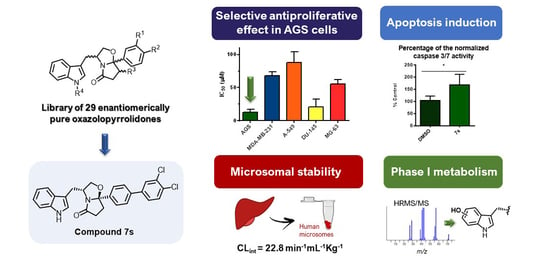
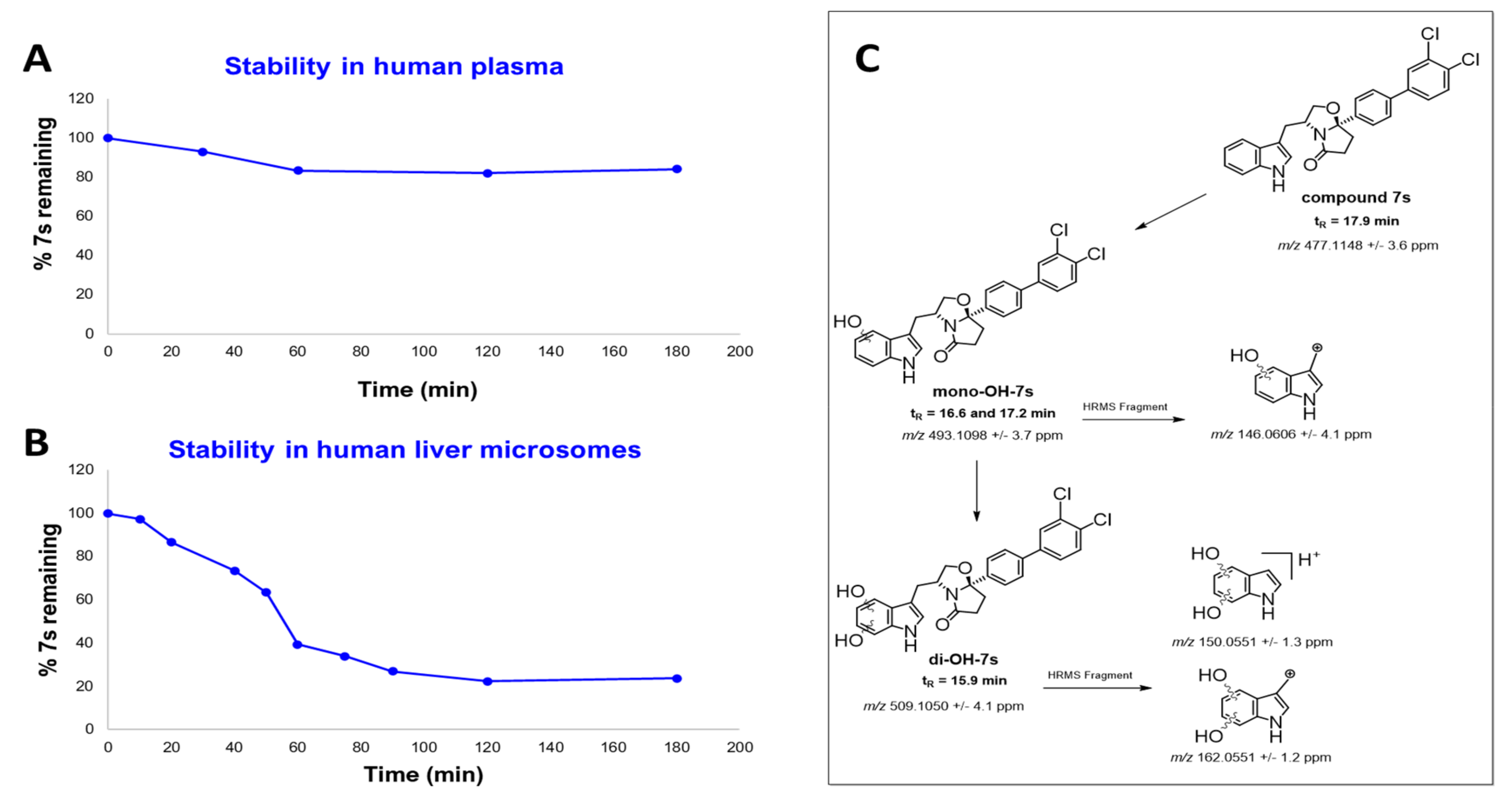
3.1. Chemistry
General information: THF was dried using sodium wire and benzophenone as indicator. (
R)-Tryptophanol was obtained by reduction of (
R)-tryptophan using lithium aluminum hydride [
28]. Other reagents were obtained from commercial suppliers (Sigma-Aldrich, Alfa Aesar, and Fluorochem). General information concerning the equipment used for the elucidation of the products’ chemical structures and product characterization (NMR, melting point, optical rotations, MS, and elemental analysis) are presented in our earlier publication [
21]. Multiplicities in
1H NMR spectra are given as: s (singlet), d (doublet), dd (double doublet), ddd (doublet of doublets of doublets), t (triplet), and m (multiplet). Compounds
7h,
7j, and
7j’ showed purity ≥ 95% by LC-MS, performed in a LaChrom HPLC constituted of a Merck Hitachi pump L-7100, Merck Hitachi autosampler L-7250, and a Merck Hitachi UV detector L-7400. Analyses were performed with a LiChrospher
®100 RP-8 (5 µm) LiChroCART
® 250-4 column at room temperature, using a mobile phase solution constituted of 65% acetonitrile and 35% Milli-Q water. Peaks were detected at λ = 254 nm.
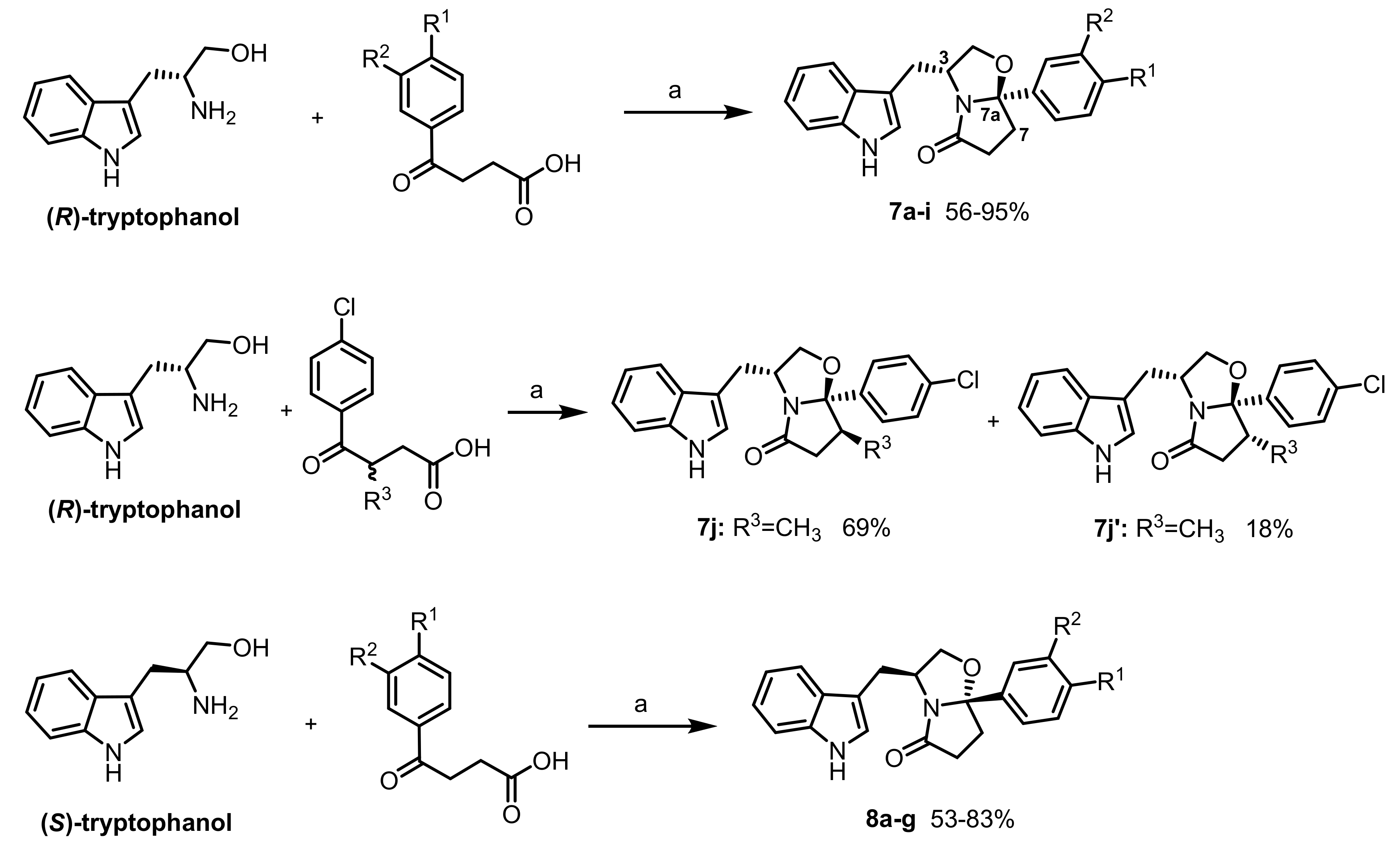
General procedure for the synthesis of compounds 7a–j, 7j’, and 8a–g: To a suspension of enantiopure tryptophanol (0.53 mmol, 1.0 eq.) in toluene (5 mL) was added the appropriate oxocarboxylic acid (0.58 mmol, 1.1 eq.). The mixture was heated at reflux for 10–25 h in a Dean–Stark apparatus. The reaction mixture was concentrated in vacuo and the residue obtained was dissolved in EtOAc (10 mL). The organic phase was washed with saturated aqueous solution of NaHCO3 (15 mL) and brine (15 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica gel flash chromatography using a mixture of EtOAc/n-hexane as eluent.
(3
R,7a
R)-3-((1
H-indol-3-yl)methyl)-7a-phenyltetrahydropyrrolo[2,1-
b]oxazol-5(6
H)-one (
7a): Following the general procedure, to a solution of (
R)-tryptophanol (0.102 g, 0.536 mmol) in toluene (5 mL) was added 3-benzoyl propionic acid (0.105 g, 0.590 mmol). Reaction time: 19 h. The compound was purified by flash chromatography (EtOAc/
n-hexane 1:1) and recrystallized from EtOAc/
n-hexane to give pale pink crystalline solid (0.166 g, 95%);
= −54.7° (
c = 2.0, MeOH);
1H NMR spectra was found to be identical to the one reported [
15] and obtained for compound 8a. Anal. Calcd. for C
21H
20N
2O
2·0.05H
2O: C, 75.67%; H, 6.09%; N, 8.41%. Found C: 75.22%; H: 5.87%; N: 8.23%. The m.p. value was found to be identical to the one reported for compound
8a.
(3
S,7a
S)-3-((1
H-indol-3-yl)methyl)-7a-phenyltetrahydropyrrolo[2,1-
b]oxazol-5(6
H)-one (
8a): Following the general procedure, to a solution of (
S)-tryptophanol (0.101 g, 0.529 mmol) in toluene (5 mL) was added 3-benzoyl propionic acid (0.104 g, 0.582 mmol). Reaction time: 24 h. The compound was purified by flash chromatography (EtOAc/
n-hexane 1:1) and recrystallized from EtOAc/
n-hexane to give pale pink crystalline solid (0.127 g, 72%);
= +40.4° (
c = 2.0, MeOH); m.p.: 153–156 °C;
1H NMR (300 MHz, CDCl
3) δ 7.99 (s, 1H, NH), 7.50 (d,
J = 6.0 Hz, 2H, ArH), 7.46–7.29 (m, 5H, ArH), 7.17 (t,
J = 7.5 Hz, 1H, ArH), 7.10–7.05 (m, 2H, ArH), 4.62–4.52 (m, 1H, H-3), 4.16 (t,
J = 8.1 Hz, 1H, H-2), 3.63–3.58 (m, 1H, H-2), 3.07 (dd,
J = 14.3, 6.2 Hz, 1H, indole-CH
2), 2.96–2.75 (m, 1H, CH
2), 2.68–2.35 (m, 3H, CH
2, and indole-CH
2), 2.34–2.14 (m, 1H, CH
2) ppm; Anal. Calcd. for C
21H
20N
2O
2: C: 75.88%; H: 6.06%; N: 8.43%. Found C: 75.95%; H: 5.76%; N: 8.36%.
1H NMR spectra was found to be identical to the one reported [
15].
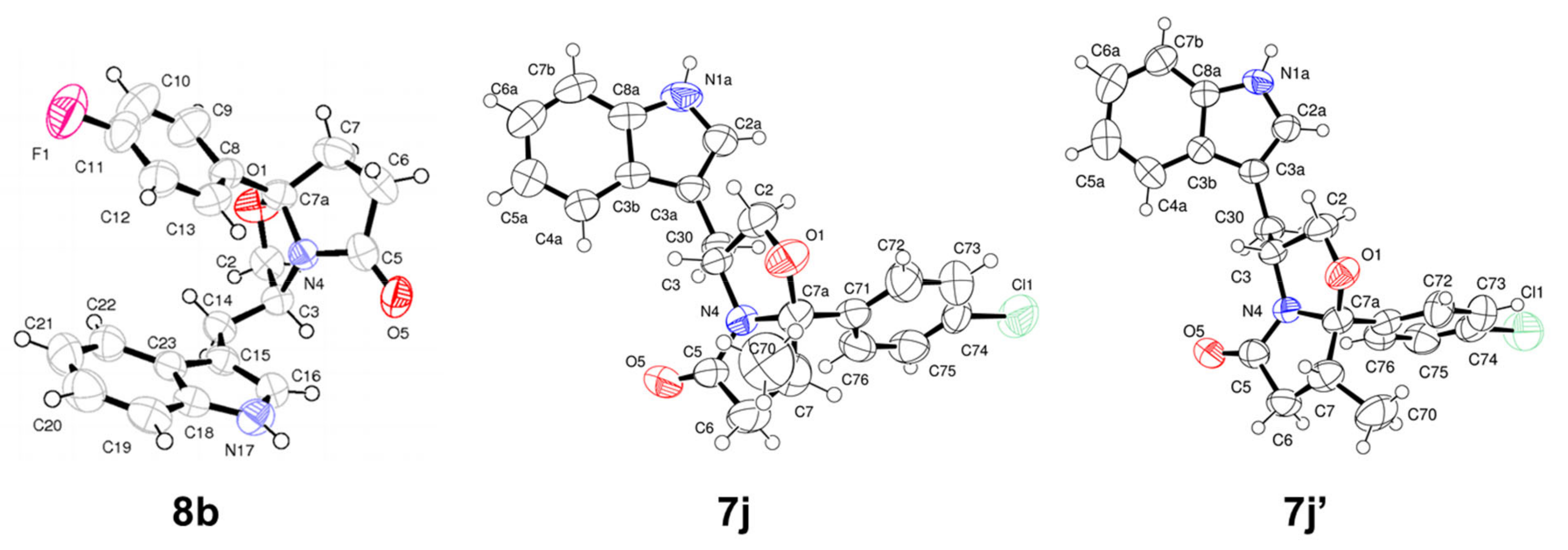
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(4-fluorophenyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7b): Following the general procedure, to a solution of (R)-tryptophanol (0.100 g, 0.526 mmol) in toluene (5 mL) was added 3-(4-fluorobenzoyl) propionic acid (0.114 g, 0.581 mmol). Reaction time: 19 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:1) and recrystallized from EtOAc/n-hexane to give pale yellow crystalline solid (0.113 g, 70%); = −48.8° (c = 2.0, MeOH); 1H NMR was found to be identical to the one obtained for compound 8b. Anal. Calcd. for C21H19FN2O2: C: 71.98%; H: 5.47%; N: 8.00%. Found C: 72.09%; H: 5.49%; N: 7.94%. The m.p. value was found to be identical to the one reported for compound 8b.
(3S,7aS)-3-((1H-indol-3-yl)methyl)-7a-(4-fluorophenyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (8b): Following the general procedure, to a solution of (S)-tryptophanol (0.102 g, 0.535 mmol) in toluene (5 mL) was added 3-(4-fluorobenzoyl) propionic acid (0.115 g, 0.588 mmol). Reaction time: 21 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:1) and recrystallized from EtOAc/n-hexane to give orange crystalline solid (0.156 g, 83%); = +39.5° (c = 2.0, MeOH); m.p.: 197-198 °C; 1H NMR (300 MHz, CDCl3) δ 8.00 (s, 1H, NH), 7.51-7.41 (m, 3H, ArH), 7.33 (d, J = 8.1 Hz, 1H, ArH), 7.21–7.15 (m, 1H, ArH), 7.11–7.03 (m, 4H, ArH), 4.62–4.53 (m, 1H, H-3), 4.17 (dd, J = 8.8 Hz, 7.4 Hz, 1H, H-2), 3.59 (dd, J = 8.8 Hz, 6.9Hz, 1H, H-2), 3.05 (dd, J = 14.7 Hz, 6.2Hz, 1H, indole-CH2), 2.90–2.78 (m, 1H, CH2), 2.65–2.43 (m, 3H, CH2, and indole-CH2), 2.24 − 2.15 (m, 1H, CH2) ppm; 13C NMR (75 MHz, CDCl3) δ 180.3 (C=O), 162.8 (d, Cq, JC-F = 245.3 Hz), 138.8 (d, Cq, J = 3.1 Hz), 136.3 (Cq), 127.5 (Cq), 126.9 (d, ArCH, J = 8.1 Hz), 122.3 (ArCH), 122.2 (ArCH), 119.6 (ArCH), 118.9 (ArCH), 115.5 (d, ArCH, J = 21.5 Hz), 111.6 (Cq), 111.3 (ArCH), 102.2 (C-7a), 72.8 (C-2), 55.8 (C-3), 35.2 (CH2), 32.7 (CH2), 29.8 (indole-CH2). Anal. Calcd. for C21H19FN2O2: C: 71.98%; H: 5.47%; N: 8.00%. Found C: 72.48%; H: 5.37%; N: 8.03%.
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(4-chlorophenyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7c): Following the general procedure, to a solution of (R)-tryptophanol (0.103 g, 0.541 mmol) in toluene (5 mL) was added 3-(4-chlorobenzoyl) propionic acid (0.127 g, 0.596 mmol). Reaction time: 18 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:1) and recrystallized from EtOAc/n-hexane to give pale yellow crystalline solid (0.133 g, 67%); = −63.1° (c = 2.0, MeOH); 1H NMR was found to be identical to the one obtained for compound 8c. Anal. Calcd. for C21H19ClN2O2: C: 68.76%; H: 5.18%; N: 7.62%. Found C: 68.76%; H: 5.22%; N: 7.64%. The m.p. value was found to be identical to the one reported for compound 8c.
(3S,7aS)-3-((1H-indol-3-yl)methyl)-7a-(4-chlorophenyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (8c): Following the general procedure, to a solution of (S)-tryptophanol (0.104 g, 0.545 mmol) in toluene (5 mL) was added 3-(4-chlorobenzoyl) propionic acid (0.128 g, 0.600 mmol). Reaction time: 23 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1/1) and recrystallized from EtOAc/n-hexane to give pale pink crystalline solid (0.164 g, 82%); = +54.5° (c = 2.0, MeOH); m.p.: 206–208 °C; 1H NMR (300 MHz, CDCl3) δ 7.98 (s, 1H, NH), 7.47–7.32 (m, 6H, ArH), 7.21–7.16 (m, 1H, ArH), 7.12–7.06 (m, 2H, ArH), 4.62–4.52 (m, 1H, H-3), 4.17 (dd, J = 8.8 Hz, 7.5 Hz, 1H, H-2), 3.59 (dd, J = 8.8 Hz, 6.9 Hz, 1H, H-2), 3.05 (dd, J = 15.1 Hz, 7.5 Hz, 1H, indole-CH2), 2.89–2.78 (m, 1H, CH2), 2.65–2.44 (m, 3H, CH2, and indole-CH2), 2.22–2.14 (m, 1H, CH2) ppm; 13C NMR (75 MHz, CDCl3) δ 180.3 (C=O), 141.5 (Cq), 136.3 (Cq), 134.3 (Cq), 129.0 (ArCH), 127.4 (Cq), 126.7 (ArCH), 122.3 (ArCH), 122.2 (ArCH), 119.5 (ArCH), 118.8 (ArCH), 111.4 (Cq), 111.3 (ArCH), 102.1 (C-7a), 72.9 (C-2), 55.8 (C-3), 35.1 (CH2), 32.6 (CH2), 29.8 (indole-CH2). Anal. Calcd. for C21H19ClN2O2: C: 68.76%; H: 5.22%; N: 7.62%. Found C: 68.94%; H: 5.06%; N: 7.60%.
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(4-bromophenyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7d): Following the general procedure, to a solution of (R)-tryptophanol (0.102 g, 0.536 mmol) in toluene (5 mL) was added 3-(4-bromobenzoyl) propionic acid (0.153 g, 0.590 mmol). Reaction time: 18 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:1) and recrystallized from EtOAc/n-hexane to give pale pink crystalline solid (0.182 g, 83%); = −53.6° (c = 2.0, MeOH); 1H NMR was found to be identical to the one obtained for compound 8d. Anal. Calcd. for C21H19BrN2O2·0.15H2O: C: 60.92%; H: 4.71%; N: 6.77%. Found C: 60.47%; H: 4.55%; N: 6.55%. The m.p. value was found to be identical to the one reported for compound 8d.
(3S,7aS)-3-((1H-indol-3-yl)methyl)-7a-(4-bromophenyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (8d): Following the general procedure, to a solution of (S)-tryptophanol (0.102 g, 0.536 mmol) in toluene (5 mL) was added 3-(4-bromobenzoyl) propionic acid (0.151 g, 0.589 mmol). Reaction time: 18 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:1) and recrystallized from EtOAc/n-hexane to give pale yellow crystalline solid (0.159 g, 72%); = +52.3° (c = 2.0, MeOH); m.p.: 207-210 °C. 1H NMR (300 MHz, CDCl3) δ 7.97 (s, 1H, NH), 7.52–7.45 (m, 3H, ArH), 7.37–7.32 (m, 3H, ArH), 7.21–7.05 (m, 3H, ArH), 4.62–4.52 (m, 1H, H-3), 4.17 (dd, J = 8.8 Hz, 7.4 Hz, 1H, H-2), 3.59 (dd, J = 8.8 Hz, 6.9 Hz, 1H, H-2), 3.05 (dd, J = 14.7 Hz, 6.1 Hz, 1H, indole-CH2), 2.89–2.78 (m, 1H, CH2), 2.65–2.44 (m, 3H, CH2, and indole-CH2), 2.22–2.14 (m, 1H, CH2) ppm; 13C NMR (75 MHz, CDCl3) δ 180.3 (C=O), 142.1 (Cq), 136.3 (Cq), 131.8 (ArCH), 127.5 (Cq), 127.1 (ArCH), 122.5 (Cq), 122.2 (ArCH), 122.1 (ArCH), 119.7 (ArCH), 118.9 (ArCH), 111.6 (Cq), 111.3 (ArCH), 102.1 (C-7a), 72.9 (C-2), 55.8 (C-3), 35.1 (CH2), 32.7 (CH2), 29.8 (indole-CH2). Anal. Calcd. for C21H19BrN2O2: C: 61.33%; H: 4.66%; N: 6.81%. Found C: 61.26%; H: 4.48%; N: 6.76%.
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(p-tolyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7e): Following the general procedure, to a solution of (R)-tryptophanol (0.103 g, 0.541 mmol) in toluene (5 mL) was added 3-(4-methylbenzoyl) propionic acid (0.114 g, 0.596 mmol). Reaction time: 19 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:1) and recrystallized from EtOAc/n-hexane to give pale pink crystalline solid (0.160 g, 86%); = −58.7° (c = 2.0, MeOH); 1H NMR was found to be identical to the one obtained for compound 8e. Anal. Calcd. for C22H22N2O2: C: 76.28%; H: 6.40%; N: 8.09%. Found C: 75.87%; H: 6.23%; N: 8.06%. The m.p. value was found to be identical to the one reported for compound 8e.
(3S,7aS)-3-((1H-indol-3-yl)methyl)-7a-(p-tolyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (8e): Following the general procedure, to a solution of (S)-tryptophanol (0.100 g, 0.526 mmol) in toluene (5 mL) was added 3-(4-methylbenzoyl) propionic acid (0.112 g, 0.583 mmol). Reaction time: 18 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:1) and recrystallized from EtOAc/n-hexane to give pale pink crystalline solid (0.097 g, 53%); = +45.1° (c = 2.0, MeOH); m.p.: 210–213 °C. 1H NMR (300 MHz, CDCl3) δ 8.00 (s, 1H, NH), 7.47–7.32 (m, 4H, ArH), 7.21–7.06 (m, 5H, ArH), 4.60–4.50 (m, 1H, H-3), 4.15 (dd, J = 8.7 Hz, 7.4 Hz, 1H, H-2), 3.61 (dd, J = 8.8 Hz, 6.9 Hz, 1H, H-2), 3.09 (dd, J = 14.7 Hz, 6.1 Hz, 1H, indole-CH2), 2.90–2.79 (m, 1H, CH2), 2.64-2.43 (m, 3H, CH2, and indole-CH2), 2.39 (s, 3H, CH3), 2.26-2.17 (m, 1H, CH2) ppm; 13C NMR (75 MHz, CDCl3) δ 180.3 (C=O), 139.9 (Cq), 138.2 (Cq), 136.3 (Cq), 129.3 (ArCH), 127.5 (Cq), 125.2 (ArCH), 122.3 (ArCH), 122.2 (ArCH), 119.6 (ArCH), 119.0 (ArCH), 111.9 (Cq), 111.2 (ArCH), 102.6 (C-7a), 72.9 (C-2), 55.7 (C-3), 35.4 (CH2), 32.9 (CH2), 29.9 (indole-CH2), 21.4 (CH3). Anal. Calcd. for C22H22N2O2: C: 76.28%; H: 6.40%; N: 8.09%. Found C: 76.49%; H: 6.27%; N: 8.16%.
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(4-methoxyphenyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7f): Following the general procedure, to a solution of (R)-tryptophanol (0.100 g, 0.526 mmol) in toluene (5 mL) was added 3-(4-methoxybenzoyl) propionic acid (0.121 g, 0.581 mmol). Reaction time: 24 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:1) and recrystallized from EtOAc/n-hexane to give pale yellow crystalline solid (0.106 g, 56%); = −43.0° (c = 1.0, MeOH); 1H NMR was found to be identical to the one obtained for compound 8f. Anal. Calcd. for C22H22N2O3·0.15H2O: C: 72.36%; H: 6.17%; N: 7.67%. Found C: 72.22%; H: 6.21%; N: 7.53%. The m.p. value was found to be identical to the one reported for compound 8f.
(3S,7aS)-3-((1H-indol-3-yl)methyl)-7a-(4-methoxyphenyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (8f): Following the general procedure, to a solution of (S)-tryptophanol (0.101 g, 0.533 mmol) in toluene (5 mL) was added 3-(4-methoxybenzoyl) propionic acid (0.122 g, 0.586 mmol). Reaction time: 25 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:1) and recrystallized from EtOAc/n-hexane to give pale yellow crystalline solid (0.134 g, 69%); = +48.1° (c = 1.0, MeOH); m.p.: 185–187 °C. 1H NMR (300 MHz, CDCl3) δ 8.00 (s, 1H, NH), 7.47–7.32 (m, 4H, ArH), 7.20–7.05 (m, 3H, ArH), 6.92–6.89 (m, 2H, ArH), 4.61–4.50 (m, 1H, H-3), 4.15 (dd, J = 8.4 Hz, 7.7 Hz, 1H, H-2), 3.84 (s, 3H, O-CH3), 3.61 (dd, J = 8.7 Hz, 7.0 Hz, 1H, H-2), 3.08 (dd, J = 14.7 Hz, 6.2 Hz, 1H, indole-CH2), 2.90–2.79 (m, 1H, CH2), 2.64–2.44 (m, 3H, CH2, and indole-CH2), 2.25–2.17 (m, 1H, CH2) ppm; 13C NMR (75 MHz, CDCl3) δ 180.2 (C=O), 159.7 (Cq), 136.3 (Cq), 134.9 (Cq), 127.5 (Cq), 126.6 (ArCH), 122.2 (ArCH), 119.5 (ArCH), 119.0 (ArCH), 114.1 (ArCH), 111.8 (Cq), 111.2 (ArCH), 102.5 (C-7a), 72.8 (C-2), 55.7 (OCH3), 55.5 (C-3), 35.3 (CH2), 32.8 (CH2), 29.8 (indole-CH2). Anal. Calcd. for C22H22N2O3: C: 72.91%; H: 6.12%; N: 7.73%. Found C: 72.73%; H: 5.76%; N: 7.73%.
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(4-(methylsulfonyl)phenyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7g): Following the general procedure, to a solution of (R)-tryptophanol (0.106 g, 0.557 mmol) in toluene (5 mL) was added 3-(4-methylsulfonylbenzoyl) propionic acid (0.157 g, 0.613 mmol). Reaction time: 22 h. The compound was purified by flash chromatography (EtOAc/n-hexane 7:3) and recrystallized from EtOAc/n-hexane to give a pale yellow crystalline solid (0.171g, 75%); = −57.2° (c = 2.0, MeOH); 1H NMR was found to be identical to the one obtained for compound 8g. Anal. Calcd. for C22H22N2O4S: C: 64.37%; H: 5.40%; N: 6.82%. Found C: 64.31%; H: 5.32%; N: 6.81%. The m.p. value was found to be identical to the one reported for compound 8g.
(3S,7aS)-3-((1H-indol-3-yl)methyl)-7a-(4-(methylsulfonyl)phenyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (8g): Following the general procedure, to a solution of (S)-tryptophanol (0.100 g, 0.526 mmol) in toluene (5 mL) was added 3-(4-methylsulfonylbenzoyl) propionic acid (0.148 g, 0.578 mmol). Reaction time: 23 h. The compound was purified by flash chromatography (EtOAc/n-hexane 7:3) and recrystallized from EtOAc/n-hexane to give yellow crystalline solid (0.131 g, 61%); = +66.9 (c = 2.0, MeOH); m.p.: 205–207 °C. 1H NMR (300 MHz, CDCl3) δ 8.01 (s, 1H, NH), 7.90–7.88 (m, 2H, ArH), 7.62–7.59 (m, 2H, ArH), 7.43 (d, J = 7.9 Hz, 1H, ArH), 7.33 (d, J = 8.1 Hz, 1H, ArH), 7.21–7.16 (m, 1H, ArH), 7.11–7.06 (m, 1H, ArH), 7.00 (d, J = 2.3 Hz, 1H, ArH), 4.66–4.56 (m, 1H, H-3), 4.22 (dd, J = 8.9, 7.4 Hz, 1H, H-2), 3.61 (dd, J = 8.9, 7.0 Hz, 1H, H-2), 3.10 (s, 3H, SO2CH3), 3.00 (dd, J = 15.5, 5.9 Hz, 1H, indole-CH2), 2.92–2.75 (m, 1H, CH2), 2.70–2.45 (m, 3H, CH2, and indole-CH2), 2.21–2.13 (m, 1H, CH2) ppm; 13C NMR (75 MHz, CDCl3) δ 180.1 (C=O), 149.2 (Cq), 140.5 (Cq), 136.3 (Cq), 128.0 (ArCH), 127.5 (Cq), 126.2 (ArCH), 122.4 (ArCH), 119.7 (ArCH), 118.7 (ArCH), 111.4 (Cq), 111.1 (ArCH), 101.8 (C-7a), 72.9 (C-2), 56.0 (C-3), 44.6 (SO2CH3), 35.1 (CH2), 32.6 (CH2), 29.5 (indole-CH2). Anal. Calcd. for C22H22N2O4S: C: 64.37%; H: 5.40%; N: 6.82%. Found C: 64.59%; H: 5.51%; N: 6.69%.
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(3-chloro-4-methylphenyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7h): Following the general procedure, to a solution of (R)-tryptophanol (0.041 g, 0.218 mmol) in toluene (2 mL) was added 4-(3-chloro-4-methylphenyl)-4-oxobutanoic acid (0.058 g, 0.254 mmol). Reaction time: 10 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:1) and recrystallized from CH2Cl2/n-hexane to give a white solid (0.077 g, 93%); = −29.6° (c = 1.0, CH2Cl2); m.p.: 171–172 °C. 1H NMR (300 MHz, CDCl3): δ 7.97 (s, 1H, NH), 7.50 (d, J = 1.4 Hz, 1H, ArH), 7.48 (d, J = 8.1 Hz, 1H, ArH), 7.33 (d, J = 8.0 Hz, 1H, ArH), 7.28–7.23 (m, 2H, ArH), 7.21–7.15 (m, 1H, ArH), 7.13–7.09 (m, 1H, ArH), 7.06 (d, J = 2.4 Hz, 1H, ArH), 4.62–4.50 (m, 1H, H-3), 4.16 (dd, J = 8.8, 7.4 Hz, 1H, H-2), 3.61 (dd, J = 8.8, 6.9 Hz, 1H, H-2), 3.10 (dd, J = 14.6, 6.0 Hz, 1H, indole-CH2), 2.85 (ddd, J = 16.3, 9.8, 8.1 Hz, 1H, CH2), 2.62 (dd, J = 10.1, 3.3 Hz, 1H, CH2), 2.50 (ddd, J = 18.4, 9.6, 6.1 Hz, 2H, CH2, and indole-CH2), 2.41 (s, 3H, CH3), 2.25–2.15 (m, 1H, CH2); 13C NMR (75 MHz, CDCl3) δ 180.1 (C=O), 160.6 (Cq), 142.3 (Cq), 136.2 (Cq), 134.9 (Cq), 131.4 (ArCH), 127.4 (Cq), 125.9 (ArCH), 123.5 (ArCH), 122.3 (ArCH), 122.2 (ArCH), 119.6 (Cq), 118.9 (ArCH), 111.6 (ArCH), 111.2 (ArCH), 101.8 (C-7a), 72.9 (C-2), 55.7 (C-3), 35.1 (CH2), 32.7 (CH2), 29.8 (indole-CH2), 20.0 (CH3). LRMS (ESI) m/z calcd for C22H21ClN2O2: 380, found 381 [M+H]+.
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(3-fluoro-4-methoxyphenyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7i): Following the general procedure, to a solution of (R)-tryptophanol (0.100 g, 0.526 mmol) in toluene (5 mL) was added 3-(3-fluoro-4-methoxybenzoyl) propionic acid (0.131 g, 0.578 mmol). Reaction time: 22 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:1) and recrystallized from EtOAc/n-hexane to give a pale yellow crystalline solid (0.139 g, 69%); = −51.3° (c = 2.0, MeOH); m.p.: 131–132 °C; 1H NMR (300 MHz, (CDCl3) δ 8.01 (s, 1H, NH), 7.47 (d, J = 7.9 Hz, 1H, ArH), 7.33 (d, J = 8.0 Hz, 1H, ArH), 7.23–7.04 (m, 5H, ArH), 6.93 (t, J = 8.4 Hz, 1H, ArH), 4.61–4.41 (m, 1H, H-3), 4.16 (dd, J = 8.7, 7.4 Hz, 1H, H-2), 3.92 (s, 3H, O-CH3), 3.61 (dd, J = 8.8, 6.9 Hz, 1H, H-2), 3.08 (dd, J = 13.9, 6.0 Hz, 1H, indole-CH2), 2.90–2.83 (m, 1H, CH2), 2.64–2.43 (m, 3H, CH2, and indole-CH2), 2.24–2.15 (m, 1H, CH2) ppm; 13C NMR (75 MHz, CDCl3) δ 180.2 (C=O), 152.2 (d, Cq, JC-F = 245.2 Hz), 147.5 (Cq), 147.3 (Cq), 136.3 (Cq), 135.7 (d, Cq, J = 5.2 Hz), 127.4 (Cq), 122.0 (d, ArCH, J = 6.3 Hz), 120.8 (d, ArCH, J = 3.5 Hz), 119.5 (ArCH), 118.8 (ArCH), 113.2 (d, ArCH, J = 1.6 Hz), 113.2 (ArCH), 111.5 (Cq), 111.3 (ArCH), 101.9 (C-7a), 72.9 (C-2), 56.5 (C-3), 55.8 (OCH3), 35.2 (CH2), 32.9 (CH2), 29.8 (indole-CH2). Anal. Calcd. for C22H21FN2O3: C: 69.46%; H: 5.56%; N: 7.36%. Found C: 69.49%; H: 5.76%; N: 7.12%.
(3R,7aR,7S)-3-((1H-indol-3-yl)methyl)-7a-(4-chlorophenyl)-7-methyltetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7j) and (3R,7aR,7R)-3-((1H-indol-3-yl)methyl)-7a-(4-chlorophenyl)-7-methyltetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7j‘): Following the general procedure, to a solution of (R)-tryptophanol (0.039 g, 0.207 mmol) in toluene (2 mL) was added 4-(4-chlorophenyl)-3-methyl-4-oxobutanoic acid (0.057 g, 0.239 mmol). Reaction time: 17 h. Two compounds were purified by flash chromatography (EtOAc/n-hexane 2:3) and recrystallized from CH2Cl2/n-hexane.
(7j): The product was obtained as a white solid (0.055 g, 69%); = −30.5° (c = 1.0, CH2Cl2); m.p.: 201–202 °C. 1H NMR (300 MHz, CDCl3): δ 8.00 (s, 1H, NH), 7.41 (d, J = 8.6 Hz, 3H, ArH), 7.34 (d, J = 8.8 Hz, 3H, ArH), 7.18 (t, J = 7.4 Hz, 1H, ArH), 7.11 (s, 1H, ArH), 7.07 (d, J = 7.4 Hz, 1H, ArH), 4.67–4.56 (m, 1H, H-3), 4.13 (t, J = 8.0 Hz, 1H, H-2), 3.58 (dd, J = 8.5, 6.6 Hz, 1H, H-2), 3.04–2.85 (m, 2H, CH2, and indole-CH2), 2.44 (td, J = 15.1, 8.1 Hz, 2H, CH2, and indole-CH2), 2.18 (dd, J = 17.3, 5.6 Hz, 1H, CH2), 1.12 (d, J = 7.1 Hz, 3H, CH3); 13C NMR (75 MHz, CDCl3) δ 181.1 (C=O), 141.4 (Cq), 136.2 (Cq), 134.2 (Cq), 128.9 (ArCH), 127.5 (Cq), 127.0 (ArCH), 122.3 (ArCH), 122.1 (ArCH), 119.6 (ArCH), 118.8 (ArCH), 111.8 (Cq), 111.2 (ArCH), 103.0 (C-7a), 72.3 (C-2), 56.5 (C-3), 40.0 (CH2), 39.7 (CH2), 29.7 (indole-CH2), 14.0 (CH3). LRMS (ESI) m/z calcd for C22H21ClN2O2: 380, found 381 [M+H]+.
(7j’): The product was obtained as white solid (0.014 g, 18%); = −45.7° (c = 1.0, CH2Cl2); m.p.: 205-206 °C. 1H NMR (300 MHz, CDCl3): δ 7.95 (s, 1H, NH), 7.48 (d, J = 7.9 Hz, 1H, ArH), 7.34 (t, J = 8.7 Hz, 5H, ArH), 7.19 (t, J = 7.3 Hz, 1H, ArH), 7.10 (t, J = 7.3 Hz, 1H, ArH), 6.99 (s, 1H, ArH), 4.55 (td, J = 12.4, 6.8 Hz, 1H, H-3), 4.24–4.17 (m, 1H, H-2), 3.64 (dd, J = 8.6, 7.1 Hz, 1H, H-2), 3.10 (dd, J = 14.7, 5.3 Hz, 1H, indole-CH2), 2.78–2.63 (m, 2H, CH2), 2.48 (dt, J = 15.8, 8.1 Hz, 2H, CH2, and indole-CH2), 0.65 (d, J = 6.5 Hz, 3H, CH3); 13C NMR (75 MHz, CDCl3) δ 177.1 (C=O), 137.9 (Cq), 136.3 (Cq), 134.4 (Cq), 128.7 (ArCH), 127.9 (ArCH), 127.5 (Cq), 122.4 (ArCH), 122.2 (ArCH), 119.7 (ArCH), 119.0 (ArCH), 111.6 (Cq), 111.2 (ArCH), 104.1 (C-7a), 73.5 (C-2), 54.8 (C-3), 42.1 (CH2), 41.3 (CH2), 29.6 (indole-CH2), 16.4 (CH3). LRMS (ESI) m/z calcd for C22H21ClN2O2: 380, found 381 [M+H]+.
General procedure for the synthesis of 7k–l: The (R)-tryptophanol-derived oxazolopyrrolidone lactam (0.129 mmol) was dissolved in dry DMF (5 mL), and the solution was cooled to 0 °C, under N2 atmosphere. Sodium hydride (NaH) in 60% dispersion in mineral oil (0.250 mmol, 2.0 eq.) was added portion wise and the mixture stirred for 15 min. The appropriate protecting reagent (0.320 mmol, 2.5 eq.) was added and the reaction mixture stirred at room temperature for 3–6 h. After reaction completion, water (10 mL) was added followed by EtOAc (10 mL). The aqueous phase was washed with EtOAc (2x10 mL); the combined organic phases were washed with brine (10 mL), dried with MgSO4, and concentrated in vacuo. The residue was purified by silica gel flash chromatography using EtOAc/n-hexane as eluent.
(3R,7aR)-7a-(4-chlorophenyl)-3-((1-ethyl-1H-indol-3-yl)methyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7k): Following the general procedure, to a solution of 7c (0.120 g, 0.327 mmol) in DMF (13.5 mL) was added NaH (0.016 g, 0.654 mmol) and ethyl iodide (65.4 µL, 0.818 mmol). Reaction time: 3 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:2) to afford the title compound as a pale yellow oil (0.101 g, 78%); 1H NMR (300 MHz, CDCl3) δ 7.39 (d, J = 7.9 Hz, 1H. ArH), 7.36–7.17 (m, 5H, ArH), 7.16–7.08 (m, 1H, ArH), 7.04–6.97 (m, 1H, ArH), 6.87 (s, 1H, ArH), 4.48 (m, 1H, H-3), 4.08 (dd, J = 8.8, 7.5 Hz, 1H, H-2), 4.01 (q, J = 7.3 Hz, 2H, CH2CH3), 3.52 (dd, J = 8.8, 7.0 Hz, 1H, H-2), 3.01 (dd, J = 14.6, 5.3 Hz, 1H, indole-CH2), 2.84–2.70 (m, 1H, CH2), 2.58–2.36 (m, 3H, CH2, and indole-CH2), 2.15–2.07 (m, 1H, CH2), 1.33 (t, J = 7.3 Hz, 3H, CH2CH3); 13C NMR (75 MHz, CDCl3) δ 180.2 (C=O), 141.6 (Cq), 136.1 (Cq), 134.3 (Cq), 129.0 (ArCH), 128.1 (Cq), 126.8 (ArCH), 125.3 (ArCH), 121.7 (ArCH), 119.2 (ArCH), 119.1 (ArCH), 110.0 (Cq), 109.4 (ArCH), 102.1 (C-7a), 72.9 (C-2), 55.9 (C-3), 40.9 (CH2CH3), 35.2 (CH2), 32.8 (CH2), 29.7 (indole-CH2), 15.6 (CH2CH3). Anal. Calcd. for C23H23ClN2O2: C: 69.96%; H: 5.87%; N: 7.09%. Found C: 70.12%; H: 6.40%; N: 6.95%.
(3R,7aR)-3-((1-acetyl-1H-indol-3-yl)methyl)-7a-(4-chlorophenyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7l): Following the general procedure, to a solution of 7c (0.094 g, 0.256 mmol) in DMF (9.5 mL) was added NaH (12.3 mg, 0.512 mmol) and acetic anhydride (60.6 µL, 0.641 mmol). Reaction time: 6 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:1) to afford the title compound as a white powder (0.072 g, 69%); m.p.: 66–67 °C; 1H NMR (300 MHz, CDCl3) δ 8.32 (d, J = 7.8 Hz, 1H, ArH), 7.56 (s, 1H, ArH), 7.28–7.11 (m, 7H, ArH), 4.61–4.45 (m, 1H, H-3), 4.20 (dd, J = 8.7, 7.6 Hz, 1H, H-2), 3.49 (dd, J = 8.7, 6.5 Hz, 1H, H-2), 2.80–2.52 (m, 3H, CH2, and indole-CH2), 2.52 (s, 3H, CH3), 2.48–2.35 (m, 2H, CH2, and indole-CH2), 2.16–2.02 (m, 1H, CH2); 13C NMR (75 MHz, CDCl3) δ 180.9 (C=O), 168.9 (C=O), 141.2 (Cq), 135.9 (Cq), 134.5 (Cq), 130.6 (Cq), 129.1 (ArCH), 126.6 (ArCH), 125.5 (ArCH), 123.7 (ArCH), 123.3 (ArCH), 118.7 (ArCH), 118.3 (Cq), 116.8 (ArCH), 102.4 (C-7a), 72.6 (C-2), 54.9 (C-3), 34.7 (CH2), 32.4 (CH2), 29.6 (indole-CH2), 24.2 (CH3). Anal. Calcd. for C23H21ClN2O3: C: 67.56%; H: 5.18%; N: 6.85%. Found C: 67.37%; H: 5.47%; N: 6.72%.
Procedure for the synthesis of tert-butyl 3-(((3R,7aR)-7a-(4-chlorophenyl)-5-oxohexahydropyrrolo[2,1-b]oxazol-3-yl)methyl)-1H-indole-1-carboxylate (7m): To a solution of 7c (0.070 g, 0.191 mmol) in THF (7.0 mL) was added anhydrous Et3N (58.6 µL, 0.420 mmol), DMAP (0.006 g, 0.048 mmol), and Boc2O (0.054 g, 0.248 mmol). The reaction mixture was stirred at room temperature for 3 h. After reaction completion, the mixture was concentrated in vacuo and the crude was dissolved in EtOAc (20 mL). The organic phase was washed with a sat. sol. of NH4Cl (2 × 15 mL), a sat. sol. of NaHCO3 (2 × 15 mL) and brine (15 mL). The combined organic phases were dried with MgSO4, concentrated in vacuo and the compound was purified by flash chromatography (EtOAc/n-hexane 2:3) to afford the title compound as a pale yellow powder (0.059 g, 66%); m.p.: 163–165 °C; 1H NMR (300 MHz, CDCl3) δ 8.01 (d, J = 7.6 Hz, 1H, ArH), 7.37–7.30 (m, 4H, ArH), 7.29–7.18 (m, 3H, ArH), 7.14 (td, J = 7.5, 1.1 Hz, 1H, ArH), 4.57–4.40 (m, 1H, H-3), 4.14 (dd, J = 8.8, 7.5 Hz, 1H, H-2), 3.51 (dd, J = 8.9, 7.0 Hz, 1H, H-2), 2.90 (ddd, J = 14.7, 5.7, 1.2 Hz, 1H, indole-CH2), 2.85–2.70 (m, 1H, CH2), 2.59–2.30 (m, 3H, CH2, and indole-CH2), 2.16–2.05 (m, 1H, CH2), 1.58 (s, 9H, C(CH3)3); 13C NMR (75 MHz, CDCl3) δ 180.3 (C=O), 149.8 (C=O), 141.4 (Cq), 134.4 (Cq), 130.4 (Cq), 129.1 (ArCH), 126.6 (ArCH), 124.7 (ArCH), 123.5 (ArCH), 122.7 (ArCH), 119.1 (ArCH), 116.1 (Cq), 115.4 (ArCH), 102.1 (C-7a), 83.8 (C(CH3)3), 72.8 (C-2), 55.2 (C-3), 35.2 (CH2), 32.7 (CH2), 29.5 (indole-CH2), 28.4 (C(CH3)3); Anal. Calcd. for C26H27ClN2O4: C: 66.88%; H: 5.83%; N: 6.00%. Found C: 66.90%; H: 6.16%; N: 5.89%.
General procedure for the synthesis of 7n–u: To a solution of the appropriate tryptophanol-derived oxazolopiperidone lactams (0.230 mmol) in dioxane (2.3 mL) was added Pd(PPh3)2Cl2 (0.023 mmol, 0.1 eq) and 1 M aq. sol. of Na2CO3 (690 µL), followed by the appropriate boronic acid (0.280 mmol,1.2 eq.). The resulting mixture was degassed and stirred at 100 °C, under N2 atmosphere, for 2–5 h. After cooling to room temperature, the reaction mixture was diluted with CH2Cl2, filtered in celite, and concentrated in vacuo. The residue was purified by silica gel flash chromatography using EtOAc/N-hexane as eluent.
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(4’-chloro-[1,1’-biphenyl]-4-yl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7n): Following the general procedure, to a solution of 7d (0.036 g, 0.088 mmol) in dioxane (1.0 mL) was added Pd(PPh3)2Cl2 (0.003 g, 8.8 µmol), 1 M aq. sol. of Na2CO3 (266 µL), and 4-chlorophenylboronic acid (0.017 g, 0.107 mmol). Reaction time: 4 h. The compound was purified by flash chromatography (EtOAc/n-hexane 2:3) to afford the title compound as a white solid (0.036 g, 94%); m.p.: 201–204 °C; 1H NMR (300 MHz, CDCl3) δ 8.08 (s, 1H, NH), 7.63–7.50 (m, 6H, ArH), 7.48–7.39 (m, 3H, ArH), 7.33 (d, J = 8.1 Hz, 1H, ArH), 7.22–7.12 (m, 1H, ArH), 7.12–7.01 (m, 2H, ArH), 4.70–4.52 (m, 1H, H-3), 4.20 (dd, J = 8.7, 7.4 Hz, 1H, H-2), 3.66 (dd, J = 8.8, 6.8 Hz, 1H, H-2), 3.11 (dd, J = 15.1, 6.6 Hz, 1H, indole-CH2), 3.01–2.78 (m, 1H, CH2), 2.73–2.44 (m, 3H, CH2, and indole-CH2), 2.38–2.17 (m, 1H, CH2); 13C NMR (75 MHz, CDCl3) δ 180.3 (C=O), 142.3 (Cq), 140.1 (Cq), 139.1 (Cq), 136.3 (Cq), 133.9 (Cq), 129.2 (ArCH), 128.5 (ArCH), 127.5 (Cq), 127.4 (ArCH), 125.9 (ArCH), 122.3 (ArCH), 122.2 (ArCH), 119.6 (ArCH), 118.9 (ArCH), 111.8 (Cq), 111.3 (ArCH), 102.4 (C-7a), 73.0 (C-2), 55.8 (C-3), 35.2 (CH2), 32.8 (CH2), 29.9 (indole-CH2); Anal. Calcd. for C27H23ClN2O2: C: 73.21%; H: 5.23%; N: 6.32%. Found C: 73.56%; H: 5.83%; N: 5.92%.
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(4’-(trifluoromethyl)-[1,1’-biphenyl]-4-yl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7o): Following the general procedure, to a solution of 7d (0.050 g, 0.122 mmol) in dioxane (1.4 mL) was added Pd(PPh3)2Cl2 (0.004 g, 12.2 µmol), 1 M aq. sol. of Na2CO3 (370 µL), and 4-(trifluoromethyl)phenylboronic acid (0.028 g, 0.148 mmol). Reaction time: 3 h. The compound was purified by flash chromatography (EtOAc/n-hexane 2:3) to afford the title compound as a white solid (0.050 g, 86%); m.p.: 201–203 °C; 1H NMR (300 MHz, CDCl3) δ 7.99 (s, 1H, NH), 7.72 (s, 4H, ArH), 7.64–7.55 (m, 4H, ArH), 7.43 (d, J = 7.5 Hz, 1H, ArH), 7.33 (d, J = 8.1 Hz, 1H, ArH), 7.21–7.14 (m, 1H, ArH), 7.12–7.03 (m, 2H, ArH), 4.67–4.53 (m, 1H, H-3), 4.20 (dd, J = 8.8, 7.4 Hz, 1H, H-2), 3.65 (dd, J = 8.8, 6.8 Hz, 1H, H-2), 3.10 (dd, J = 15.0, 6.5 Hz, 1H, indole-CH2), 2.91–2.83 (m, 1H, CH2), 2.67–2.48 (m, 3H, CH2, and indole-CH2), 2.33–2.22 (m, 1H, CH2); 13C NMR (75 MHz, CDCl3) δ 180.3 (C=O), 143.9 (Cq), 142.7 (Cq), 139.8 (Cq), 136.0 (Cq), 129.5 (q, Cq, J = 32.3 Hz), 127.8 (ArCH), 127.6 (ArCH), 127.1 (Cq), 125.8 (q, ArCH, J = 3.7 Hz), 125.7 (ArCH), 124.2 (q, Cq, JC-F = 270.2 Hz), 122.3 (ArCH), 122.2 (ArCH), 119.6 (ArCH), 118.1 (ArCH), 111.7 (Cq), 111.2 (ArCH), 102.4 (C-7a), 72.9 (C-2), 55.8 (C-3), 35.2 (CH2), 32.8 (CH2), 29.9 (indole-CH2); Anal. Calcd. for C28H23F3N2O2: C: 70.58%; H: 4.87%; N: 5.88%. Found C: 70.09%; H: 5.19%; N: 5.83%.
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(4’-hydroxy-[1,1’-biphenyl]-4-yl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7p): Following the general procedure, to a solution of 7d (0.050 g, 0.122 mmol) in dioxane (1.4 mL) was added Pd(PPh3)2Cl2 (0.004 g, 12.2 µmol), 1 M aq. sol. of Na2CO3 (370 µL), and 4-hydroxyphenylboronic acid (0.021 g, 0.148 mmol). Reaction time: 2 h. The compound was purified by flash chromatography (EtOAc/n-hexane 3:2) to afford the title compound as a white solid (0.044 g, 85%); m.p.: 223–225 °C; 1H NMR (300 MHz, CDCl3) δ 7.98 (s, 1H, NH), 7.62–7.43 (m, 7H, ArH), 7.34 (d, J = 7.8 Hz, 1H, ArH), 7.18 (t, J = 7.6 Hz, 1H, ArH), 7.10–7.05 (m, 2H, ArH), 6.95 (d, J = 8.5 Hz, 2H, ArH), 5.21 (s, 1H, OH), 4.68–4.51 (m, 1H, H-3), 4.20 (dd, J = 8.5, 7.8 Hz, 1H, H-2), 3.66 (dd, J = 8.6, 6.8 Hz, 1H, H-2), 3.13 (dd, J = 14.9, 6.3 Hz, 1H, indole-CH2), 2.98–2.76 (m, 1H, CH2), 2.75–2.46 (m, 3H, CH2, and indole-CH2), 2.33–2.24 (m, 1H, CH2); 13C NMR (75 MHz, (CD3)2SO) δ 179.5 (C=O), 157.2 (Cq), 140.8 (Cq), 139.9 (Cq), 136.0 (Cq), 130.1 (Cq), 127.7 (ArCH), 126.9 (Cq), 126.0 (ArCH), 125.5 (ArCH), 122.9 (ArCH), 121.0 (ArCH), 118.3 (ArCH), 117.9 (ArCH), 115.8 (ArCH), 111.4 (ArCH), 109.9 (Cq), 101.7 (C-7a), 72.3 (C-2), 55.3 (C-3), 40.4 (CH2), 32.8 (CH2), 29.8 (indole-CH2); Anal. Calcd. for C27H24N2O3·0.15H2O: C: 75.91%; H: 5.75%; N: 6.56%. Found C: 75.74%; H: 5.85%; N: 6.57%.
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(4’-(hydroxymethyl)-[1,1’-biphenyl]-4-yl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7q): Following the general procedure, to a solution of 7d (0.070 g, 0.170 mmol) in dioxane (2.0 mL) was added Pd(PPh3)2Cl2 (0.005 g, 17.0 µmol), 1 M aq. sol. of Na2CO3 (520 µL), and 4-(hydroxymethyl)phenylboronic acid (0.032 g, 0.208 mmol). Reaction time: 3 h. The compound was purified by flash chromatography (EtOAc/n-hexane 3:2) to afford the title compound as a pale yellow solid (0.053 g, 71%); m.p.: 213–215 °C; 1H NMR (300 MHz, CDCl3) δ 8.00 (s, 1H, NH), 7.65–7.44 (m, 9H, ArH), 7.33 (d, J = 8.0 Hz, 1H, ArH), 7.17 (t, J = 7.3 Hz, 1H, ArH), 7.11–7.02 (m, 2H, ArH), 4.77 (s, 2H, CH2), 4.67–4.53 (m, 1H, H-3), 4.19 (t, J = 8.0 Hz, 1H, H-2), 3.66 (t, J = 8.0 Hz, 1H, H-2), 3.11 (dd, J = 14.7, 6.0 Hz, 1H, indole-CH2), 2.95–2.78 (m, 1H, CH2), 2.69–2.47 (m, 3H, CH2, and indole-CH2), 2.38–2.20 (m, 1H, CH2); 13C NMR (75 MHz, CDCl3) δ 180.3 (C=O), 142.0 (Cq), 140.9 (Cq), 140.4 (Cq), 140.1 (Cq), 136.3 (Cq), 127.5 (ArCH), 127.4 (Cq), 127.3 (ArCH), 127.2 (ArCH), 125.8 (ArCH), 122.3 (ArCH), 122.2 (ArCH), 119.6 (ArCH), 119.0 (ArCH), 111.9 (Cq), 111.3 (ArCH), 102.5 (C-7a), 73.0 (C-2), 65.3 (CH2OH), 55.8 (C-3), 35.3 (CH2), 32.8 (CH2), 29.9 (indole-CH2); Anal. Calcd. (C28H26N2O3·0.40H2O): C: 75.44%; H: 6.07%; N: 6.29%. Found C: 75.18%; H: 6.21%; N: 6.14%.
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(3’-chloro-[1,1’-biphenyl]-4-yl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7r): Following the general procedure, to a solution of 7d (0.070 g, 0.170 mmol) in dioxane (2.0 mL) was added Pd(PPh3)2Cl2 (0.005 g, 17.0 µmol), 1 M aq. sol. of Na2CO3 (520 µL), and 3-chlorophenylboronic acid (0.033 g, 0.208 mmol). Reaction time: 4 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:1) to afford the title compound as a pale yellow solid (0.059 g, 78%); m.p.: 204–206 °C; 1H NMR (300 MHz, CDCl3) δ 7.98 (s, 1H, NH), 7.47 (t, J = 1.6 Hz, 1H, ArH), 7.43 (s, 4H, ArH), 7.36 (dt, J = 7.4 Hz, 1.6 Hz, 1H, ArH), 7.30 (d, J = 7.7 Hz, 1H, ArH), 7.25–7.17 (m, 3H, ArH), 7.06–7.00 (m, 1H, ArH), 6.95–6.91 (m, 2H, ArH), 4.51–4.42 (m, 1H, H-3), 4.05 (dd, J = 8.9, 6.9 Hz, 1H, H-2), 3.51 (dd, J = 8.8, 6.8 Hz, 1H, H-2), 2.95 (dd, J = 14.4, 6.6 Hz, 1H, indole-CH2), 2.80–2.66 (m, 1H, CH2), 2.53–2.34 (m, 3H, CH2, and indole-CH2), 2.22–2.08 (m, 1H, CH2); 13C NMR (75 MHz, CDCl3) δ 180.3 (C=O), 142.51 (Cq), 142.46 (Cq), 139.9 (Cq), 136.3 (Cq), 134.9 (Cq), 130.3 (ArCH), 127.8 (ArCH), 127.6 (ArCH), 127.6 (Cq), 127.5 (ArCH), 125.9 (ArCH), 125.4 (ArCH), 122.3 (ArCH), 119.6 (ArCH), 118.9 (ArCH), 111.7 (Cq), 111.3 (ArCH), 102.4 (C-7a), 72.9 (C-2), 55.7 (C-3), 35.2 (CH2), 32.8 (CH2), 29.8 (indole-CH2); Anal. Calcd. for C27H23ClN2O2: C: 73.21%; H: 5.23%; N: 6.32%. Found C: 72.91%; H: 5.70%; N: 6.24%.
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(3’,4’-dichloro-[1,1’-biphenyl]-4-yl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7s): Following the general procedure, to a solution of 7d (0.070 g, 0.170 mmol) in dioxane (2.0 mL) was added Pd(PPh3)2Cl2 (0.005 g, 17.0 µmol), 1 M aq. sol. of Na2CO3 (520 µL), and 3,4-dichlorophenylboronic acid (0.040 g, 0.208 mmol). Reaction time: 4 h. The compound was purified by flash chromatography (EtOAc/n-hexane 1:1) to afford the title compound as a pale yellow solid (0.069 g, 85%); m.p.: 176–178 °C; 1H NMR (300 MHz, CDCl3) δ 7.98 (s, 1H, NH), 7.71 (d, J = 2.1 Hz, 1H, ArH), 7.49 (s, 5H, ArH), 7.38 (dd, J = 8.4 Hz, 2.2 Hz, 2H, ArH), 7.27 (d, J = 8.1 Hz, 1H, ArH), 7.11 (d, J = 8.1 Hz, 1H, ArH), 7.06–6.97 (m, 2H, ArH), 4.65–4.45 (m, 1H, H-3), 4.14 (dd, J = 8.7, 7.4 Hz, 1H, H-2), 3.58 (dd, J = 8.8, 6.8 Hz, 1H, H-2), 3.02 (dd, J = 15.0, 6.6 Hz, 1H, indole-CH2), 2.87–2.76 (m, 1H, CH2), 2.65–2.38 (m, 3H, CH2,, and indole-CH2), 2.26–2.15 (m, 1H, CH2); 13C NMR (75 MHz, (CD3)2SO) δ 180.3 (C=O), 142.9 (Cq), 140.7 (Cq), 138.9 (Cq), 136.2 (Cq), 132.9 (Cq), 131.7 (Cq), 131.0 (ArCH), 128.9 (ArCH), 127.5 (Cq), 127.4 (ArCH), 126.3 (ArCH), 126.0 (ArCH), 122.3 (ArCH), 122.2 (ArCH), 119.6 (ArCH), 118.8 (ArCH), 111.7 (Cq), 111.3 (ArCH), 102.3 (C-7a), 72.8 (C-2), 55.8 (C-3), 35.2 (CH2), 32.9 (CH2), 29.9 (indole-CH2); Anal. Calcd. for C27H22Cl2N2O2: C: 67.93%; H: 4.65%; N: 5.87%. Found C: 67.96%; H: 4.90%; N: 5.73%. HRMS-ESI m/z calcd for C27H22Cl2N2O2: 476.1058, found 477.1143 ± 3.6 ppm [M+H]+.
(3R,7aR)-3-((1H-indol-3-yl)methyl)-7a-(4-(pyridin-4-yl)phenyl)tetrahydropyrrolo[2,1-b]oxazol-5(6H)-one (7t): Following the general procedure, to a solution of 7d (0.070 g, 0.170 mmol) in dioxane (2.0 mL) was added Pd(PPh3)2Cl2 (0.005 g, 17.0 µmol), 1 M aq. sol. of Na2CO3 (520 µL), and 4-pyridinylboronic acid (0.026 g, 0.208 mmol). Reaction time: 2 h. The compound was purified by flash chromatography (EtOAc/n-hexane 3:1) to afford the title compound as a pale yellow solid (0.068 g, 97%); m.p.: 214–215 °C; 1H NMR (300 MHz, CDCl3) δ 8.69 (d, J = 5.6 Hz, 2H, ArH), 8.23 (s, 1H, NH), 7.63 (q, J = 8.4 Hz, 4H, ArH), 7.54 (d, J = 5.9 Hz, 2H, ArH), 7.43 (d, J = 7.8 Hz, 1H, ArH), 7.32 (d, J = 8.0 Hz, 1H, ArH), 7.17 (t, J = 7.4 Hz, 1H, ArH), 7.12–7.01 (m, 2H, ArH), 4.71–4.55 (m, 1H, H-3), 4.21 (t, J = 8.1 Hz, 1H, H-2), 3.65 (dd, J = 8.6, 7.0 Hz, 1H, H-2), 3.09 (dd, J = 14.7, 6.1 Hz, 1H, indole-CH2), 2.88 (ddd, J = 24.1, 12.1, 6.3 Hz, 1H, CH2), 2.72–2.45 (m, 3H, CH2, and indole-CH2), 2.37–2.20 (m, 1H, CH2); 13C NMR (75 MHz, CDCl3) δ 180.3 (C=O), 150.5 (ArCH), 147.9 (Cq), 144.0 (Cq), 138.2 (Cq), 136.3 (Cq), 127.5 (ArCH), 126.1 (ArCH), 122.4 (ArCH), 122.3 (ArCH), 121.8 (ArCH), 119.6 (ArCH), 118.9 (ArCH), 111.6 (Cq), 111.3 (ArCH), 102.3 (C-7a), 73.0 (C-2), 55.81 (C-3), 35.19 (CH2), 32.75 (CH2), 29.85 (indole-CH2). Anal. Calcd. for C26H23N3O2·0.20H2O: C: 75.59%; H: 5.72%; N: 10.17%. Found C: 75.90%; H: 5.81%; N: 9.73%.
(3
R,7a
R)-3-((1
H-indol-3-yl)methyl)-7a-(4-(2,3-dihydrobenzo[
b][
1,
4]dioxin-6-yl)phenyl)tetrahydropyrrolo[2,1-
b]oxazol-5(6
H)-one (
7u): Following the general procedure, to a solution of
7d (0.070 g, 0.170 mmol) in dioxane (2.0 mL) was added Pd(PPh
3)
2Cl
2 (0.005 g, 17.0 µmol), 1 M aq. sol. of Na
2CO
3 (520 µL), and 1,4-benzodioxane-6-boronic acid (0.037 g, 0.208 mmol). Reaction time: 5 h. The compound was purified by flash chromatography (EtOAc/
n-hexane 1:1) to afford the title compound as a pale yellow solid (0.022 g, 28%); m.p.: 286–288 °C;
1H NMR (300 MHz, CDCl
3) δ 7.97 (s, 1H, NH), 7.56–7.44 (m, 5H, ArH), 7.33 (d,
J = 8.0 Hz, 1H, ArH), 7.19–7.08 (m, 5H, ArH), 6.95 (d,
J = 8.3 Hz, 1H, ArH), 4.64–4.54 (m, 1H, H-3), 4.31 (s, 4H, CH2), 4.18 (dd,
J = 8.7, 7.5 Hz, 1H, H-2), 3.65 (dd,
J = 8.8, 6.9 Hz, 1H, H-2), 3.11 (dd,
J = 14.7, 6.1 Hz, 1H, indole-CH2), 2.93–2.82 (m, 1H, CH2), 2.67–2.45 (m, 3H, CH2, and indole-CH2), 2.37–2.19 (m, 1H, CH2);
13C NMR (75 MHz, CDCl
3) δ 180.4 (C=O), 143.9 (Cq), 143.6 (Cq), 141.4 (Cq), 140.7 (Cq), 136.3 (Cq), 134.2 (Cq), 127.3 (Cq), 126.9 (ArCH), 125.4 (ArCH), 122.3 (ArCH), 122.0 (ArCH), 120.4 (ArCH), 119.4 (ArCH), 118.8 (ArCH), 117.6 (ArCH), 115.8 (ArCH), 111.8 (Cq), 111.2 (ArCH), 102.4 (C-7a), 72.9 (C-2), 64.7 (CH
2), 55.7 (C-3), 35.2 (CH
2), 32.9 (CH
2), 29.9 (indole-CH
2). Anal. Calcd. for C
29H
26N
2O
4: C: 74.66%; H: 5.68%; N: 6.00%. Found C: 74.65%; H: 5.70%; N: 5.67%.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


