
Reuse of Molecules for Glioblastoma Therapy

 , , ,
, , , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Molecular Classification
1.2. Cellular Pathways in GBMs
1.3. Current Treatment Options
1.4. Barriers to Identifying Effective Treatment
1.5. Repurposing and Repositioning Drugs
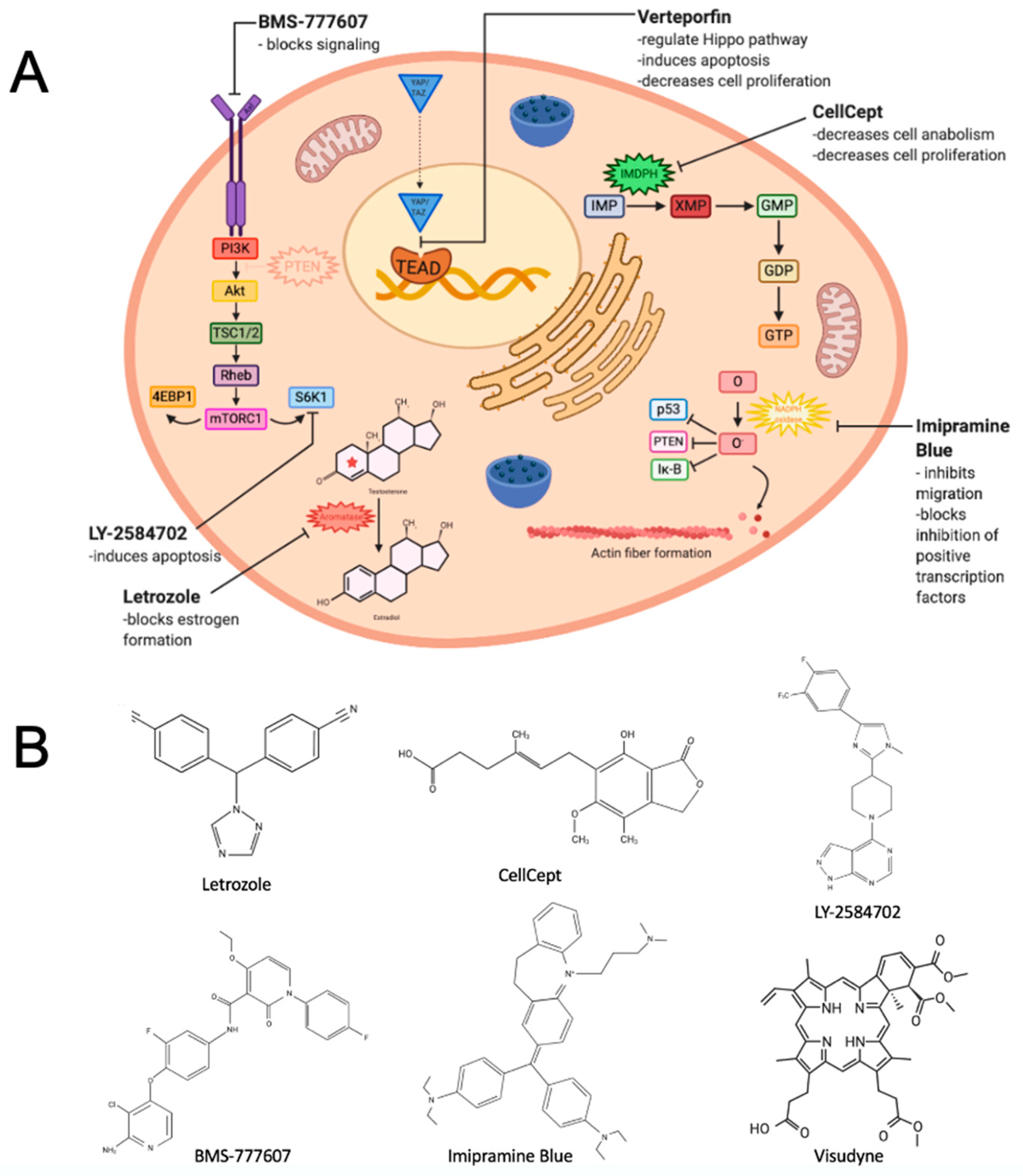
2. Repositioning a Small Molecule Inhibitor Used in Breast Cancer That Targets Estrogen Biosynthesis
3. Repurposing an Immunosuppressive Agent That Inhibits an Enzyme Critical to Guanosine Nucleotide Synthesis
4. Repositioning Two Agents to Target a Kinase Receptor Signaling Pathway
5. Repurposing a Derivative of an Antidepressant Found to Inhibit Cancer Cell Migration
6. Repurposing a Drug for Macular Degeneration
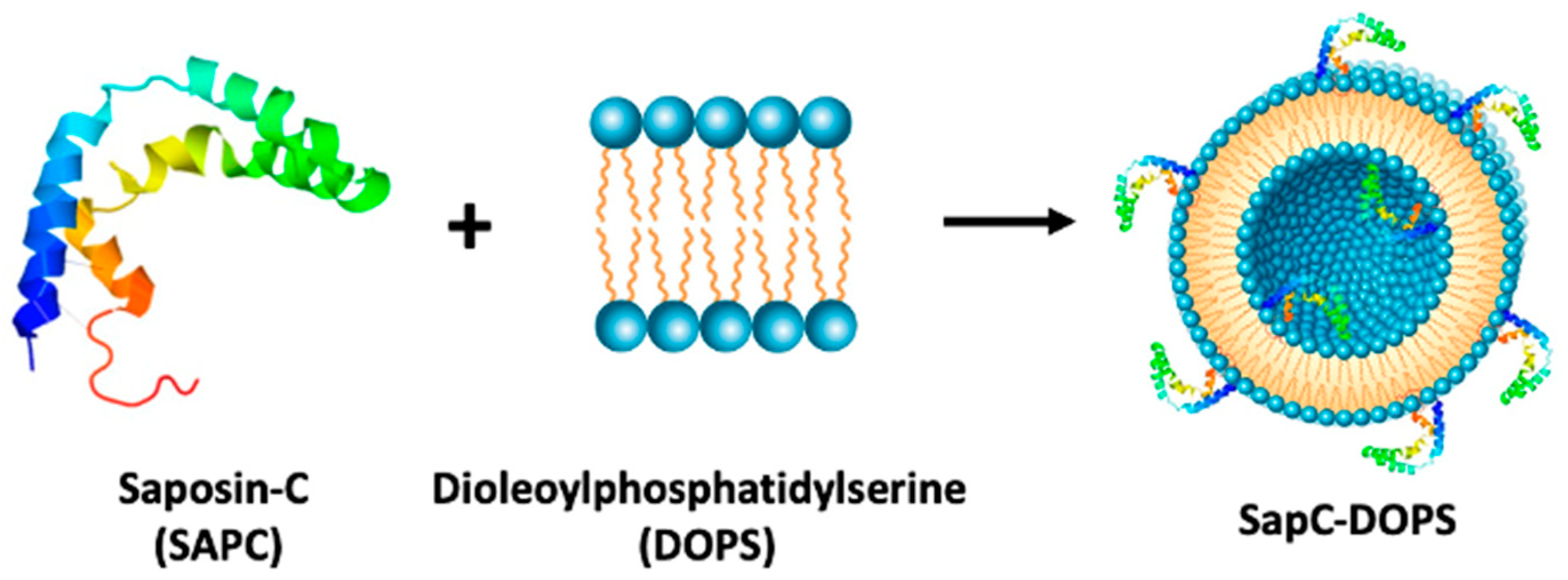
7. Targeting a Phospholipid with a Saposin C Embedded Nanoparticle
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Aldape, K.; Zadeh, G.; Mansouri, S.; Reifenberger, G.; von Deimling, A. Glioblastoma: Pathology, molecular mechanisms and markers. Acta Neuropthol. 2015, 129, 829–848. [Google Scholar] [CrossRef] [PubMed]
- Glioblastoma Multiforme. Available online: https://www.aans.org/en/Patients/Neurosurgical-Conditions-and-Treatments/Glioblastoma-Multiforme (accessed on 16 October 2020).
- Zhang, P.; Xia, Q.; Liu, L.; Li, S.; Dong, L. Current Opinion on Molecular Characterization for GBM Classification in Guiding Clinical Diagnosis, Prognosis, and Therapy. Front. Mol. Biosci. 2020, 7, 562798. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Yeom, J.; Cho, H.J.; Kim, J.-H.; Yoon, S.-J.; Kim, H.; Sa, J.K.; Ju, S.; Lee, H.; Oh, M.J.; et al. Integrated pharmaco-proteogenomics defines two subgroups in isocitrate dehydrogenase wild-type glioblastoma with prognostic and therapeutic opportunities. Nat. Commun. 2020, 11, 3288. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golbuc, T.; Mesirov, J.P.; et al. An integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR and NF1. Cancer Cell 2010, 10, 98. [Google Scholar] [CrossRef] [Green Version]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Sig. Transduct. Target Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol. 2009, 11, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Bahadur, S.; Sahu, A.K.; Baghel, P.; Saha, S. Current promising treatment strategy for glioblastoma multiform: A review. Oncol. Rev. 2019, 13, 417. [Google Scholar] [CrossRef] [Green Version]
- Fabian, D.; Del Pilar Guillermo Prieto Eibl, M.; Alnahhas, I.; Sebastian, N.; Giglio, P.; Puduvalli, V.; Gonzalez, J.; Palmer, J.D. Treatment of Glioblastoma (GBM) with the Addition of Tumor-Treating Fields (TTF): A Review. Cancers 2019, 11, 174. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Debinski, W. Receptor-Targeted Glial Brain Tumor Therapies. Int. J. Mol. Sci. 2018, 19, 3326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadavalli, S.; Yenugonda, V.M.; Kesari, S. Repurposed Drugs in Treating Glioblastoma Multiforme: Clinical Trials Update. Cancer J. 2019, 25, 139–146. [Google Scholar] [CrossRef]
- Tan, S.K.; Jermakowicz, A.; Mookhtiar, A.K.; Nemeroff, C.B.; Schürer, S.C.; Ayad, N.G. Drug Repositioning in Glioblastoma: A Pathway Perspective. Front. Pharmacol. 2018, 9, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pounds, R.; Leonard, S.; Dawson, C.; Kehoe, S. Repurposing itraconazole for the treatment of cancer. Oncol. Lett. 2017, 14, 2587–2597. [Google Scholar] [CrossRef] [Green Version]
- Thyparambil, S.P.; Liao, W.-L.; An, E.; Bhalkikar, A.; Heaton, R.; Sylvester, K.G.; Ling, X.B. Proteomic profiling to identify therapeutics targets in glioblastoma (GBM). J. Clin. Oncol. 2020, 38, 2555. [Google Scholar] [CrossRef]
- Lubanska, D.; Porter, L. Revisiting CDK Inhibitors for Treatment of Glioblastoma Multiforme. Drugs R D 2017, 17, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Breckenridge, A.; Jacob, R. Overcoming the legal and regulatory barriers to drug repurposing. Nat. Rev. Drug Discov. 2019, 18, 1–2. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Kato, S.; Moulder, S.L.; Ueno, N.T.; Wheler, J.J.; Meric-Bernstam, F.; Kurzrock, R.; Janku, F. Challenges and perspective of drug repurposing strategies in early phase clinical trials. Oncoscience 2015, 2, 576–580. [Google Scholar] [CrossRef]
- Femara [Package Insert]; Novartis Pharmaceuticals Corp.: East Hanover, NJ, USA, 2001.
- Connolly, R.M.; Stearns, V. Current approaches for neoadjuvant chemotherapy in breast cancer. Eur. J. Pharmacol. 2013, 717, 58–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Segura, L.M. Aromatase in the brain: Not just for reproduction anymore. J. Neuroendocr. 2008, 20, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Navarro Silvera, S.A.; Miler, A.B.; Rohan, T.E. Hormonal and reproductive factors and risk of glioma: A prospective cohort study. Int. J. Cancer 2006, 118, 1321–1324. [Google Scholar] [CrossRef] [PubMed]
- Duenas Jimenez, J.M.; Candanedi Arellano, A.; Santerre, A.; Orozco Suarez, S.; Sandoval Sanchez, H.; Feria Romero, I.; Lopez-Elizalde, R.; Venegas, M.A.; Netel, B.; de la Torre Valdovinos, B.; et al. Aromatase and estrogen receptor alpha mRNA expression as prognostic biomarkers in patients with astrocytomas. J. Nuerooncol. 2014, 119, 275–284. [Google Scholar] [CrossRef]
- Dave, N.; Chow, L.M.L.; Gudelsky, G.A.; LaSance, K.; Qi, X.; Desai, P.B. Preclinical pharmacological evaluation of letrozole as a novel treatment for gliomas. Mol. Cancer Ther. 2015, 14, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Dave, N.; Gudelsky, G.A.; Desai, P.B. The pharmacokinetics of letrozole in brain and brain tumor in rats with orthotopically implanted C6 glioma, assessed using intracerebral microdialysis. Cancer Chemother. Pharmacol. 2013, 72, 349–357. [Google Scholar] [CrossRef]
- Cohen, M.H.; Johnson, J.R.; Li, N.; Chen, G.; Pazdur, R. Approval summary: Letrozole in the treatment of postmenopausal women with advanced breast cancer. Clin. Cancer Res. 2002, 8, 665–669. [Google Scholar]
- Traut, T. Physiological concentrations of purines and pyrimidines. Mol. Cell. Biochem. 1994, 140, 1–22. [Google Scholar] [CrossRef]
- Kofuji, S.; Hirayama, A.; Eberhardt, A.O.; Kawaguchi, R.; Sugiura, Y.; Sampetrean, O.; Ikeda, Y.; Warren, M.; Naoya, S.; Kitahara, S.; et al. IMP dehydrogenase-2 drives aberrant nucleolar activity and promotes tumorigenesis in glioblastoma. Nat. Cell Biol. 2019, 21, 1003–1014. [Google Scholar] [CrossRef]
- Kofuji, S.; Sasaki, A.T. GTP Metabolic Reprogramming by IMPDH2: Unlocking Cancer Cells’ Fueling Mechanism. J. Biochem. 2020, 168, 319–328. [Google Scholar] [CrossRef]
- Rao, S.; Morris, R.; Rice, Z.P.; Arbiser, J.L. Regression of diffuse B-cell lymphoma of the leg with intralesional gentian violet. Exp. Derm. 2018, 27, 93–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majd, N.; Sumita, K.; Yoshino, H.; Chen, D.; Terakawa, J.; Daikoku, T.; Kofuji, S.; Curry, R.; Wise-Draper, T.M.; Warnick, R.E.; et al. A Review of the Potential Utility of Mycophenolate Mofetil as a Cancer Therapeutic. J. Cancer Res. 2014, 2014, 423401. [Google Scholar] [CrossRef] [Green Version]
- Naffouje, R.; Grover, P.; Yu, H.; Sendilnathan, A.; Wolfe, K.; Majd, N.; Smith, E.P.; Takeuchi, K.; Senda, T.; Kofuji, S.; et al. Anti-Tumor Potential of IMP Dehydrogenase Inhibitors: A Century-Long Story. Cancers 2019, 11, 1346. [Google Scholar] [CrossRef] [Green Version]
- Tressler, R.J.; Garvin, L.J.; Slate, D.L. Anti-tumor activity of mycophenolate mofetil against human and mouse tumors in vivo. Int. J. Cancer 1994, 57, 568–573. [Google Scholar] [CrossRef]
- Bacus, S.S.; Kiguchi, K.; Chin, D.; King, C.R.; Huberman, E. Differentiation of cultured human breast cancer cells (AU-565 and MCF-7) associated with loss of cell surface HER-2/neu antigen. Mol. Carcinog. 1990, 3, 350–362. [Google Scholar] [CrossRef]
- Floryk, D.; Huberman, E. Mycophenolic acid-induced replication arrest, differentiation markers and cell death of androgen-independent prostate cancer cells DU145. Cancer Lett. 2006, 231, 20–29. [Google Scholar] [CrossRef]
- Kiguchi, K.; Collart, F.R.; Henning-Chubb, C.; Huberman, E. Induction of cell differentiation in melanoma cells by inhibitors of IMP dehydrogenase: Altered patterns of IMP dehydrogenase expression and activity. Cell Growth Differ. 1990, 1, 259–270. [Google Scholar]
- Collart, F.R.; Huberman, E. Expression of IMP dehydrogenase in differentiating HL-60 cells. Blood 1990, 75, 570–576. [Google Scholar] [CrossRef] [Green Version]
- Messina, E.; Micheli, V.; Giacomello, A. Guanine nucleotide depletion induces differentiation and aberrant neurite outgrowth in human dopaminergic neuroblastoma lines: A model for basal ganglia dysfunction in Lesch-Nyhan disease. Neurosci. Lett. 2005, 375, 97–100. [Google Scholar] [CrossRef]
- Takebe, N.; Cheng, X.; Wu, S.; Bauer, K.; Goloubeva, O.G.; Fenton, R.G.; Heyman, M.; Rapoport, A.P.; Badros, A.; Shaughnessy, J.; et al. Phase I clinical trial of the inosine monophosphate dehydrogenase inhibitor mycophenolate mofetil (cellcept) in advanced multiple myeloma patients. Clin. Cancer Res. 2004, 10, 8301–8308. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, D.; Von Hoff, D.D. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2007, 6, 1186–1197. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci. 2017, 20, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Yao, Y.; Scott, A.J.; Wilder-Romans, K.; Dresser, J.J.; Werner, C.K.; Sun, H.; Pratt, P.; Sajjakulnuki, P.; Zhao, S.G.; et al. Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat. Commun. 2020, 11, 3811–3814. [Google Scholar] [CrossRef]
- Shireman, J.M.; Atashi, F.; Lee, G.; Ali, E.S.; Saathoff, M.R.; Park, C.H.; Baisiwala, S.; Miska, J.; Lesniak, M.S.; David, J.C.; et al. De-novo purine biosynthesis is a major driver of chemoresistance in glioblastoma. bioRXiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Murayi, R.; Chittiboina, P. Glucocorticoids in the management of peritumoral brain edema: A review of molecular mechanisms. Child’s Nerv. Syst. 2016, 32, 2293–2302. [Google Scholar] [CrossRef]
- Dietrich, J.; Rao, K.; Pastorino, S.; Kesari, S. Corticosteroids in brain cancer patients: Benefits and pitfalls. Expert Rev. Clin. Pharmacol. 2014, 4, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Stamatovic, S.M.; Dimitrijevic, O.B.; Keep, R.F.; Andjelkovic, A.V. Inflammation and brain edema: New insights into the role of chemokines and their receptors. Acta Neurochir. Suppl. 2006, 96, 444–450. [Google Scholar] [CrossRef]
- Abbott, N.J. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell. Mol. Neurobiol. 2000, 20, 131–147. [Google Scholar] [CrossRef]
- Kermode, A.G.; Thompson, A.J.; Tofts, P.; MacManus, D.G. Breakdown of the blood-brain barrier precedes symptoms and other MRI signs of new lesions in multiple sclerosis. Brain 1990, 113, 1477–1489. [Google Scholar] [CrossRef]
- Brightman, M.W.; Klatzo, I.; Olsson, Y.; Reese, T.S. The blood-brain barrier to proteins under normal and pathological conditions. J. Neurol. Sci. 1970, 10, 215–239. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Saadoun, S.; Binder, D.K.; Manley, G.T.; Krishna, S.; Verkman, A.S. Molecular mechanisms of brain tumor edema. NSC 2004, 129, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W. Mechanisms of tumor-related brain edema. Neurosurg. Focus 2007, 22, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Xue, Y.; Duan, Q.; Sun, B.; Lin, H.; Huang, X.; Chen, X. The role of cerebral blood flow gradient in peritumoral edema for differentiation of glioblastomas from solitary metastatic lesions. Oncotarget 2016, 7, 69051–69059. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.-X.; Lin, G.-S.; Lin, Z.-X.; Zhang, J.-D.; Liu, S.-Y.; Zhou, C.-F. Peritumoral edema shown by MRI predicts poor clinical outcome in glioblastoma. World J. Surg. Oncol. 2015, 13, 97–99. [Google Scholar] [CrossRef] [Green Version]
- Schiff, D.; Lee, E.Q.; Nayak, L.; Norden, A.D.; Reardon, D.A.; Wen, P.Y. Medical management of brain tumors and the sequelae of treatment. Neuro Oncol. 2015, 17, 488–504. [Google Scholar] [CrossRef] [Green Version]
- Arvold, N.D.; Armstrong, T.S.; Warren, K.E.; Chang, S.M.; DeAngelis, L.M.; Blakeley, J.; Chamberlain, M.C.; Dunbar, E.; Loong, H.H.; Macdonald, D.R.; et al. Corticosteroid use endpoints in neuro-oncology: Response Assessment in Neuro-Oncology Working Group. Neuro Oncol. 2018, 20, 897–906. [Google Scholar] [CrossRef]
- Pitter, K.L.; Tamagno, I.; Alikhanyan, K.; Hosni-Ahmed, A.; Pattwell, S.S.; Donnola, S.; Dai, C.; Ozawa, T.; Chang, M.; Chan, T.A.; et al. Corticosteroids compromise survival in glioblastoma. Brain 2016, 139, 1458–1471. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Chinot, O.L.; Wick, W.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Carpentier, A.F.; Hoang-Xuan, K.; Kavan, P.; Cernea, D.; et al. Bevacizumab plus Radiotherapy–Temozolomide for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 709–722. [Google Scholar] [CrossRef] [Green Version]
- Wick, W.; Chinot, O.L.; Bendszus, M.; Mason, W.; Henriksson, R.; Saran, F.; Nishikawa, R.; Revil, C.; Kerloeguen, Y.; Cloughesy, T. Evaluation of pseudoprogression rates and tumor progression patterns in a phase III trial of bevacizumab plus radiotherapy/temozolomide for newly diagnosed glioblastoma. Neuro Oncol. 2016, 18, 1434–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, G.M.; Sahin, S.; Kantarci, G.; Ergin, H. Mycophenolate mofetil treatment for therapy-resistant glomerulopathies. Nephrology 2007, 12, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.; Appel, G.; Dooley, M.A.; Ginzler, E.; Isenberg, D.; Jayne, D.; Wofsy, D.; Solomons, N. Mycophenolate mofetil as induction and maintenance therapy for lupus nephritis: Rationale and protocol for the randomized, controlled Aspreva Lupus Management Study (ALMS). Lupus 2007, 16, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Heatwole, C.; Ciafaloni, E. Mycophenolate mofetil for myasthenia gravis: A clear and present controversy. Neuropsychiatr. Dis. Treat. 2008, 4, 1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orvis, A.K.; Wesson, S.K.; Breza Jr, T.S.; Church, A.A.; Mitchell, C.L.; Watkins, S.W. Mycophenolate mofetil in dermatology. J. Am. Dermatol. 2009, 60, 183–199. [Google Scholar] [CrossRef]
- Aggarwal, R.; Oddis, C.V. Therapeutic Approaches in Myositis. Curr. Rheumatol. Rep. 2011, 13, 182–191. [Google Scholar] [CrossRef]
- Rodriguez-Pascual, J.; Sha, P.; Garcia-Garcia, E.; Rajeshkumar, N.V.; De Vicente, E.; Quijano, Y.; Cubillo, A.; Angulo, B.; Hernando, O.; Hidalgo, M. A preclinical and clinical study of mycophenolate mofetil in pancreatic cancer. Investig. New Drugs 2013, 31, 14–19. [Google Scholar] [CrossRef]
- Hur, E.; Bozkurt, D.; Timur, O.; Bicak, S.; Sarsik, B.; Akcicek, F.; Duman, S. The effects of mycophenolate mofetil on encapsulated peritoneal sclerosis model in rats. Clin. Nephrol. 2012, 77, 1–7. [Google Scholar] [CrossRef]
- Cherikh, W.S.; Kauffman, H.M.; McBride, M.A.; Maghirang, J.; Swinnen, L.J.; Hanto, D.W. Association of the type of induction immunosuppression with posttransplant lymphoproliferative disorder, graft survival, and patient survival after primary kidney transplantation. Transplantation 2003, 76, 1289–1293. [Google Scholar] [CrossRef]
- Végso, G.; Sebestyén, A.; Paku, S.; Barna, G.; Hajdu, M.; Tóth, M.; Járay, J.; Kopper, L. Antiproliferative and apoptotic effects of mycophenolic acid in human B-cell non-Hodgkin lymphomas. Leuk. Res. 2007, 31, 1003–1008. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wedl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Arman Aksoy, B.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Onur Sumer, S.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Onur Sumer, S.; Arman Aksoy, B.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.M.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.-F.; et al. Randomized phase II trial of erlotinib versus temozolomide or Carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. 2009, 10, 1268–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Wen, P.Y.; Mellinghoff, I.K. Targeted molecular therapies against epidermal growth factor receptor: Past experiences and challenges. Neuro Oncol. 2014, 8, viii7–viii13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaoka, T.; Kusumoto, S.; Ando, K.; Ohba, M.; Ohmori, T. Receptor Tyrosine Kinase-Targeted Cancer Therapy. Int. J. Mol. Sci. 2018, 19, 3491. [Google Scholar] [CrossRef] [Green Version]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Drappatz, J.; de Groot, J.; Prados, M.D.; Reardon, D.A.; Schiff, D.; Chamberlain, M.; Mikkelsen, T.; Desjardins, A.; Ping, J.; et al. Phase II study of cabozantinib in patients with progressive glioblastoma: Subset analysis of patients with prior antiangiogenic therapy. Neuro Oncol. 2018, 20, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Feng, X.; Ennis, K.N.; Behrmann, C.A.; Sarma, P.; Jiang, T.T.; Kofuji, S.; Niu, L.; Stratton, Y.; Thomas, H.E.; et al. Pharmacologic Targeting of S6K1 in PTEN-Deficient Neoplasia. Cell Rep. 2017, 18, 2088–2095. [Google Scholar] [CrossRef]
- Schroeder, G.M.; An, Y.; Cai, Z.W.; Chen, X.T.; Clark, C.; Cornelius, L.A.; Dai, J.; Gullo-Brown, J.; Gupta, A.; Henley, B.; et al. Discovery of N-(4-(2-amino-3-chloropyridin-4-yloxy)-3-fluorophenyl)-4-ethoxy-1-(4-fluorophenyl)-2-oxo-1,2-dihydropyridine-3-carboxamide (BMS-777607), a selective and orally efficacious inhibitor of the Met kinase superfamily. J. Med. Chem. 2009, 52, 1251–1254. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Mu, Q.; Bao, Z.; Chen, Y.; Liu, Y.; Chen, J.; Wang, K.; Wang, Z.; Nam, Y.; Jiang, B.; et al. Mutational Landscape of Secondary Glioblastoma Guides MET-Targeted Trial in Brain Tumor. Cell 2018, 175, 1665–1678.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maley, A.M.; Arbiser, J.L. Gentian Violet: A 19th Century Drug Re-Emerges in the 21st Century. Exp. Derm. 2013, 22, 775–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munson, J.M.; Fried, L.; Rowson, S.A.; Bonner, M.Y.; Karumbaiah, L.; Diaz, B.; Courtneidge, S.A.; Knaus, U.G.; Brat, D.J.; Arbiser, J.L.; et al. Anti-invasive adjuvant therapy with imipramine blue enhances chemotherapeutic efficacy against glioma. Sci. Transl. Med. 2012, 4, 127ra36. [Google Scholar] [CrossRef] [PubMed]
- Rajamanickam, S.; Panneerdoss, S.; Gorthi, A.; Timilsina, S.; Onyeagucha, B.; Kovalsky, D.; Ivanov, D.; Hanes, M.A.; Vadlamudi, R.K.; Chen, Y.; et al. Inhibition of FoxM1-Mediated DNA Repair by Imipramine Blue Suppresses Breast Cancer Growth and Metastasis. Clin. Cancer Res. 2016, 22, 3524–3536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metts, J.; Bradley, H.L.; Wang, Z.; Shah, N.P.; Kapur, R.; Arbiser, J.L.; Bunting, K.D. Imipramine blue sensitively and selectively targets FLT3-ITD positive acute myeloid leukemia cells. Sci. Rep. 2017, 7, 4447. [Google Scholar] [CrossRef] [PubMed]
- Laidlaw, K.M.E.; Berhan, S.; Liu, S.; Silvestri, G.; Holyoake, T.L.; Frank, D.A.; Aggarwal, B.; Bonner, M.Y.; Perrotti, D.; Jorgensen, H.G.; et al. Cooperation of imipramine blue and tyrosine kinase blockade demonstrates activity against chronic myeloid leukemia. Oncotarget 2016, 7, 51651–51664. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.H.; Su, Y.H.; Hsu, W.H.; Wang, C.C.; Arbiser, J.L.; Yang, M.H. Imipramine blue halts head and neck cancer invasion through promoting F-box and leucine-rich repeat protein 14-mediated Twist1 degradation. Oncogene 2016, 35, 2287–2298. [Google Scholar] [CrossRef] [Green Version]
- Ammar, D.A.; Kahook, M.Y. In vitro effects of vertepofin on ocular cells. Mol. Vis. 2013, 19, 424–429. [Google Scholar]
- Zheng, Y.; Pan, D. The Hippo Signaling Pathway in Development and Disease. Dev. Cell 2019, 50, 264–282. [Google Scholar] [CrossRef]
- Bhat, K.P.L.; Salazar, K.L.; Balasubramaniyan, V.; Wani, K.; Heathcock, L.; Hollingsworth, F.; James, J.D.; Gumin, J.; Diefes, K.L.; Kim, S.H.; et al. The transcriptional coactivator TAZ regulates mesenchymal differentiation in malignant glioma. Genes Dev. 2011, 25, 2594–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, B.A.; Bai, H.; Odia, Y.; Jain, D.; Anders, R.A.; Eberhart, C.G. Yes-associated protein 1 is widely expressed in human brain tumors and promotes glioblastoma growth. J. Neuropathol. Exp. Neurol. 2011, 70, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.; Bardet, A.F.; Roma, G.; Bergling, S.; Clay, I.; Ruchti, A.; Agarinis, C.; Schmelzle, T.; Bouwmeester, T.; Schubeler, D.; et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet 2015, 11, e1005465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eales, K.L.; Wilkinson, E.A.; Cruickshank, G.; Tucker, J.H.R.; Tennant, D.A. Verteporfin selectively kills hypoxic glioma cells through iron-binding and increased production of reactive oxygen species. Sci. Rep. 2018, 8, 14358. [Google Scholar] [CrossRef]
- Al-Moujahed, A.; Brodowska, K.; Stryjewski, T.P.; Efstathiou, N.E.; Vasilikos, I.; Cichy, J.; Miller, J.W.; Gragoudas, E.; Vavvas, D.G. Verteporfin inhibits growth of human glioma in vitro without light activation. Sci. Rep. 2017, 7, 7602. [Google Scholar] [CrossRef]
- Zhang, H.; Ramakrishnan, S.K.; Triner, D.; Centofanti, B.; Maitra, D.; Gyorffy, B.; Sebolt-Leopold, J.S.; Dame, M.K.; Varani, J.; Brenner, D.E.; et al. Tumor-selective proteotoxicity of verteporfin inhibits colon cancer progression independently of YAP1. Sci. Signal. 2015, 8, ra98. [Google Scholar] [CrossRef] [Green Version]
- Donohue, E.; Tovey, A.; Vogl, A.W.; Arns, S.; Sternberg, E.; Young, R.N.; Roberge, M. Inhibition of autophagosome formation by the benzoporphyrin derivative verteporfin. J. Biol. Chem. 2011, 286, 7290–7300. [Google Scholar] [CrossRef] [Green Version]
- Dasari, V.R.; Mazack, V.; Feng, W.; Nash, J.; Carey, D.J.; Gogoi, R. Verteporfin exhbits YAP-independent anti-proliferative and cytoxic effects in endometrial cancer cells. Oncotarget 2017, 8, 28628–28640. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.-W.; Pan, J.-W. Verteporfin induces apoptosis and eliminates cancer stem-like cells in uveal melanoma in the absence of light activation. Am. J. Cancer Res. 2016, 6, 2816–2830. [Google Scholar]
- Yin, L.; Chen, G. Verteporfin Promotes the Apoptosis and Inhibits the Proliferation, Migration, and Invasion of Cervical Cancer Cells by Downregulating SULT2B1 Expression. Med. Sci. Monit. 2020, 26, e926780-1–e926780-11. [Google Scholar] [CrossRef]
- Wei, C.; Li, X. Verteporfin inhibits cell proliferation and induces apoptosis in different subtypes of breast cancer cell lines without light activation. BMC Cancer 2020, 20, 1042. [Google Scholar] [CrossRef]
- Hill, J.S.; Kahl, S.B.; Kaye, A.H.; Stylli, S.S.; Koo, M.S.; Gonzales, M.F.; Vardaxis, N.J.; Johnson, C.I. Selective tumor uptake of a boronated porphyrin in an animal model of cerebral glioma. Proc. Natl. Acad. Sci. USA 1992, 89, 1785–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, J.S.; Kaye, A.H.; Sawyer, W.H.; Morstyn, G.; Megison, P.D.; Stylli, S.S. Selective uptake of hematoporphyrin derivative into human cerebral glioma. Neurosurgery 1990, 26, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Stummer, W.; Pichlmeier, U.; Meinel, T.; Wiestler, O.D.; Zanella, F.; Reulen, H.J.; ALA-Glioma Study Group. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: A randomised controlled multicentre phase III trial. Lancet Oncol. 2006, 7, 392–401. [Google Scholar] [CrossRef]
- Vigneswaran, K.; Oh, S.Y.; Zhang, Z.; Olsen, J.; Read, R. Characterizing the over-expression of Yki/YAP/TAZ transcription factors in gliomagenesis and a proposed novel treatment of glioblastoma. Neuro Oncol. 2017, 19, vi56. [Google Scholar] [CrossRef] [Green Version]
- Pellosi, D.S.; Paula, L.B.; de Melo, M.T.; Tedesco, A.C. Targeted and Synergic Glioblastoma Treatment: Multifunctional Nanoparticles Delivering Verteporfin as Adjuvant Therapy for Temozolomide Chemotherapy. Mol. Pharm. 2019, 16, 1009–1024. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.R.; Kim, J.; Schiapparelli, P.; Vazquez-Ramos, C.A.; Martinez-Gutierrez, J.C.; Ruiz-Valls, A.; Inman, K.; Shamul, J.G.; Green, J.J.; Quinones-Hinojosa, A. Verteporfin-Loaded Polymeric Microparticles for Intratumoral Treatment of Brain Cancer. Mol. Pharm. 2019, 16, 1433–1443. [Google Scholar] [CrossRef]
- Qi, X.; Leonova, T.; Grabowski, G.A. Functional human saposins expressed in Escherichia coli (evidence for binding and activation properties of saposins C with acid β-glucosidase. J. Biol. Chem. 1994, 269, 16746–16753. [Google Scholar] [CrossRef]
- Davis, H.W.; Hussain, N.; Qi, X. Detection of cancer cells using SapC-DOPS nanovesicles. Mol. Cancer 2016, 10, 33. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Grabowski, G.A. Acid β-glucosidase: Intrinsic fluorescence and conformational changes induced by phospholipids and saposin C. Biochemistry 1998, 31, 11544–11554. [Google Scholar] [CrossRef]
- Qi, X.; Qin, W.; Sun, Y.; Kondoh, K.; Grabowski, G.A. Functional organization of saposin C: Definition of the neurotrophic and acid β-glucosidase activation regions. J. Biol. Chem. 1996, 271, 6874–6880. [Google Scholar] [CrossRef] [Green Version]
- Parks, S.K.; Pouyssegur, J. Targeting pH regulating proteins for cancer therapy-Progress and limitations. Semin. Cancer Biol. 2017, 43, 66–73. [Google Scholar] [CrossRef]
- Qi, X.; Chu, Z.; Mahller, Y.Y.; Stringer, K.F.; Witte, D.P.; Cripe, T.P. Cancer-selective targeting and cytotoxicity by liposomal-couples lysosomal saposin C protein. Clin. Cancer Res. 2009, 15, 5840–5851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Liou, B.; Chu, Z.; Fannin, V.; Blackwood, R.; Peng, Y.; Grabowski, G.A.; Davis, H.W.; Qi, X. Systemic Enzyme Delivery by Blood-Brain Barrier-Penetrating SapC-DOPS Nanovesicles for Treatment for Neuronopathic Gaucher Disease. EBioMedicine 2020, 55, 102735. [Google Scholar] [CrossRef] [PubMed]
- N’Guessan, K.F.; Patel, P.H.; Qi, X. SapC-DOPS—A phosphatidylserine-targeted nanovesicle for selective cancer therapy. Cell Commun. Signal. 2020, 18, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vallabhapurapu, S.D.; Blanco, V.M.; Sulaiman, M.K.; Vallabhapurapu, S.L.; Chu, Z.; Franco, R.S.; Qi, X. Variation in human cancer cell external phosphatidylserine is regulated by flippase activity and intracellular calcium. Oncotarget 2015, 6, 34375–34388. [Google Scholar] [CrossRef] [Green Version]
- Chu, Z.; Abu-Baker, S.; Palascak, M.B.; Ahmad, S.A.; Franco, R.S.; Qi, X. Targeting cytotoxicity of SapC-DOPS nanovesicles in pancreatic cancer. PLoS ONE 2013, 8, e75507. [Google Scholar] [CrossRef] [Green Version]
- Sulaiman, M.K.; Chu, Z.; Blanco, V.M.; Vallabhapurapu, S.D.; Franco, R.S.; Qi, X. SapC-DOPS nanovesicles induce Smac- and Bax-dependent apotosis through mitochondrial activation in neuroblastomas. Mol. Cancer 2015, 14, 78. [Google Scholar] [CrossRef] [Green Version]
- Wojton, J.; Meisen, W.H.; Jacob, N.K.; Thorne, A.H.; Hardcastle, J.; Denton, N.; Chu, Z.; Dmitrieva, N.; Marsh, R.; Van Meir, E.G.; et al. SapC-DOPS-induced lysosomal cell death synergizes with TMZ in glioblastoma. Oncotarget 2014, 5, 9703–9709. [Google Scholar] [CrossRef] [Green Version]
- Wojton, J.; Chu, Z.; Mathsyaraja, H.; Meisen, W.H.; Denton, N.; Kwon, C.H.; Chow, L.M.L.; Palascak, M.; Franco, R.; Bourdeau, T.; et al. Systemic delivery of SapC-DOPS has antiangiogenic and antitumor effects against glioblastoma. Mol. Ther. 2013, 21, 1517–1525. [Google Scholar] [CrossRef] [Green Version]
- Winter, P.M.; Pearce, J.; Chu, Z.; McPherson, C.M.; Takigiku, R.; Lee, J.H.; Qi, X. Imaging of brain tumors with paramagnetic vesicles targeted to phosphatidylserine. J. Magn. Res. Imaging 2015, 41, 1079–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bexion Pharmaceuticals. Available online: https://www.bexionpharma.com (accessed on 31 October 2020).


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koehler, A.; Karve, A.; Desai, P.; Arbiser, J.; Plas, D.R.; Qi, X.; Read, R.D.; Sasaki, A.T.; Gawali, V.S.; Toukam, D.K.; et al. Reuse of Molecules for Glioblastoma Therapy. Pharmaceuticals 2021, 14, 99. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020099
Koehler A, Karve A, Desai P, Arbiser J, Plas DR, Qi X, Read RD, Sasaki AT, Gawali VS, Toukam DK, et al. Reuse of Molecules for Glioblastoma Therapy. Pharmaceuticals. 2021; 14(2):99. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020099
Chicago/Turabian StyleKoehler, Abigail, Aniruddha Karve, Pankaj Desai, Jack Arbiser, David R. Plas, Xiaoyang Qi, Renee D. Read, Atsuo T. Sasaki, Vaibhavkumar S. Gawali, Donatien K. Toukam, and et al. 2021. "Reuse of Molecules for Glioblastoma Therapy" Pharmaceuticals 14, no. 2: 99. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14020099