
High-Efficacy α,β-Dehydromonacolin S Improves Hepatic Steatosis and Suppresses Gluconeogenesis Pathway in High-Fat Diet-Induced Obese Rats

 ,
,
Abstract
:
1. Introduction
2. Results
2.1. C5 Improved the General Characteristics and Biochemical Parameters of HFD-Induced Obese Rats
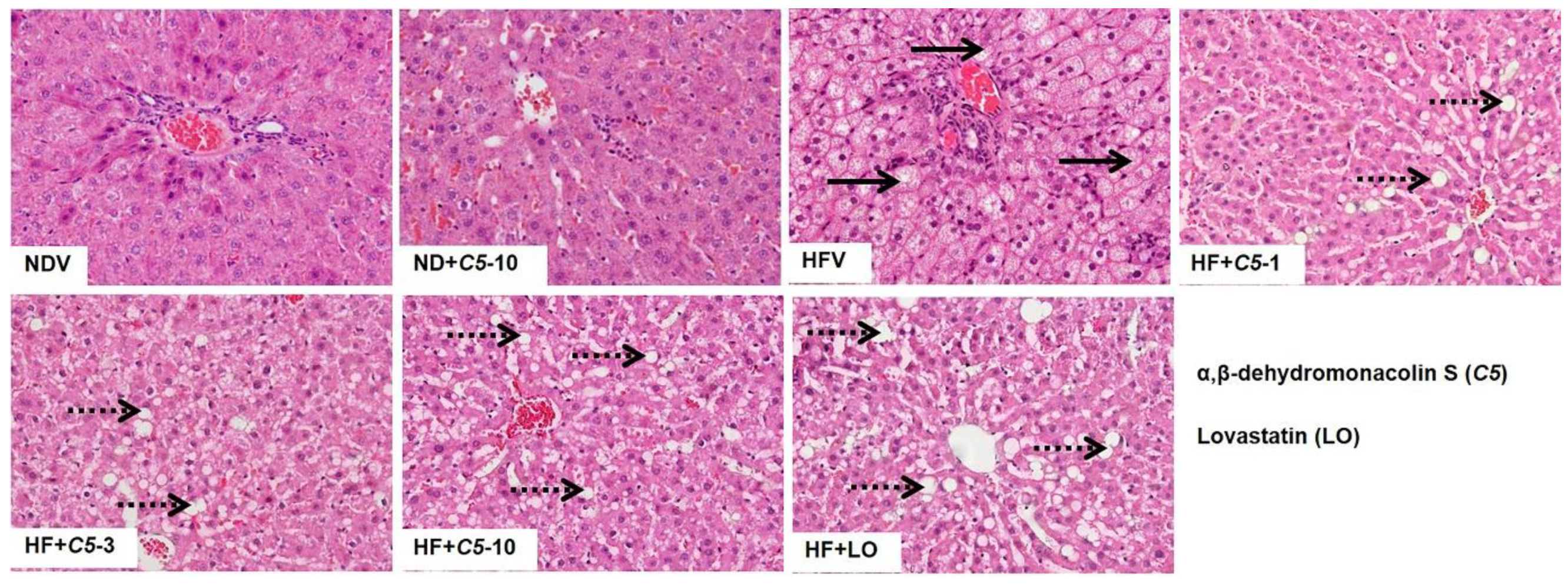
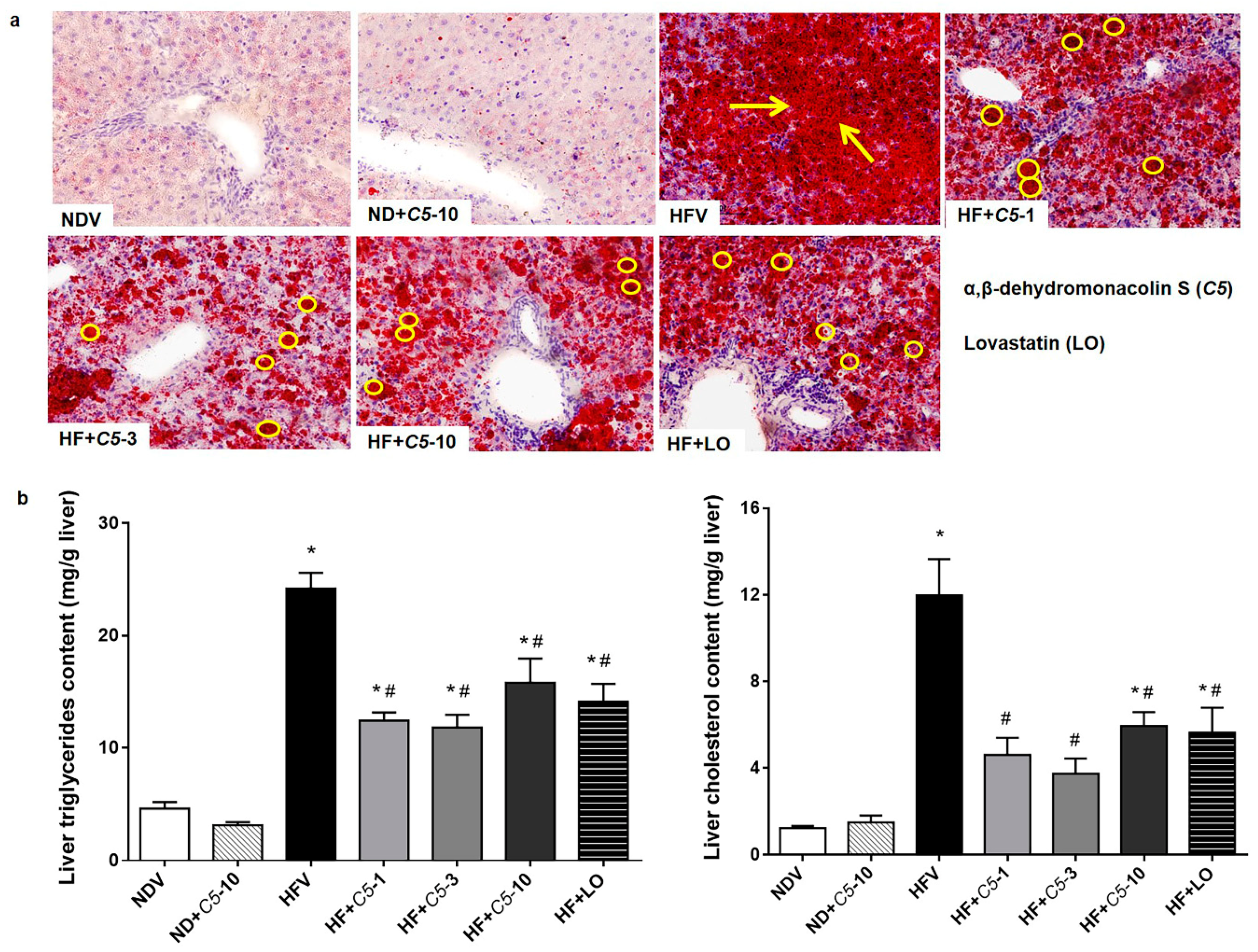
2.2. C5 Improved Hepatic Histological Features and Exhibited Anti-Hepatic Steatosis
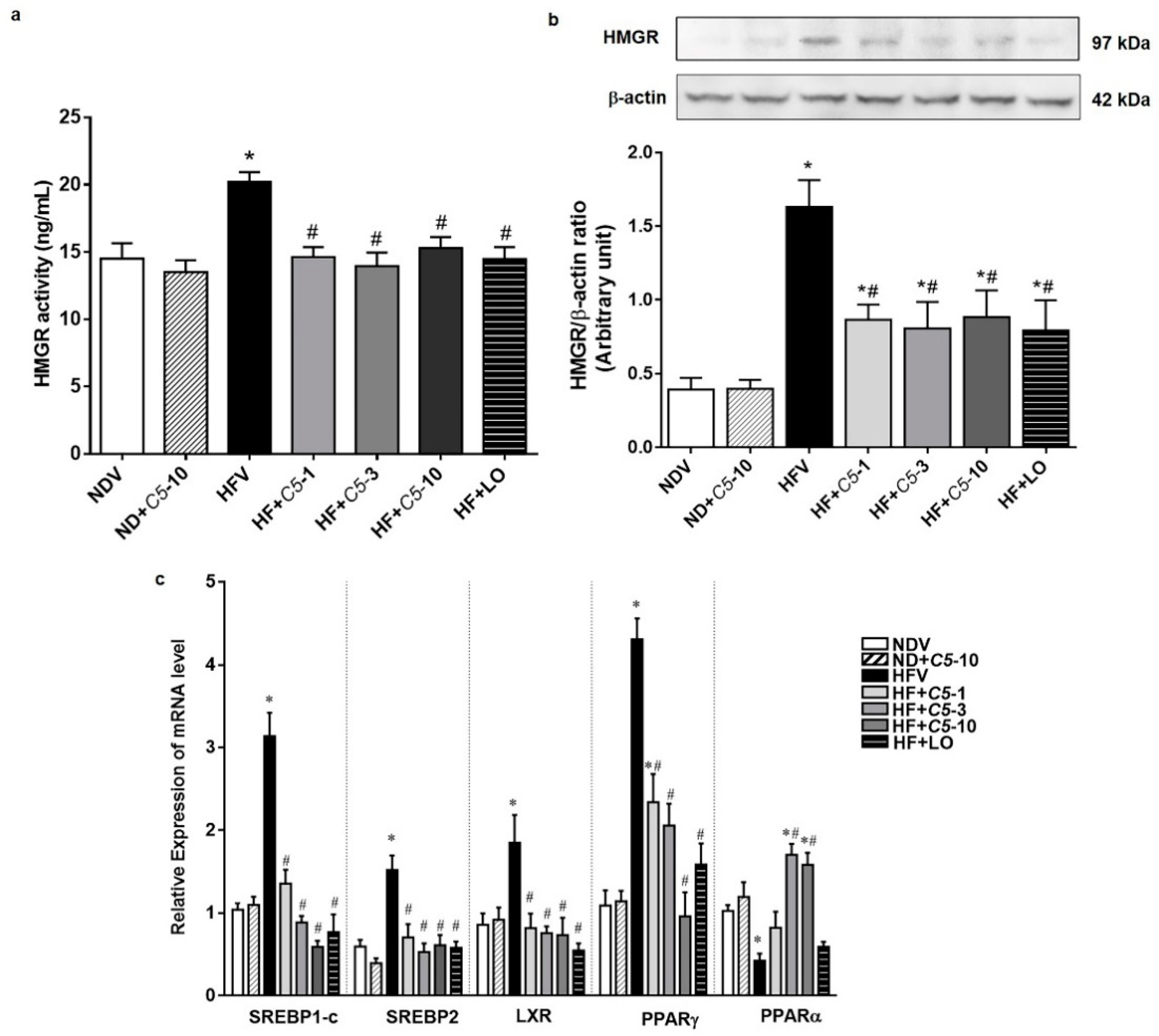
2.3. C5 Inhibited HMG-CoA Reductase Activity and Expression and Modulated the Lipid Metabolic Gene Expression
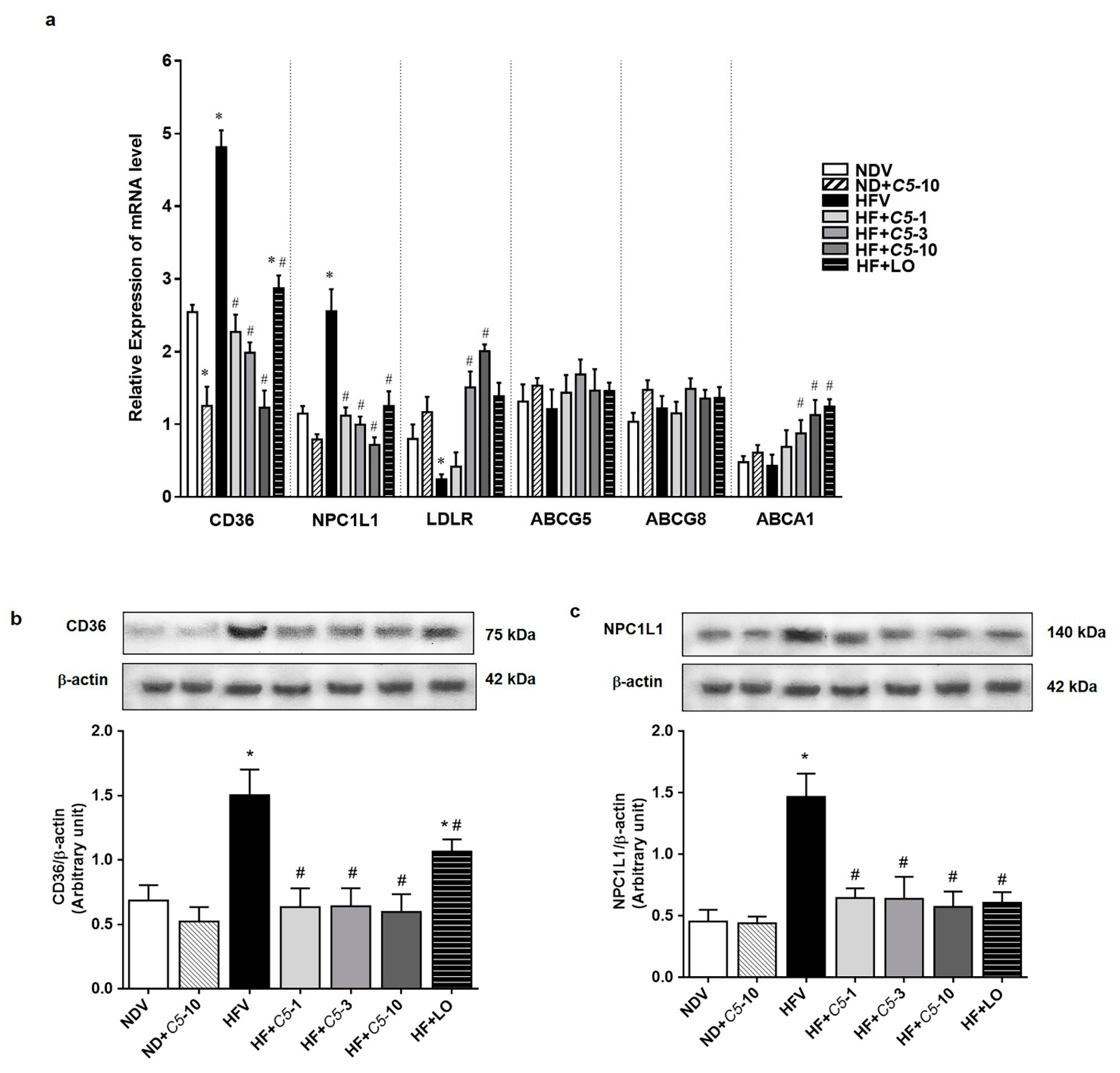
2.4. C5 Modulated Hepatic Lipid Transporter Gene and Protein Expression
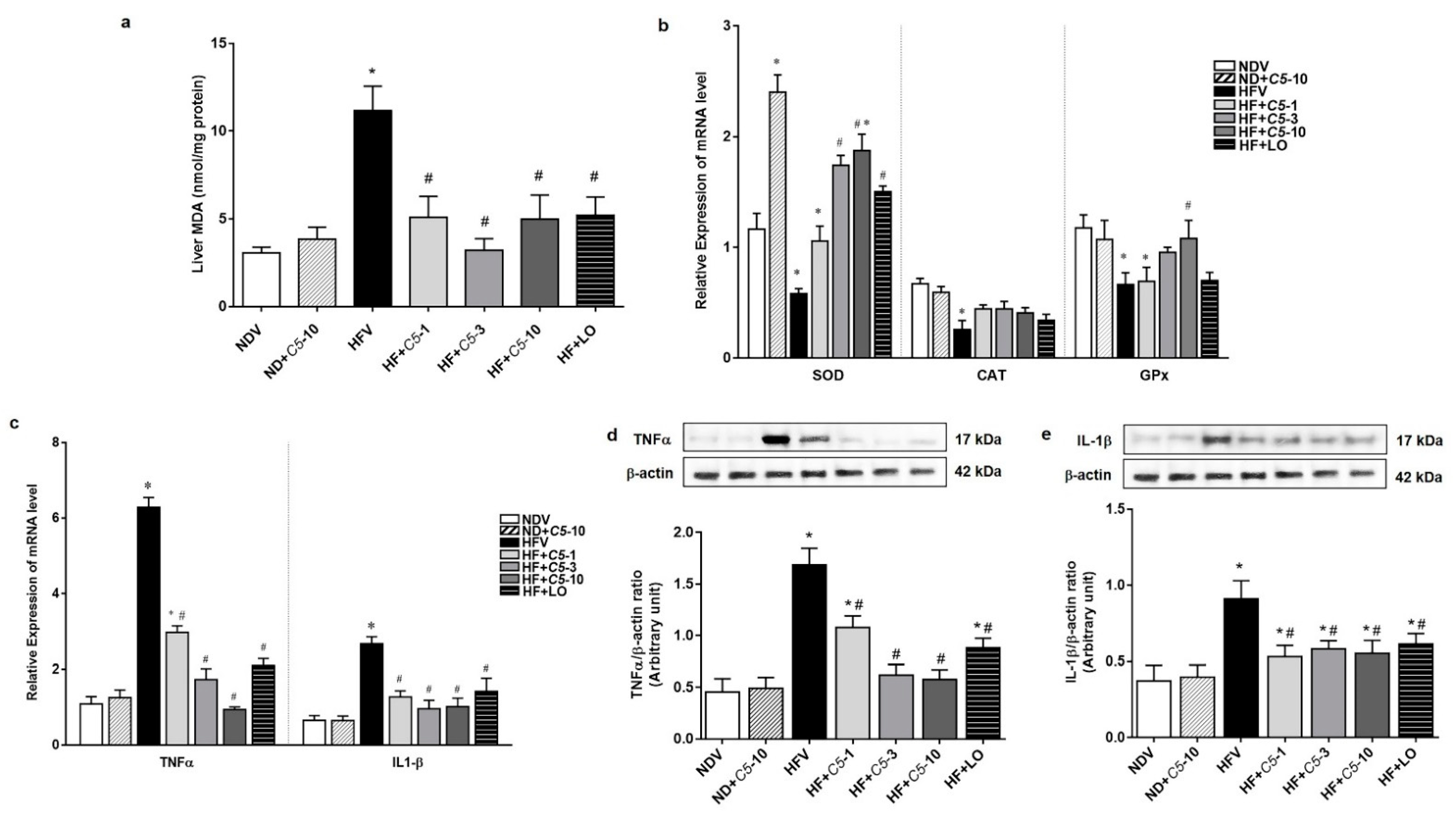
2.5. Antioxidant and Anti-Inflammatory Effects of C5
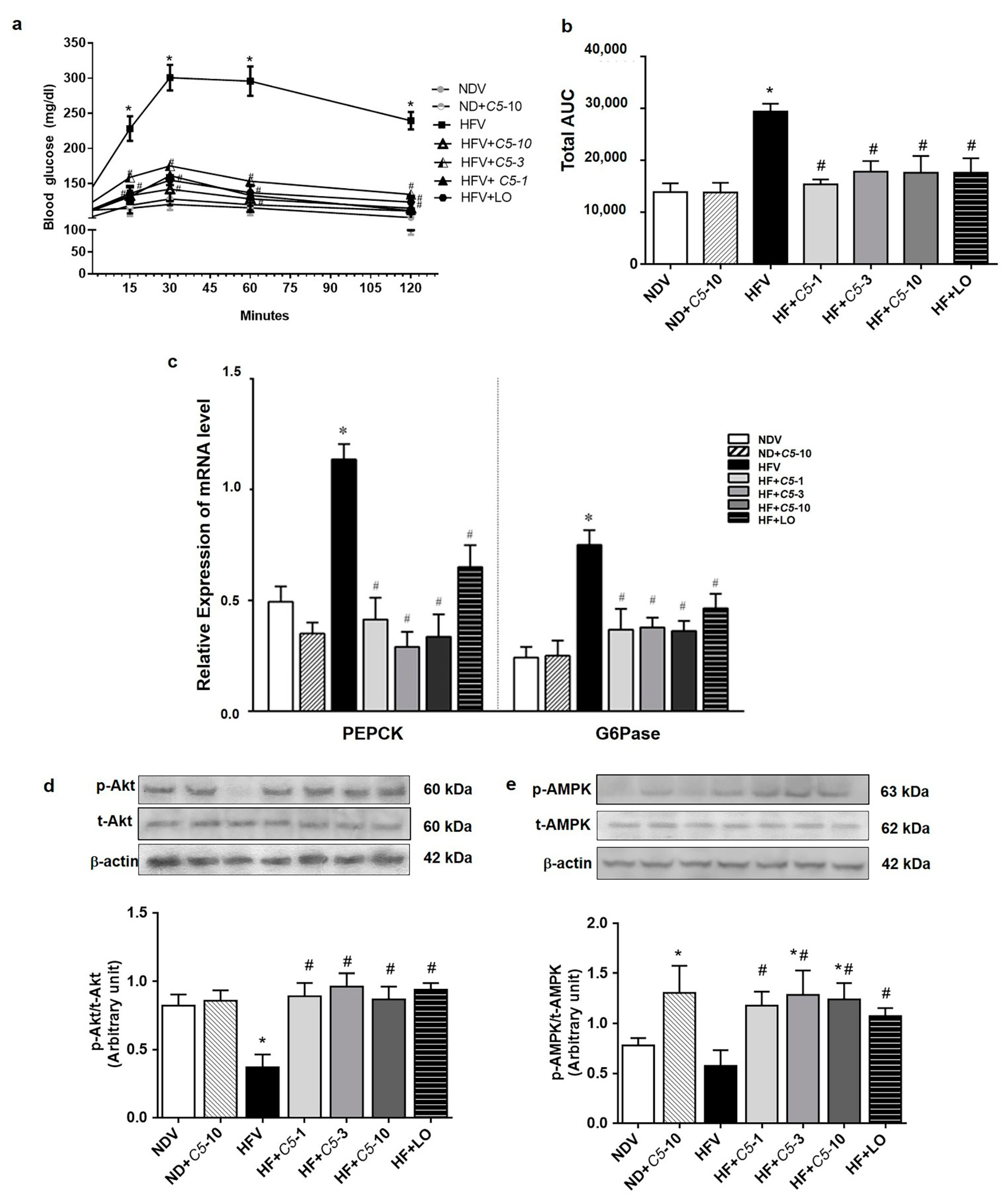
2.6. C5 Suppressed Hepatic Gluconeogenesis and Improved Insulin Signaling
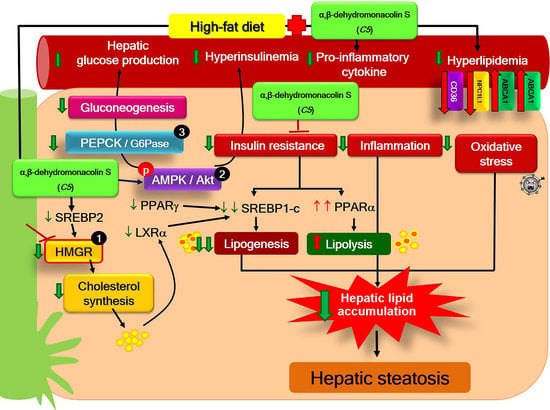
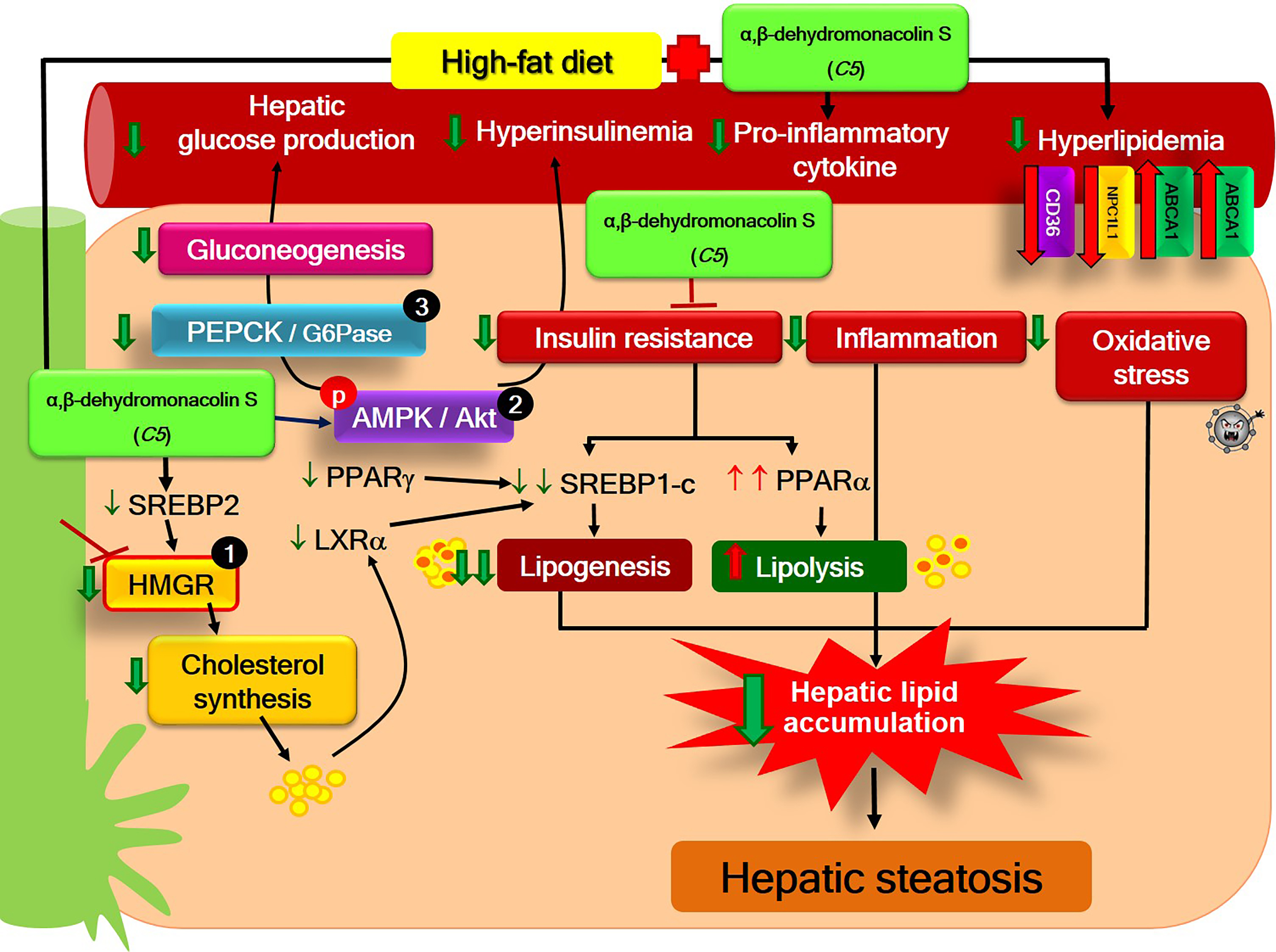
3. Discussion
4. Methods
4.1. Chemicals
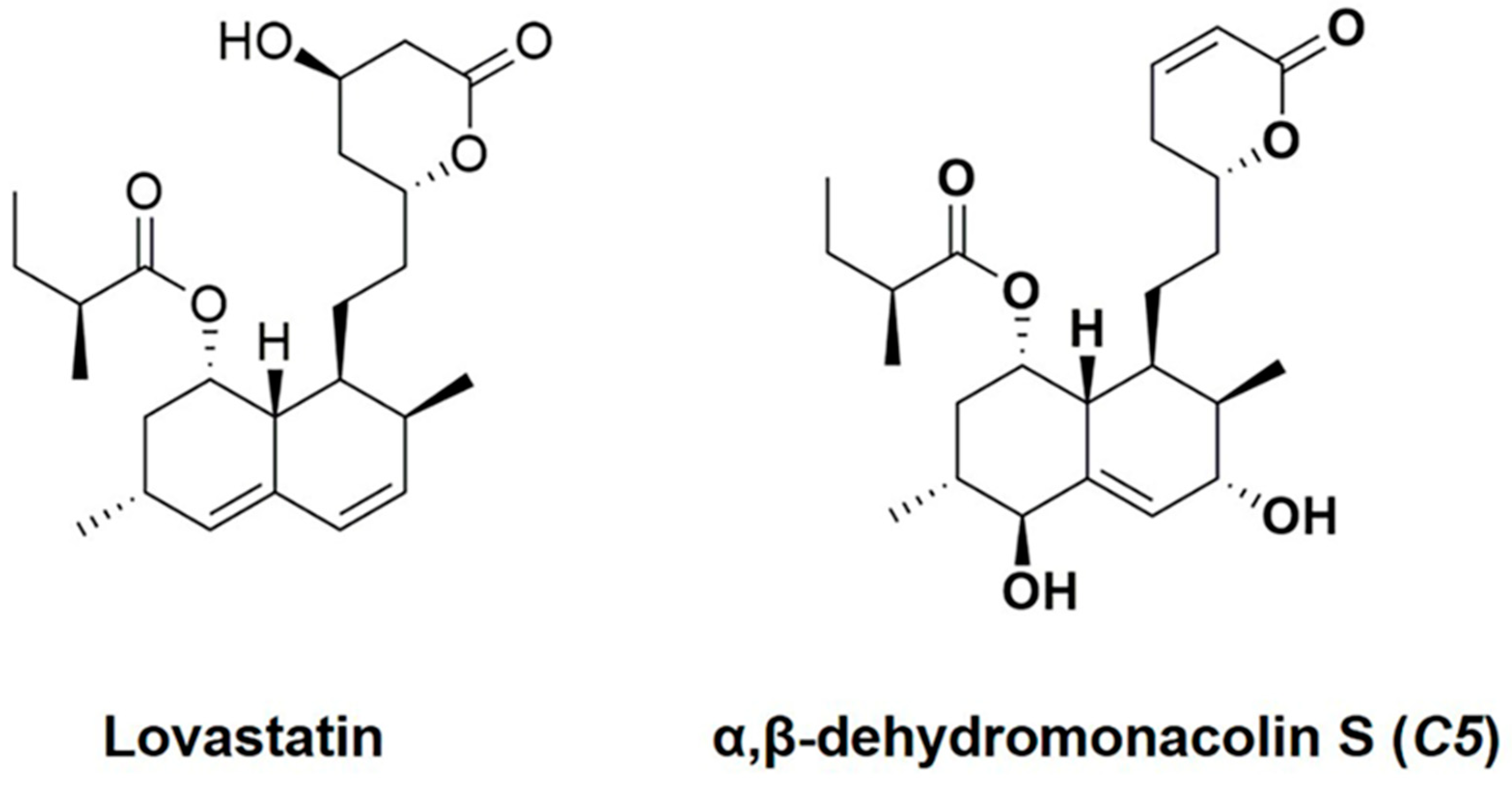
4.2. Preparation of Lovastatin and α,β-Dehydromonacolin S
4.3. Animal Induction
4.4. Determination of Plasma Metabolic Parameters
4.5. Determination of the Liver Morphology, Lipid Accumulation, and Lipid Content
4.6. Determination of Hepatic HMGR Activity and Lipid Peroxidation
4.7. Expression of Genes Involved Hepatic Lipid Metabolism, Lipid Transporters, Antioxidant, and In-Flammation Using the Quantitative Polymerase Chain Reaction (qPCR) Technique
4.8. Expression of Protein Involved in Hepatic Lipid Transporters and Insulin Signaling Using Western Blotting Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Seebacher, F.; Zeigerer, A.; Kory, N.; Krahmer, N. Hepatic lipid droplet homeostasis and fatty liver disease. Semin. Cell Dev. Biol. 2020, 108, 72–81. [Google Scholar] [CrossRef]
- Chen, Q.; Xiong, C.; Jia, K.; Jin, J.; Li, Z.; Huang, Y.; Liu, Y.; Wang, L.; Luo, H.; Li, H.; et al. Hepatic transcriptome analysis from HFD-fed mice defines a long noncoding RNA regulating cellular cholesterol levels. J. Lipid Res. 2019, 60, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, N.; Sarna, L.K.; Hwang, S.Y.; Zhu, Q.; Wang, P.; Siow, Y.L.; O, K. Activation of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase during high fat diet feeding. Biochim. Biophys. Acta 2013, 1832, 1560–1568. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, S.; Mo, Z.; Sun, S.; Yin, K.; Lv, Y. Emerging role of Insig-1 in lipid Metabolism and lipid disorders. Clin. Chim. Acta 2020, 508, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Ducheix, S.; Lobaccaro, J.M.; Martin, P.G.; Guillou, H. Liver X Receptor: An oxysterol sensor and a major player in the control of lipogenesis. Chem. Phys. Lipids 2011, 164, 500–514. [Google Scholar] [CrossRef]
- Mourikis, P.; Zako, S.; Dannenberg, L.; Nia, A.M.; Heinen, Y.; Busch, L.; Richter, H.; Hohlfeld, T.; Zeus, T.; Kelm, M.; et al. Lipid lowering therapy in cardiovascular disease: From myth to molecular reality. Pharmacol. Ther. 2020, 213, 107592. [Google Scholar] [CrossRef]
- Meor Anuar Shuhaili, M.F.R.; Samsudin, I.N.; Stanslas, J.; Hasan, S.; Thambiah, S.C. Effects of Different Types of Statins on Lipid Profile: A Perspective on Asians. Int. J. Endocrinol. Metab. 2017, 15, e43319. [Google Scholar] [CrossRef] [Green Version]
- McFarland, A.J.; Anoopkumar-Dukie, S.; Arora, D.S.; Grant, G.D.; McDermott, C.M.; Perkins, A.V.; Davey, A.K. Molecular mechanisms underlying the effects of statins in the central nervous system. Int. J. Mol. Sci. 2014, 15, 20607–20637. [Google Scholar] [CrossRef] [Green Version]
- Kavalipati, N.; Shah, J.; Ramakrishan, A.; Vasnawala, H. Pleiotropic effects of statins. Indian J. Endocrinol. Metab. 2015, 19, 554–562. [Google Scholar]
- Wang, H.J.; Park, J.Y.; Kwon, O.; Choe, E.Y.; Kim, C.H.; Hur, K.Y.; Lee, M.S.; Yun, M.; Cha, B.S.; Kim, Y.B.; et al. Chronic HMGCR/HMG-CoA reductase inhibitor treatment contributes to dysglycemia by upregulating hepatic gluconeogenesis through autophagy induction. Autophagy 2015, 11, 2089–2101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.Y.; Bang, I.H.; Han, C.Y.; Lee, D.H.; Park, B.H.; Bae, E.J. Statin suppresses sirtuin 6 through miR-495, increasing FoxO1-dependent hepatic gluconeogenesis. Theranostics 2020, 10, 11416–11427. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Garcia, U.; Jebari, S.; Larrea-Sebal, A.; Uribe, K.B.; Siddiqi, H.; Ostolaza, H.; Benito-Vicente, A.; Martín, C. Statin Treatment-Induced Development of Type 2 Diabetes: From Clinical Evidence to Mechanistic Insights. Int. J. Mol. Sci. 2020, 21, 4725. [Google Scholar] [CrossRef]
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 484–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Srivastava, N.; Gomes, J. The effect of lovastatin on oxidative stress and antioxidant enzymes in hydrogen peroxide intoxicated rat. Food Chem. Toxicol. 2011, 49, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.W.; Shin, P.G.; Lee, J.H.; Choi, W.S.; Kang, M.J.; Kong, W.S.; Oh, M.J.; Seo, Y.B.; Kim, G.D. Anti-inflammatory effect of lovastatin is mediated via the Modulation of NF-kappaB and inhibition of HDAC1 and the PI3K/Akt/mTOR pathway in RAW264.7 macrophages. Int. J. Mol. Med. 2018, 41, 1103–1109. [Google Scholar]
- Vásquez-Bochm, L.X.; Velázquez-Paniagua, M.; Castro-Vázquez, S.S.; Guerrero-Rodríguez, S.L.; Mondragon-Peralta, A.; De La Fuente-Granada, M.; Pérez-Tapia, S.M.; González-Arenas, A.; Velasco-Velázquez, M.A. Transcriptome-based identification of lovastatin as a breast cancer stem cell-targeting drug. Pharmacol. Rep. 2019, 71, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Sharkawi, S.M.Z.; El-Shoura, E.A.M.; Abo-Youssef, A.M.; Hemeida, R.A.M. The potential hepatoprotective effects of lovastatin combined with oral hypoglycemic agents in streptozotocin-induced diabetes in rats. ImmunoPharmacol. Immunotoxicol. 2020, 42, 165–173. [Google Scholar] [CrossRef]
- Phainuphong, P.; Rukachaisirikul, V.; Saithong, S.; Phongpaichit, S.; Bowornwiriyapan, K.; Muanprasat, C.; Srimaroeng, C.; Duangjai, A.; Sakayaroj, J. Lovastatin Analogues from the Soil-Derived Fungus Aspergillus sclerotiorum PSU-RSPG178. J. Nat. Prod. 2016, 79, 1500–1507. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, W.H.; Zhao, Q.B.; Wang, X.Y.; Bao, B. Inhibition of HMG-CoA reductase by MFS, a purified extract from the fermentation of marine fungus Fusarium solani FG319, and optimization of MFS production using response surface methodology. Chin. J. Nat. Med. 2015, 13, 346–354. [Google Scholar]
- Rahman, M.A.; Abdullah, N.; Aminudin, N. Inhibitory effect on in vitro LDL Oxidation and HMG Co-A reductase activity of the liquid-liquid partitioned fractions of Hericium erinaceus (Bull.) Persoon (lion’s mane mushroom). Biomed. Res. Int. 2014, 2014, 828149. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, D.; Khan, M.S.; Khan, M.S.; Ahmad, S.; Hussain, M.S.; Ali, M. Bioactivity guided fractionation and hypolipidemic property of a novel HMG-CoA reductaseinhibitor from Ficus virens Ait. Lipids Health Dis. 2015, 14, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Viscarra, J.; Kim, S.J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013, 5, 1544–1560. [Google Scholar] [CrossRef] [PubMed]
- Alkhatatbeh, M.J.; Lincz, L.F.; Thorne, R.F. Low simvastatin concentrations reduce oleic acid-induced steatosis in HepG2 cells: An in vitro model of non-alcoholic fatty liver disease. Exp. Ther. Med. 2016, 11, 1487–1492. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.; Budoff, M.J.; Saab, S.; Ahmadi, N.; Gordon, C.; Guerci, A.D. Atorvastatin and antioxidants for the treatment of nonalcoholic fatty liver disease: The St Francis Heart Study randomized clinical trial. Am. J. Gastroenterol. 2011, 106, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Arazi, S.S.; Genvigir, F.D.; Willrich, M.A.; Hirata, M.H.; Dorea, E.L.; Bernik, M.; Hirata, R.D. Atorvastatin effects on SREBF1a and SCAP gene expression in mononuclear cells and its relation with lowering-lipids response. Clin. Chim. Acta 2008, 393, 119–124. [Google Scholar] [CrossRef]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Jing, F.; Yu, C.; Gao, L.; Qin, Y.; Zhao, J. AICAR-Induced Activation of AMPK inhibits TSH/SREBP-2/HMGCR pathway in liver. PLoS ONE 2015, 10, e0124951. [Google Scholar] [CrossRef]
- Tang, H.; Yu, R.; Liu, S.; Huwatibieke, B.; Li, Z.; Zhang, W. Irisin Inhibits Hepatic Cholesterol Syn-thesis via AMPK-SREBP2 Signaling. EBioMedicine 2016, 6, 139–148. [Google Scholar] [CrossRef] [Green Version]
- Sanguino, E.; Roglans, N.; Alegret, M.; Sanchez, R.M.; Vazquez-Carrera, M.; Laguna, J.C. Atorvastatin reverses age-related reduction in rat hepatic PPARalpha and HNF-4. Br. J. Pharmacol. 2005, 145, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.S.; Chen, J.H. The mechanism of enterohepatic circulation in the formation of gallstone disease. J. Membr. Biol. 2014, 247, 1067–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Zhou, X.; Yokoyama, T.; Hajjar, D.P.; Gotto, A.M., Jr.; Nicholson, A.C. Pitavastatin down-regulates expression of the macrophage type B scavenger receptor, CD36. Circulation 2004, 109, 790–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, G.G.; Xiao, H.B.; Lu, X.Y.; Sun, Z.L. Naringin Activates AMPK Resulting in Altered Expression of SREBPs, PCSK9, and LDLR To Reduce Body Weight in Obese C57BL/6J Mice. J. Agric. Food Chem. 2018, 66, 8983–8990. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Deng, Y.; Xiao, J.; Liu, X.; Zhou, M.; Ren, Z.; Peng, J.; Tang, Y.; Jiang, Z.; Tang, Z.; et al. Three Musketeers for Lowering Cholesterol: Statins, Ezetimibe and Evolocumab. Curr. Med. Chem. 2020, 28, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Lindeman, J.H.; Menke, A.L.; Koonen, D.P.; Morrison, M.; Havekes, L.M.; van den Hoek, A.M.; Kleemann, R. Metabolically induced liver inflammation leads to NASH and differs from LPS- or IL-1beta-induced chronic inflammation. Lab Investig. 2014, 94, 491–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozguler, I.M.; Burma, O.; Uysal, A.; Akbulut, H. Rosuvastatin Lowers Systemic Inflammatory Response in Coronary Artery Bypass Graft Accompanied by Cardiopulmonary Bypass Surgery: A Randomized Controlled Study. Clin. Investig. Med. 2015, 38, E154–E163. [Google Scholar] [CrossRef] [Green Version]
- Ascer, E.; Bertolami, M.C.; Venturinelli, M.L.; Buccheri, V.; Souza, J.; Nicolau, J.C.; Ramires, J.A.; Serra-no, C.V., Jr. Atorvastatin reduces proinflammatory markers in hypercholesterolemic patients. Atherosclerosis 2004, 177, 161–166. [Google Scholar] [CrossRef]
- Rezaie-Majd, A.; Maca, T.; Bucek, R.A.; Valent, P.; Muller, M.R.; Husslein, P.; Kashanipour, A.; Minar, E.; Baghestanian, M. Simvastatin reduces expression of cytokines interleukin-6, interleukin-8, and monocyte chemoattractant protein-1 in circulating monocytes from hypercholesterolemic patients. Arterioscler Thromb. Vasc. Biol. 2002, 22, 1194–1199. [Google Scholar] [CrossRef]
- Sun, W.; Lee, T.S.; Zhu, M.; Gu, C.; Wang, Y.; Zhu, Y.; Shyy, J.Y. Statins activate AMP-activated protein kinase in vitro and in vivo. Circulation 2006, 114, 2655–2662. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Meng, S.; Chang, E.; Beckwith-Fickas, K.; Xiong, L.; Cole, R.N.; Radovick, S.; Wondisford, F.E.; He, L. Low concentrations of metformin suppress glucose production in hepatocytes through AMP-activated protein kinase (AMPK). J. Biol. Chem. 2014, 289, 20435–20446. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, K.; Viswanad, B.; Asrat, L.; Kaul, C.L.; Ramarao, P. Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: A model for type 2 diabetes and pharmacological screening. Pharmacol. Res. 2005, 52, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Ontawong, A.; Boonphang, O.; Pasachan, T.; Duangjai, A.; Pongchaidecha, A.; Phatsara, M.; Jinakote, M.; Amornlerdpison, D.; Srimaroeng, C. Hepatoprotective effect of coffee pulp aqueous extract combined with simvastatin against hepatic steatosis in high-fat diet-induced obese rats. J. Funct. Foods 2019, 54, 568–577. [Google Scholar] [CrossRef]
- Cheng, O.; Tian, X.; Luo, Y.; Mai, S.; Yang, Y.; Kuang, S.; Chen, Q.; Ma, J.; Chen, B.; Li, R.; et al. Liver X receptors agonist promotes differentiation of rat bone marrow derived mesenchymal stem cells into dopaminergic neuron-like cells. Oncotarget 2018, 9, 576–590. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, H.; Sato, M.; Yakushiji, M.; Koshiguchi, M.; Hirai, S.; Egashira, Y. Regulation of rat hepatic α-amino-β-carboxymuconate-ε-semialdehyde decarboxylase, a key enzyme in the tryptophan- NAD pathway, by dietary cholesterol and sterol regulatory element-binding protein-2. Eur. J. Nutr. 2014, 53, 469–477. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Hu, J.; Shen, W.J.; Singh, M.; Hou, X.; Bittner, A.; Bittner, S.; Cortez, Y.; Tabassum, J.; et al. Effect of Creosote Bush-Derived NDGA on Expression of Genes Involved in Lipid Metabolism in Liver of High-Fructose Fed Rats: Relevance to NDGA Amelioration of Hypertriglyceridemia and Hepatic Steatosis. PLoS ONE 2015, 10, e0138203. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | NDV | ND+C5-10 | HFV | HF+C5-1 | HF+C5-3 | HF+C5-10 | HF+LO |
|---|---|---|---|---|---|---|---|
| Food intake | 30.46 ± 1.87 | 28.46 ± 2.18 | 29.73 ± 1.86 | 28.76 ± 1.46 | 27.65 ± 2.25 | 30.12 ± 2.83 | 29.75 ± 2.14 |
| Calories intake (kcal/day) | 122.92 ± 1.45 | 115.27 ± 2.45 | 159.05 ± 1.75 * | 146.39 ± 2.17 * | 147.92 ± 1.45 * | 161.142 ± 2.13 * | 159.16 ± 1.97 * |
| Water intake (ml/day) | 20.84 ± 3.15 | 20.12 ± 1.56 | 20.46 ± 2.46 | 19.65 ± 2.86 | 20.46 ± 2.73 | 21.78 ± 3.45 | 20.56 ± 2.16 |
| BW (g) | 546.43 ± 7.31 | 510.00 ± 9.91 | 637.50 ± 20.24 * | 633.57 ± 26.90 * | 610.00 ± 16.22 | 648.57 ± 37.30 * | 615.00 ± 24.86 |
| BW gain (g) | 371.43 ± 5.61 | 340.83 ± 10.60 | 467.50 ± 19.86 * | 457.86 ± 25.89 * | 436.43 ± 15.65 | 475.00 ± 37.64 * | 441.43 ± 25.21 |
| LW (g) | 13.07 ± 0.62 | 14.34 ± 0.59 | 29.48 ± 2.61 * | 23.78 ± 2.44 | 21.14 ± 1.32 | 24.63 ± 2.43 | 22.51 ± 2.19 |
| Liver index * | 2.40 ± 0.12 | 2.81 ± 0.10 | 4.72 ± 0.58 * | 3.81 ± 0.45 * | 3.49 ± 0.27 | 3.56 ± 0.22 | 3.68 ± 0.37 * |
| Visceral fat weight (g) | 25.66 ± 1.32 | 27.61 ± 3.86 | 65.32 ± 2.26 * | 43.48 ± 3.38 *# | 30.64 ± 1.18 # | 32.89 ± 1.48 # | 41.15 ± 3.29 *# |
| Plasma Parameters | NDV | ND+C5-10 | HFV | HF+C5-1 | HF+C5-3 | HF+C5-10 | HF+LO |
|---|---|---|---|---|---|---|---|
| Cholesterol (mg/dL) | 73.26 ± 3.21 | 73.48 ± 4.60 | 115.10 ± 8.41 * | 83.43 ± 4.18 # | 70.57 ± 3.60 # | 86.22 ± 4.12 # | 79.39 ± 3.19 # |
| Triglyceride (mg/dL) | 43.86 ± 2.94 | 51.04 ± 3.10 | 80.29 ± 4.40 * | 60.69 ± 5.61 # | 55.54 ± 3.09 # | 64.39 ± 5.17 # | 58.72 ± 6.67 # |
| HDL (mg/dL) | 31.70 ± 2.99 | 28.47 ± 1.89 | 14.69 ± 1.05 * | 26.26 ± 1.90 # | 28.71 ± 2.02 # | 31.36 ± 2.00 # | 30.39 ± 1.68 # |
| LDL (mg/dL) | 32.78 ± 5.42 | 34.80 ± 5.33 | 84.35 ± 8.42 * | 45.03 ± 4.78 # | 30.76 ± 5.39 # | 41.99 ± 6.11 # | 36.71 ± 2.84 # |
| Glucose (mg/dL) | 117.48 ± 9.21 | 117.96 ± 15.77 | 137.75 ± 13.73 | 116.41 ± 10.32 | 120.88 ± 10.35 | 122.69 ± 9.63 | 124.27 ± 12.01 |
| Insulin (ng/mL) | 1.60 ± 0.41 | 1.51 ± 0.12 | 3.94 ± 0.37 * | 1.47 ± 0.29 # | 1.95 ± 0.35 # | 1.86 ± 0.30 # | 1.63 ± 0.27 # |
| HOMA index | 11.01 ± 2.12 | 10.89 ± 2.13 | 32.41 ± 3.62 * | 11.41 ± 3.07 # | 14.26 ± 3.35 # | 14.05 ± 2.57 # | 11.55 ± 1.62 # |
| AST (U/L) | 153.71 ± 13.42 | 142.67 ± 16.79 | 222.00 ± 21.32 * | 123.29 ± 13.51 # | 133 ± 9.88 # | 166.43 ± 11.90 | 128.14 ± 7.22 # |
| ALT (U/L) | 86.57 ± 15.53 | 79.83 ± 15.06 | 253.00 ± 15.62 * | 66.43 ± 9.12 # | 70.57 ± 13.71 # | 79.29 ± 10.63 # | 81.14 ± 9.36 # |
| BUN (mg/dL) | 21.18 ± 2.50 | 23.71 ± 1.51 | 43.83 ± 13.04 * | 23.64 ± 2.00 # | 23.03 ± 2.35 # | 27.60 ± 1.85 # | 26.24 ± 2.77 # |
| Creatinine (mg/dL) | 0.81 ± 0.03 | 0.80 ± 10.03 | 1.39 ± 10.07 * | 0.80 ± 0.06 # | 0.92 ± 0.03 # | 1.12 ± 0.05 *# | 0.97 ± 0.07 # |
| GFR (ml/min) | 2.36 ± 0.19 | 2.10 ± 10.24 | 3.39 ± 10.23 * | 2.31 ± 0.19 | 2.42 ± 0.25 | 2.54 ± 0.26 | 2.68 ± 0.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaewmalee, J.; Ontawong, A.; Duangjai, A.; Tansakul, C.; Rukachaisirikul, V.; Muanprasat, C.; Srimaroeng, C. High-Efficacy α,β-Dehydromonacolin S Improves Hepatic Steatosis and Suppresses Gluconeogenesis Pathway in High-Fat Diet-Induced Obese Rats. Pharmaceuticals 2021, 14, 375. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14040375
Kaewmalee J, Ontawong A, Duangjai A, Tansakul C, Rukachaisirikul V, Muanprasat C, Srimaroeng C. High-Efficacy α,β-Dehydromonacolin S Improves Hepatic Steatosis and Suppresses Gluconeogenesis Pathway in High-Fat Diet-Induced Obese Rats. Pharmaceuticals. 2021; 14(4):375. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14040375
Chicago/Turabian StyleKaewmalee, Jutatip, Atcharaporn Ontawong, Acharaporn Duangjai, Chittreeya Tansakul, Vatcharin Rukachaisirikul, Chatchai Muanprasat, and Chutima Srimaroeng. 2021. "High-Efficacy α,β-Dehydromonacolin S Improves Hepatic Steatosis and Suppresses Gluconeogenesis Pathway in High-Fat Diet-Induced Obese Rats" Pharmaceuticals 14, no. 4: 375. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14040375