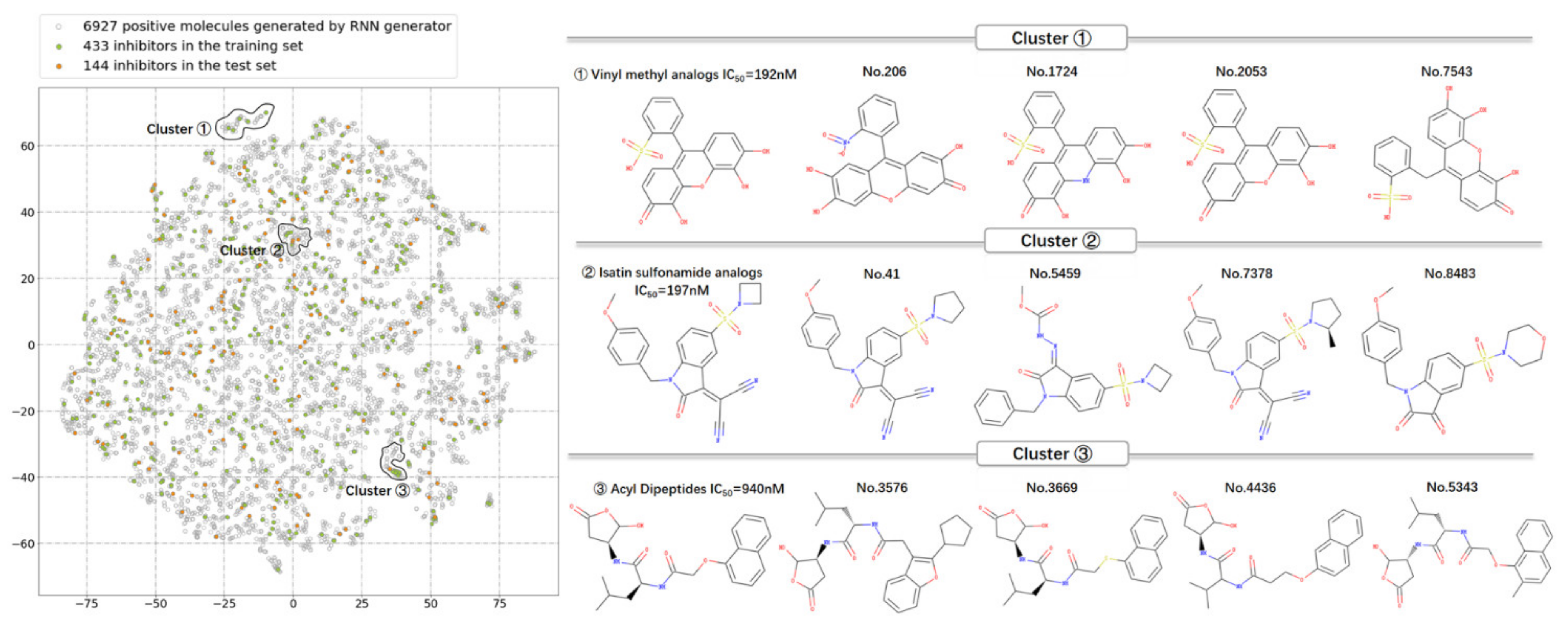
3.3. The Distribution in Chemical Space of the Potential Caspase-6 Inhibitors
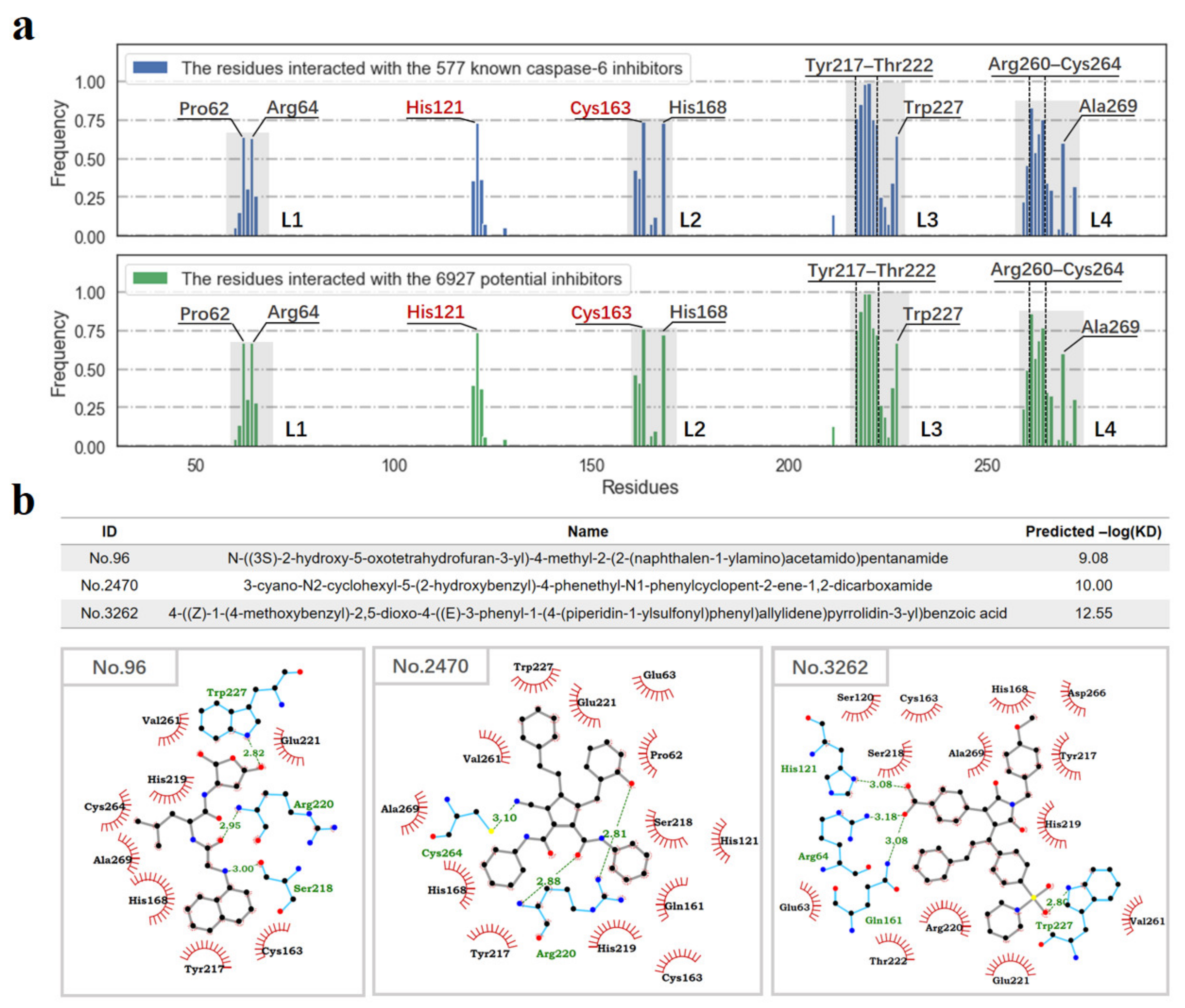
3.4. Molecular Docking-Based Ligand Screening
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clark, A.C. Caspase Allostery and Conformational Selection. Chem. Rev. 2016, 116, 6666–6706. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner caspase-3,-6, and-7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a008656. [Google Scholar] [CrossRef]
- Denecker, G.; Ovaere, P.; Vandenabeele, P.; Declercq, W. Caspase-14 reveals its secrets. J. Cell Biol. 2008, 180, 451–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.J.; Cao, Q.; Zhang, Y.; Su, X.D. Activation and Regulation of Caspase-6 and Its Role in Neurodegenerative Diseases. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 553–572. [Google Scholar] [CrossRef] [Green Version]
- LeBlanc, A.; Liu, H.; Goodyer, C.; Bergeron, C.; Hammond, J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer’s disease. J. Biol. Chem. 1999, 274, 23426–23436. [Google Scholar] [CrossRef] [Green Version]
- Klaiman, G.; Petzke, T.L.; Hammond, J.; LeBlanc, A.C. Targets of Caspase-6 activity in human neurons and Alzheimer disease. Mol. Cell. Proteom. 2008, 7, 1541–1555. [Google Scholar] [CrossRef] [Green Version]
- Sexton, K.B.; Kato, D.; Berger, A.B.; Fonovic, M.; Verhelst, S.H.L.; Bogyo, M. Specificity of aza-peptide electrophile activity-based probes of caspases. Cell Death Differ. 2007, 14, 727–732. [Google Scholar] [CrossRef] [Green Version]
- Linton, S.D.; Karanewsky, D.S.; Ternansky, R.J.; Wu, J.C.; Pham, B.; Kodandapani, L.; Smidt, R.; Diaz, J.-L.; Fritz, L.C.; Tomaselli, K.J. Acyl Dipeptides as reversible caspase inhibitors. Part 1: Initial lead optimization. Bioorg. Med. Chem. Lett. 2002, 12, 2969–2971. [Google Scholar] [CrossRef]
- Linton, S.D.; Karanewsky, D.S.; Ternansky, R.J.; Chen, N.; Guo, M.; Jahangiri, K.G.; Kalish, V.J.; Meduna, S.P.; Robinson, E.D.; Ullman, B.R.; et al. Acyl Dipeptides as reversible caspase inhibitors. Part 2: Further optimization. Bioorg. Med. Chem. Lett. 2002, 12, 2973–2975. [Google Scholar] [CrossRef]
- Chu, W.H.; Rothfuss, J.; d’Avignon, A.; Zeng, C.B.; Zhou, D.; Hotchkiss, R.S.; Mach, R.H. Isatin sulfonamide analogs containing a michael addition acceptor: A new class of caspase 3/7 inhibitors. J. Med. Chem. 2007, 50, 3751–3755. [Google Scholar] [CrossRef]
- Chu, W.H.; Rothfuss, J.; Chu, Y.X.; Zhou, D.; Mach, R.H. Synthesis and in Vitro Evaluation of Sulfonamide Isatin Michael Acceptors as Small Molecule Inhibitors of Caspase-6. J. Med. Chem. 2009, 52, 2188–2191. [Google Scholar] [CrossRef]
- Chu, W.H.; Rothfuss, J.; Zhou, D.; Mach, R.H. Synthesis and evaluation of isatin analogs as caspase-3 inhibitors: Introduction of a hydrophilic group increases potency in a whole cell assay. Bioorg. Med. Chem. Lett. 2011, 21, 2192–2197. [Google Scholar] [CrossRef] [Green Version]
- Limpachayaporn, P.; Schafers, M.; Schober, O.; Kopka, K.; Haufe, G. Synthesis of new fluorinated, 2-substituted 5-pyrrolidinylsulfonyl isatin derivatives as caspase-3 and caspase-7 inhibitors: Nonradioactive counterparts of putative PET-compatible apoptosis imaging agents. Bioorg. Med. Chem. 2013, 21, 2025–2036. [Google Scholar] [CrossRef]
- Limpachayaporn, P.; Wagner, S.; Kopka, K.; Schober, O.; Schafers, M.; Haufe, G. Synthesis of 7-Halogenated Isatin Sulfonamides: Nonradioactive Counterparts of Caspase-3/-7 Inhibitor-Based Potential Radiopharmaceuticals for Molecular Imaging of Apoptosis. J. Med. Chem. 2014, 57, 9383–9395. [Google Scholar] [CrossRef] [PubMed]
- Leyva, M.J.; Degiacomo, F.; Kaltenbach, L.S.; Holcomb, J.; Zhang, N.; Gafni, J.; Park, H.; Lo, D.C.; Salvesen, G.S.; Ellerby, L.M.; et al. Identification and evaluation of small molecule pan-caspase inhibitors in Huntington’s disease models. Chem. Biol. 2010, 17, 1189–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pakavathkumar, P.; Sharma, G.; Kaushal, V.; Foveau, B.; LeBlanc, A.C. Methylene Blue Inhibits Caspases by Oxidation of the Catalytic Cysteine. Sci. Rep. 2015, 5, 13730. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Shin, E.A.; Lee, J.H.; Ahn, D.; Kim, C.G.; Kim, J.H.; Kim, S.H. Caspase inhibitors: A review of recently patented compounds (2013–2015). Expert Opin. Ther. Pat. 2018, 28, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Pakavathkumar, P.; Noel, A.; Lecrux, C.; Tubeleviciute-Aydin, A.; Hamel, E.; Ahlfors, J.E.; LeBlanc, A.C. Caspase vinyl sulfone small molecule inhibitors prevent axonal degeneration in human neurons and reverse cognitive impairment in Caspase-6-overexpressing mice. Mol. Neurodegener. 2017, 12, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heise, C.E.; Murray, J.; Augustyn, K.E.; Bravo, B.; Chugha, P.; Cohen, F.; Giannetti, A.M.; Gibbons, P.; Hannoush, R.N.; Hearn, B.R.; et al. Mechanistic and Structural Understanding of Uncompetitive Inhibitors of Caspase-6. PLoS ONE 2012, 7, e50864. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, S.H.; Schipper, J.L.; Clark, A.C. The potential for caspases in drug discovery. Curr. Opin. Drug Discov. Dev. 2010, 13, 568–576. [Google Scholar]
- Jing, Y.; Bian, Y.; Hu, Z.; Wang, L.; Xie, X.Q. Deep Learning for Drug Design: An Artificial Intelligence Paradigm for Drug Discovery in the Big Data Era. AAPS J. 2018, 20, 58. [Google Scholar] [CrossRef] [PubMed]
- Gawehn, E.; Hiss, J.A.; Schneider, G. Deep Learning in Drug Discovery. Mol. Inform. 2016, 35, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Sellwood, M.A.; Ahmed, M.; Segler, M.H.S.; Brown, N. Artificial intelligence in drug discovery. Future Med. Chem. 2018, 10, 2025–2028. [Google Scholar] [CrossRef] [Green Version]
- Xue, D.Y.; Gong, Y.K.; Yang, Z.Y.; Chuai, G.H.; Qu, S.; Shen, A.Z.; Yu, J.; Liu, Q. Advances and challenges in deep generative models for de novo molecule generation. Wiley Interdiscip. Res. Comput. Mol. Sci. 2019, 9, e1395. [Google Scholar] [CrossRef]
- Grisoni, F.; Moret, M.; Lingwood, R.; Schneider, G. Bidirectional Molecule Generation with Recurrent Neural Networks. J. Chem. Inf. Model. 2020, 60, 1175–1183. [Google Scholar] [CrossRef]
- Amabilino, S.; Pogany, P.; Pickett, S.D.; Green, D.V.S. Guidelines for Recurrent Neural Network Transfer Learning-Based Molecular Generation of Focused Libraries. J. Chem. Inf. Model. 2020, 60, 5699–5713. [Google Scholar] [CrossRef]
- Gomez-Bombarelli, R.; Wei, J.N.; Duvenaud, D.; Hernandez-Lobato, J.M.; Sanchez-Lengeling, B.; Sheberla, D.; Aguilera-Iparraguirre, J.; Hirzel, T.D.; Adams, R.P.; Aspuru-Guzik, A. Automatic Chemical Design Using a Data-Driven Continuous Representation of Molecules. ACS Cent. Sci. 2018, 4, 268–276. [Google Scholar] [CrossRef]
- Winter, R.; Montanari, F.; Noe, F.; Clevert, D.A. Learning continuous and data-driven molecular descriptors by translating equivalent chemical representations. Chem. Sci. 2019, 10, 1692–1701. [Google Scholar] [CrossRef] [Green Version]
- Olivecrona, M.; Blaschke, T.; Engkvist, O.; Chen, H.M. Molecular de-novo design through deep reinforcement learning. J. Cheminform. 2017, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Jaques, N.; Gu, S.; Bahdanau, D.; Hernández-Lobato, J.M.; Turner, R.E.; Eck, D. Sequence Tutor: Conservative Fine-Tuning of Sequence Generation Models with KL-control. arXiv 2017, arXiv:1611.02796. [Google Scholar]
- Benhenda, M. ChemGAN challenge for drug discovery: Can AI reproduce natural chemical diversity? arXiv 2017, arXiv:1708.08227. [Google Scholar]
- Sousa, T.; Correia, J.; Pereira, V.; Rocha, M. Generative Deep Learning for Targeted Compound Design. J. Chem. Inf. Model. 2021, 61, 5343–5361. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, J.C.; Zhou, Z.L.; Yang, W.; Guastella, J.; Drewe, J.; Cai, S.X. Dipeptidyl aspartyl fluoromethylketones as potent caspase-3 inhibitors: SAR of the P-2 amino acid. Bioorg. Med. Chem. Lett. 2004, 14, 1269–1272. [Google Scholar] [CrossRef] [PubMed]
- Choong, I.C.; Lew, W.; Lee, D.; Pham, P.; Burdett, M.T.; Lam, J.W.; Wiesmann, C.; Luong, T.N.; Fahr, B.; DeLano, W.L.; et al. Identification of potent and selective small-molecule inhibitors of caspase-3 through the use of extended tethering and structure-based drug design. J. Med. Chem. 2002, 45, 5005–5022. [Google Scholar] [CrossRef] [PubMed]
- Asgian, J.L.; James, K.E.; Li, Z.Z.; Carter, W.; Barrett, A.J.; Mikolajczyk, J.; Salvesen, G.S.; Powers, J.C. Aza-peptide epoxides: A new class of inhibitors selective for clan CD cysteine proteases. J. Med. Chem. 2002, 45, 4958–4960. [Google Scholar] [CrossRef]
- Lee, D.; Long, S.A.; Murray, J.H.; Adams, J.L.; Nuttall, M.E.; Nadeau, D.P.; Kikly, K.; Winkler, J.D.; Sung, C.-M.; Ryan, M.D.; et al. Potent and selective nonpeptide inhibitors of caspases 3 and 7. J. Med. Chem. 2001, 44, 2015–2026. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guan, L.F.; Jia, S.J.; Tseng, B.; Drewe, J.; Cai, S.X. Dipeptidyl aspartyl fluoromethylketones as potent caspase inhibitors: Peptidomimetic replacement of the P-2 alpha-amino acid by a alpha-hydroxy acid. Bioorg. Med. Chem. Lett. 2005, 15, 1379–1383. [Google Scholar] [CrossRef]
- Han, Y.X.; Giroux, A.; Colucci, J.; Bayly, C.I.; Mckay, D.J.; Roy, S.; Xanthoudakis, S.; Vaillancourt, J.; Rasper, D.M.; Tam, J.; et al. Novel pyrazinone mono-amides as potent and reversible caspase-3 inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, S.J.; Tseng, B.; Drewe, J.; Cai, S.X. Dipeptidyl aspartyl fluoromethylketones as potent caspase inhibitors: Peptidomimetic replacement of the P-2 amino acid by 2-aminoaryl acids and other non-natural amino acids. Bioorg. Med. Chem. Lett. 2007, 17, 6178–6182. [Google Scholar] [CrossRef]
- Thompson, C.M.; Quinn, C.A.; Hergenrother, P.J. Total Synthesis and Cytoprotective Properties of Dykellic Acid. J. Med. Chem. 2009, 52, 117–125. [Google Scholar] [CrossRef]
- Mott, B.T.; Ferreira, R.S.; Simeonov, A.; Jadhav, A.; Ang, K.K.H.; Leister, W.; Shen, M.; Silveira, J.T.; Doyle, P.S.; Arkin, M.R.; et al. Identification and Optimization of Inhibitors of Trypanosomal Cysteine Proteases: Cruzain, Rhodesain, and TbCatB. J. Med. Chem. 2010, 53, 52–60. [Google Scholar] [CrossRef] [Green Version]
- Rosse, G. Irreversible Inhibitors of Cysteine Proteases. ACS Med. Chem. Lett. 2013, 4, 163–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause-Heuer, A.M.; Howell, N.R.; Matesic, L.; Dhand, G.; Young, E.L.; Burgess, L.; Jiang, C.D.; Lengkeek, N.A.; Fookes, C.J.R.; Pham, T.Q.; et al. A new class of fluorinated 5-pyrrolidinylsulfonyl isatin caspase inhibitors for PET imaging of apoptosis. MedChemComm 2013, 4, 347–352. [Google Scholar] [CrossRef]
- Landrum, G. RDKit: Open-Source Cheminformatics. 2006. Available online: https://www.rdkit.org/ (accessed on 30 November 2020).
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Powers, D.M. Evaluation: From precision, recall and F-measure to ROC, informedness, markedness and correlation. arXiv 2020, arXiv:2010.16061. [Google Scholar]
- Lučić, B.; Batista, J.; Bojović, V.; Lovrić, M.; Kržić, A.S.; Bešlo, D.; Nadramija, D.; Vikić-Topić, D. Estimation of Random Accuracy and its Use in Validation of Predictive Quality of Classification Models within Predictive Challenges. Croat. Chem. Acta 2019, 92, 379–391. [Google Scholar] [CrossRef] [Green Version]
- Batista, J.; Vikić-Topić, D.; Lučić, B. The Difference Between the Accuracy of Real and the Corresponding Random Model is a Useful Parameter for Validation of Two-State Classification Model Quality. Croat. Chem. Acta 2016, 89, 527–534. [Google Scholar] [CrossRef]
- Kingma, D.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Clark, M.; Cramer, R.D.; Vanopdenbosch, N. Validation of the General-Purpose Tripos 5.2 Force-Field. J. Comput. Chem. 1989, 10, 982–1012. [Google Scholar] [CrossRef]








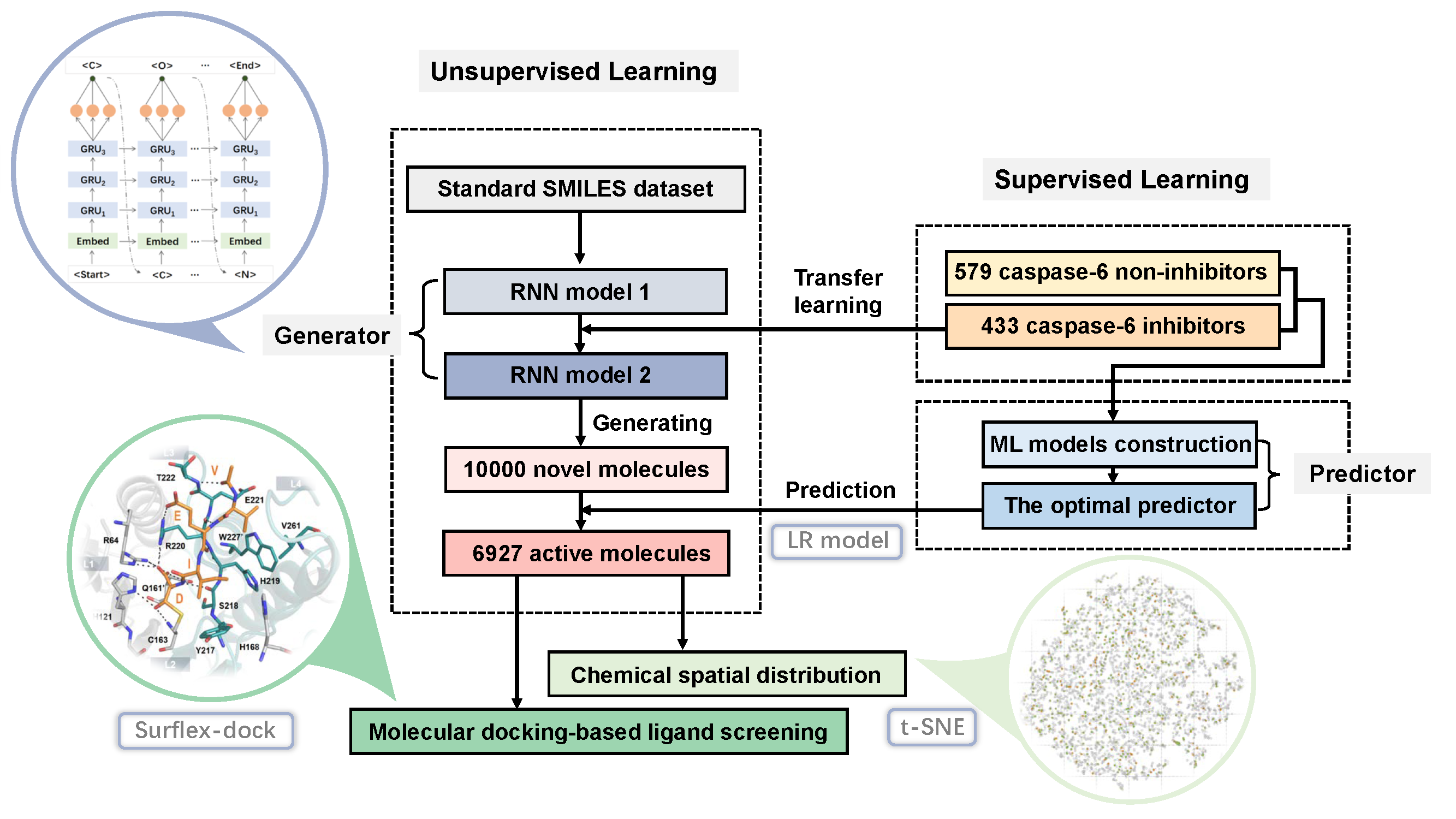{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Confusion Matrix | Performance | |||||||
|---|---|---|---|---|---|---|---|---|
| CP | CN | Acc | Spe | Sen | MCC | Random Acc | ||
| Independent test set | PCP | 102 | 49 | 0.86 | 0.90 | 0.71 | 0.60 | 0.647 |
| PCN | 42 | 451 | ||||||
| Sampling Process | I | II | III | IV | V | VI | VII | VIII | IX | X |
|---|---|---|---|---|---|---|---|---|---|---|
| No. of SMILES strings | 1000 | 2000 | 3000 | 4000 | 5000 | 10,000 | 20,000 | 30,000 | 40,000 | 50,000 |
| The predicted positive samples (%) | 76.0 | 72.7 | 71.4 | 70.7 | 70.6 | 69.3 | 67.1 | 66.2 | 65.5 | 65.0 |
| Recall (%) | 2.08 | 2.08 | 3.47 | 5.55 | 6.94 | 8.33 | 10.41 | 11.80 | 13.19 | 13.19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.; Mei, H.; Lu, L.; Qiu, M.; Liang, X.; Xu, L.; Kuang, Z.; Heng, Y.; Pan, X. De Novo Molecular Design of Caspase-6 Inhibitors by a GRU-Based Recurrent Neural Network Combined with a Transfer Learning Approach. Pharmaceuticals 2021, 14, 1249. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14121249
Huang S, Mei H, Lu L, Qiu M, Liang X, Xu L, Kuang Z, Heng Y, Pan X. De Novo Molecular Design of Caspase-6 Inhibitors by a GRU-Based Recurrent Neural Network Combined with a Transfer Learning Approach. Pharmaceuticals. 2021; 14(12):1249. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14121249
Chicago/Turabian StyleHuang, Shuheng, Hu Mei, Laichun Lu, Minyao Qiu, Xiaoqi Liang, Lei Xu, Zuyin Kuang, Yu Heng, and Xianchao Pan. 2021. "De Novo Molecular Design of Caspase-6 Inhibitors by a GRU-Based Recurrent Neural Network Combined with a Transfer Learning Approach" Pharmaceuticals 14, no. 12: 1249. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14121249