
Potential Tamoxifen Repurposing to Combat Infections by Multidrug-Resistant Gram-Negative Bacilli

 , , and
, , and
Abstract
:1. Introduction
2. Results
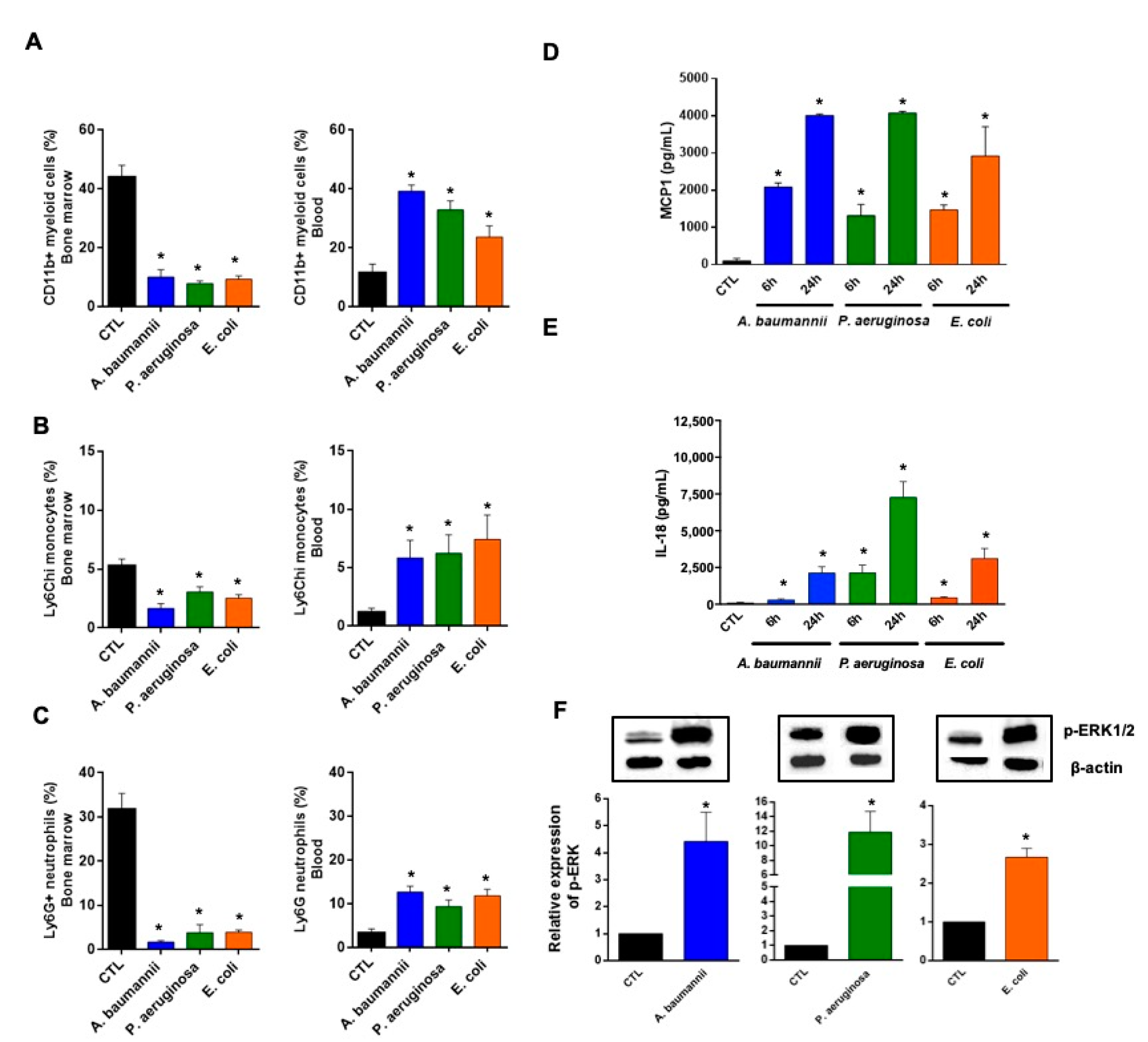
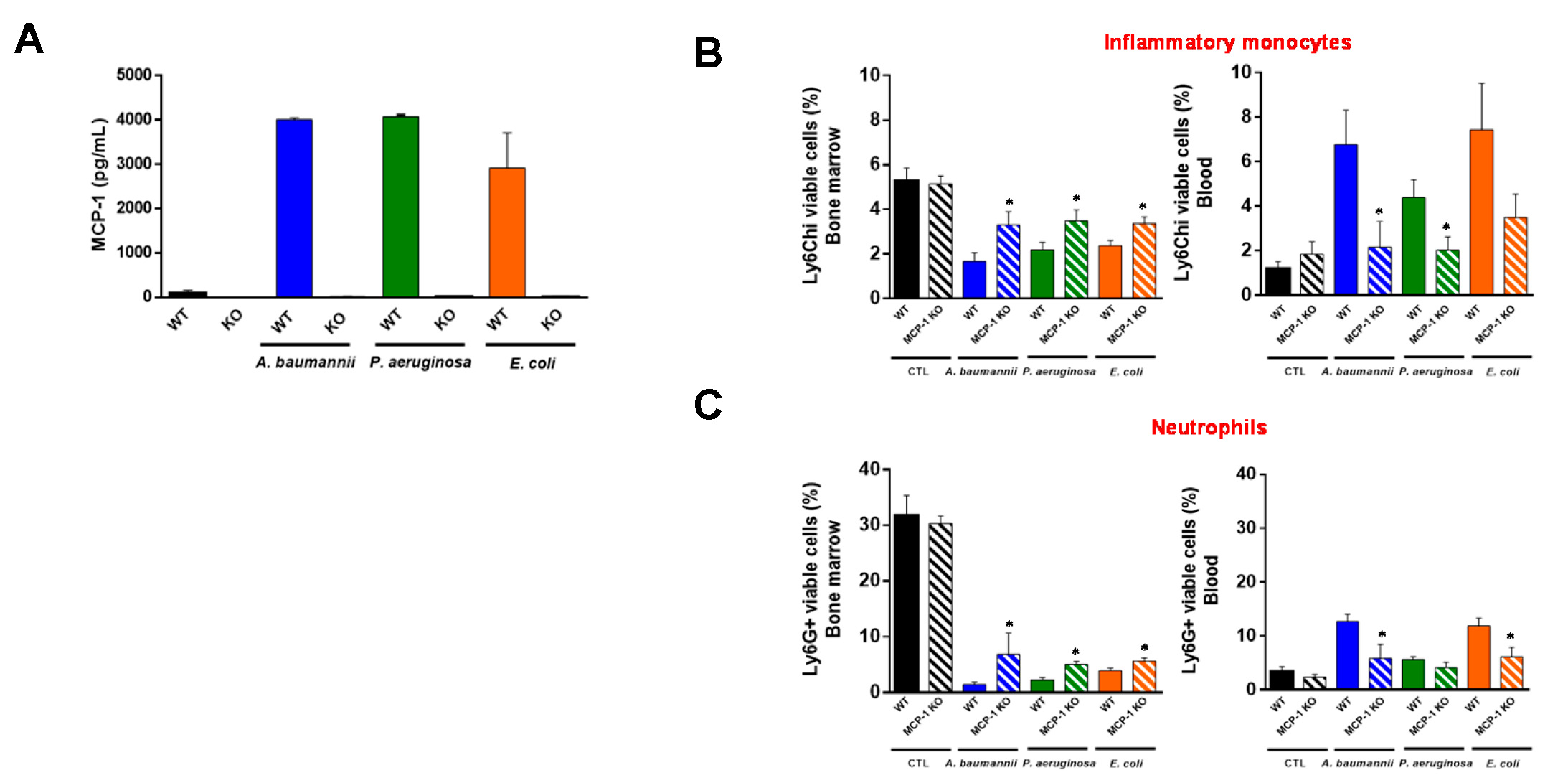
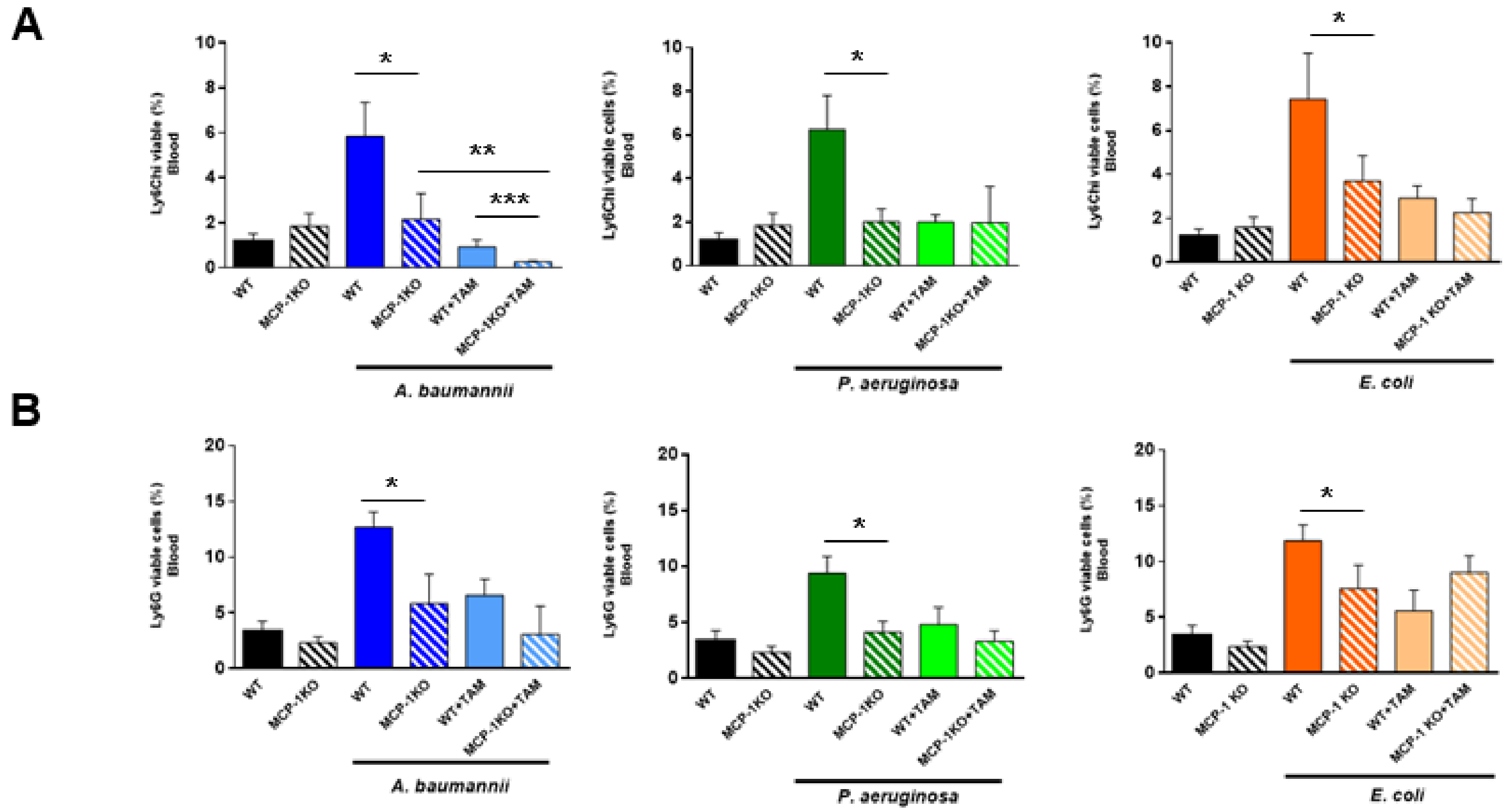
2.1. Bone Marrow Immune Cells Migrate in Response to Monocyte Chemotactic Protein-1 (MCP-1) and Interleukin-18 (IL-18) during Bacterial Infection
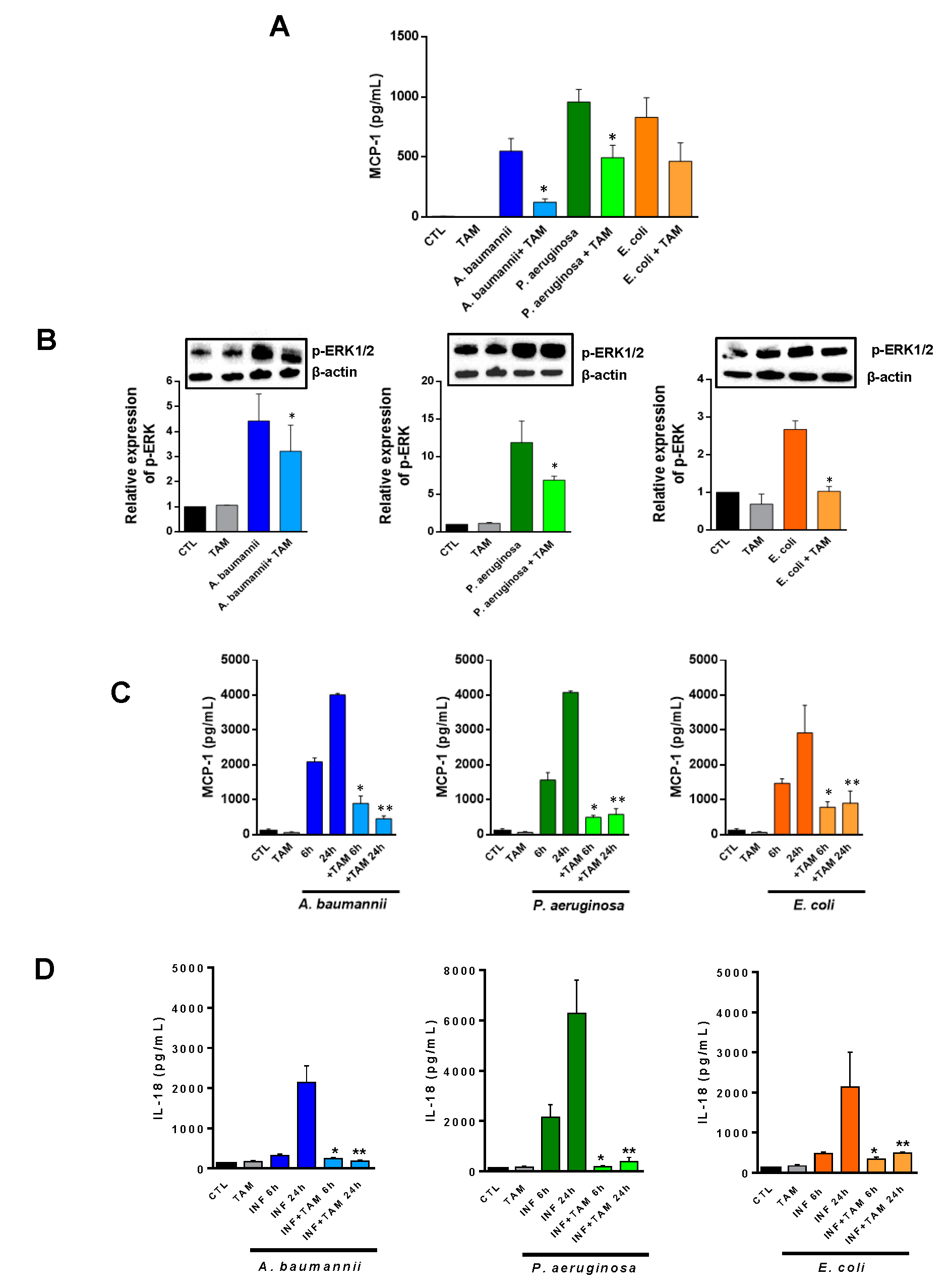
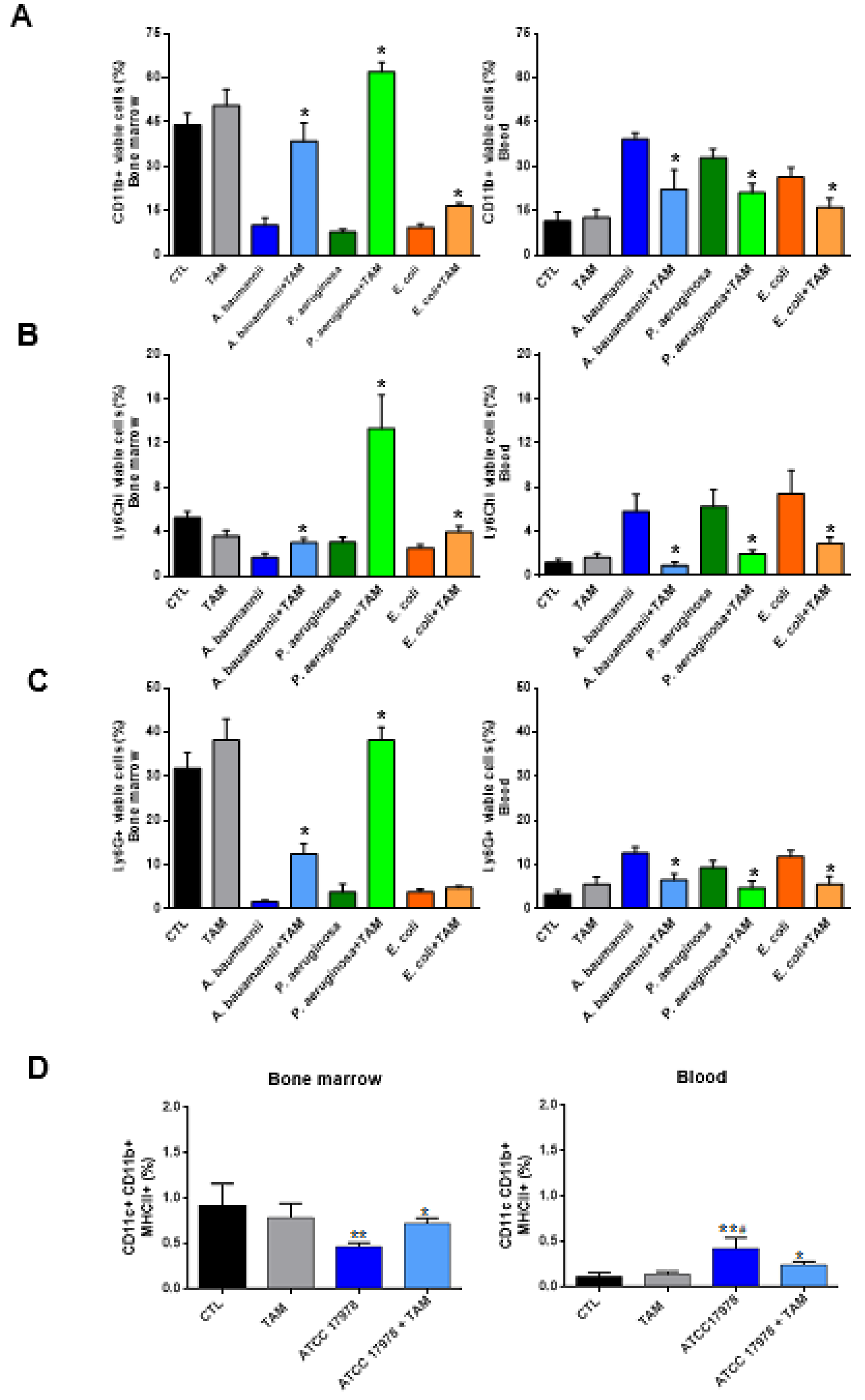
2.2. Tamoxifen Impairs the Migration of Immune Cells from Bone Marrow to Blood
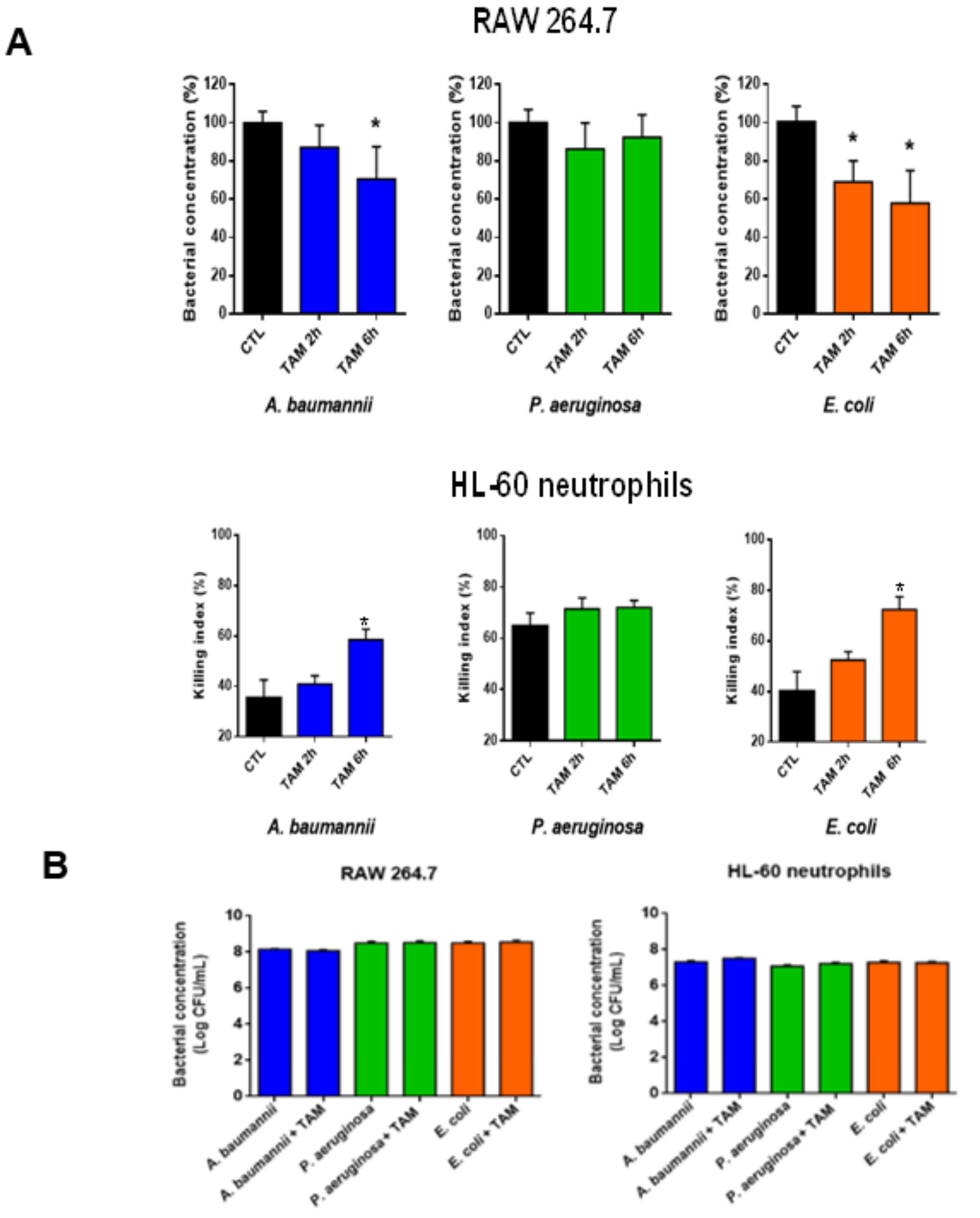
2.3. Tamoxifen Enhances Bacterial Killing by Macrophages and Neutrophils In Vitro
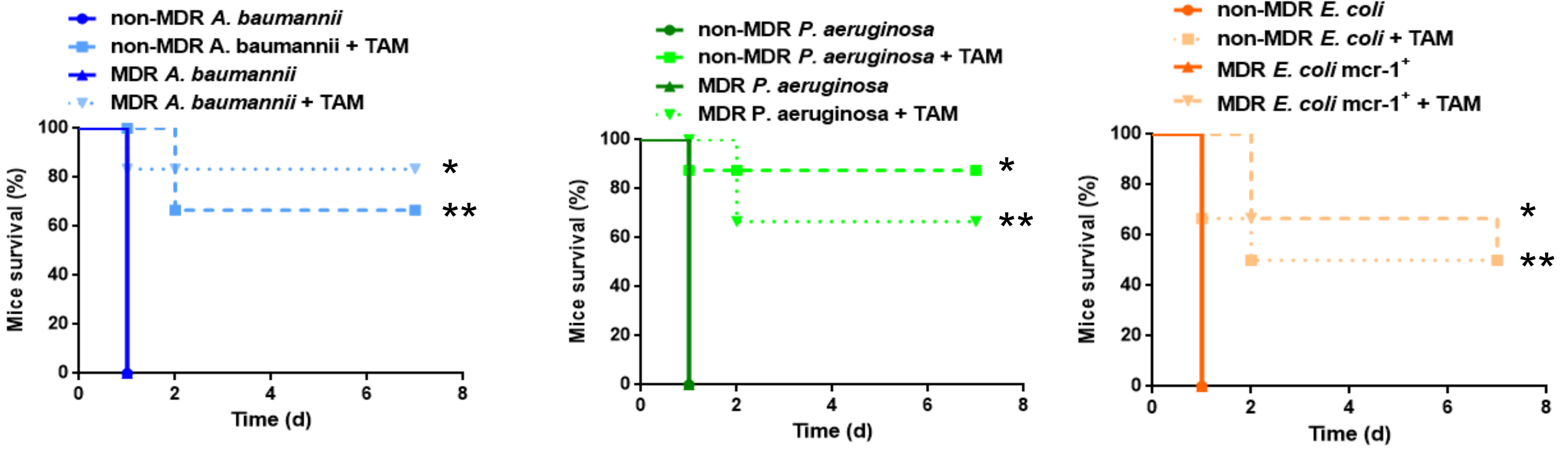
2.4. Tamoxifen Increases Mice Survival and Decreases the Bacterial Burden in a Murine Sepsis Model of A. Baumannii, P. Aeruginosa, and E. Coli
2.5. Direct Treatment with Tamoxifen Increases Mice Survival and Decreases the Bacterial Burden in a Murine Sepsis Model of A. Baumannii
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Bacterial Strains
4.3. Animals
4.4. A. Baumannii, P. Aeruginosa, and E. Coli Peritoneal Sepsis Models
4.5. Therapeutic Effect of Tamoxifen in Immunocompetent Murine Models of Peritoneal Sepsis
4.6. Flow Cytometry
4.7. Cytokine Assays
4.8. Cell Culture and Infection
4.9. Western Blot Immunoblotting
4.10. Macrophages Adhesion Assay
4.11. Neutrophil Killing Assay
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valero, C.; Garcia Palomo, J.D.; Mattoras, P.; Fernández-Mazarrasa, C.; Gonzales-Fernández, C.; Farinas, M.C. Acinetobacter bacteraemia in a teaching hospital, 1989–1998. Eur. J. Int. Med. 2001, 12, 425–429. [Google Scholar] [CrossRef]
- Annane, D.; Bellissant, E.; Cavaillon, J.M. Septic shock. Lancet 2005, 365, 63–78. [Google Scholar] [CrossRef]
- Martin, G.S.; Mannino, D.M.; Eaton, S.; Moss, M. The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med. 2003, 348, 1546–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkey, A.J.; Lagu, T.; Lindenauer, P.K. Trends in sepsis and infection sources in the US. A population-based study. Ann. Am. Thorac. Soc. 2015, 12, 216–220. [Google Scholar] [CrossRef]
- Garnacho-Montero, J.; Garcia-Garmendia, J.L.; Barrero-Almodovar, A.; Jiménez-Jiménez, F.J.; Pérez-Paredes, C.; Ortiz-Leyba, C. Impact of adequate empirical antibiotic therapy on the outcome of patients admitted to the intensive care unit with sepsis. Crit. Care Med. 2003, 31, 2742–2751. [Google Scholar] [CrossRef]
- Rasko, D.V.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. Drug Discov. 2010, 9, 117–128. [Google Scholar] [CrossRef]
- Vila-Farrés, X.; Parra-Millán, R.; Sánchez-Encinales, V.; Varese, M.; Ayerbe-Algaba, R.; Bayó, N.; Guardiola, S.; Pachón-Ibáñez, M.E.; Kotev, M.; García, J.; et al. Combating virulence of Gram-negative bacilli by OmpA inhibition. Sci. Rep. 2017, 7, 14683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smani, Y.; Domínguez-Herrera, J.; Ibáñez-Martínez, J.; Pachón, J. Therapeutic efficacy of lysophosphatidylcholine in severe infections caused by Acinetobacter baumannii. Antimicrob. Agents Chemother. 2015, 59, 3920–3924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, G.D. Antibiotic adjuvants: rescuing antibiotics from resistance. Trends Microbiol. 2016, 24, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Parra Millán, R.; Jiménez Mejías, M.E.; Sánchez Encinales, V.; Ayerbe Algaba, R.; Gutiérrez Valencia, A.; Pachón Ibáñez, M.E.; Díaz, C.; Pérez Del Palacio, J.; López Cortés, L.F.; Pachón, J.; et al. Efficacy of lysophosphatidylcholine in combination with antimicrobial agents against Acinetobacter baumannii in experimental murine peritoneal sepsis and pneumonia models. Antimicrob. Agents Chemother. 2016, 60, 4464–4470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, H.; Midorikawa, N.; Fujimoto, S.; Miyoshi, N.; Yoshida, H.; Matsumoto, T. Antimicrobial effects of lysophosphatidylcholine on methicillin-resistant Staphylococcus aureus. Ther. Adv. Infect. Dis. 2017, 4, 89–94. [Google Scholar] [PubMed] [Green Version]
- Zhao, L.; Kuo Lee, R.; Harris, G.; Tram, K.; Yan, H.; Chen, W. c-di-GMP protects against intranasal Acinetobacter baumannii infection in mice by chemokine induction and enhanced neutrophil recruitment. Int. Immunopharmacol. 2011, 11, 1378–1383. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Jia, T.; Mendez-Ferrer, S.; Hohl, T.M.; Serbina, N.V.; Lipuma, L.; Leiner, I.; Li, M.O.; Frenette, P.S.; Pamer, E.G. Bone marrow mesenchymal stem and progenitor cells induce monocyte emigration in response to circulating toll-like receptor ligands. Immunity 2011, 34, 590–601. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.K.; Kwon, H.; Khil, L.Y.; Zhang, L.; Jun, H.S.; Yoon, J.W. IL-18 induces monocyte chemotactic protein-1 production in macrophages through the phosphatidylinositol 3-kinase/Akt and MEK/ERK1/2 pathways. J. Immunol. 2005, 175, 8280–8286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bossink, A.W.; Paemen, L.; Jansen, P.M.; Hack, C.E.; Thijs, L.G.; Van Damme, J. Plasma levels of the chemokines monocyte chemotactic proteins-1 and -2 are elevated in human sepsis. Blood 1995, 86, 3841–3847. [Google Scholar] [CrossRef] [Green Version]
- Yong, K.K.; Chang, J.H.; Chien, M.H.; Tsao, S.M.; Yu, M.C.; Bai, K.J.; Tsao, T.C.; Yang, S.F. Plasma monocyte chemoattractant protein-1 level as a predictor of the severity of community-acquired pneumonia. Int. J. Mol. Sci. 2016, 17, E179. [Google Scholar] [CrossRef] [Green Version]
- Behjati, S.; Frank, M.H. The effects of tamoxifen on immunity. Curr. Med. Chem. 2009, 16, 3076–3080. [Google Scholar] [CrossRef] [Green Version]
- Seli, E.; Pehlivan, T.; Selam, B.; Garcia-Velasco, J.A.; Arici, A. Estradiol down-regulates MCP-1 expression in human coronary artery endothelial cells. Fertil. Steril. 2002, 77, 542–547. [Google Scholar] [CrossRef]
- Wang, L.; Zheng, W.; Zhang, S.; Chen, X.; Hornung, D. Expression of monocyte chemotactic protein-1 in human endometrial cancer cells and the effect of treatment with tamoxifen or buserelin. J. Int. Med. Res. 2006, 34, 284–290. [Google Scholar] [CrossRef]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Robbins, C.S.; Chudnovskiy, A.; Rauch, P.J.; Figueiredo, J.L.; Iwamoto, Y.; Gorbatov, R.; Etzrodt, M.; Weber, G.F.; Ueno, T.; van Rooijen, N.; et al. Extramedullary hematopoiesis generates Ly-6C(high) monocytes that infiltrate atherosclerotic lesions. Circulation 2012, 125, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Handel, T.M.; Johnson, Z.; Crown, S.E.; Lau, E.K.; Proudfoot, A.E. Regulation of protein function by glycosaminoglycans-as exemplified by chemokines. Annu. Rev. Biochem. 2005, 74, 385–410. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Corriden, R.; Hollands, A.; Olson, J.; Derieux, J.; Lopez, J.; Chang, J.T.; Gonzalez, D.J.; Nizet, V. Tamoxifen augments the innate immune function of neutrophils through modulation of intracellular ceramide. Nat. Commun. 2015, 6, 8369. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Jiang, M.; Chen, Y.; Zhang, S.; Zhang, W.; Yang, X.; Li, X.; Li, Y.; Duan, S.; Han, J.; et al. Inhibition of macrophage CD36 expression and cellular oxidized low density lipoprotein (oxLDL) accumulation by tamoxifen: a peroxisome proliferator-activated receptor (PPAR)gamma-dependent mechanism. J. Biol. Chem. 2016, 291, 16977–16989. [Google Scholar] [CrossRef] [Green Version]
- Miro-Canturri, A.; Ayerbe-Algaba, R.; Smani, Y. Drugs repurposing for the treatment of bacterial and fungal infections. Front. Microbiol. 2019, 10, 41. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Chen, X.H.; Yin, Y.J.; Zhang, J.X. Sepsis and immune response. World J. Emerg. Med. 2011, 2, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Delano, M.J.; Ward, P.A. The immune system’s role in sepsis progression, resolution and long-term outcome. Immunol. Rev. 2016, 274, 330–353. [Google Scholar] [CrossRef] [PubMed]
- Serbina, N.V.; Hohl, T.M.; Cherny, M.; Pamer, E.G. Selective expansion of the monocytic lineage directed by bacterial infection. J. Immunol. 2009, 183, 1900–1910. [Google Scholar] [CrossRef]
- Serbina, N.V.; Kuziel, W.; Flavell, R.; Akira, S.; Rollins, B.; Pamer, E.G. Sequential MyD88-independent and -dependent activation of innate immune responses to intracellular bacterial infection. Immunity 2003, 19, 891–901. [Google Scholar] [CrossRef] [Green Version]
- Balamayooran, G.; Batra, S.; Balamayooran, T.; Cai, S.; Jeyaseelan, S. Monocyte chemoattractant protein 1 regulates pulmonary host defense via neutrophil recruitment during Escherichia coli infection. Infect. Immun. 2011, 79, 2567–2577. [Google Scholar] [CrossRef] [Green Version]
- Jia, T.; Serbina, N.V.; Brandl, K.; Zhong, M.X.; Leiner, I.M.; Charo, I.F.; Pamer, E.G. Additive roles for MCP-1 and MCP-3 in CCR2-mediated recruitment of inflammatory monocytes during Listeria monocytogenes infection. J. Immunol. 2008, 180, 6846–6853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsou, C.L.; Peters, W.; Si, Y.; Slaymaker, S.; Aslanian, A.M.; Weisberg, S.P.; Mack, M.; Charo, I.F. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Investig. 2007, 117, 902–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdon, P.C.; Martin, C.; Rankin, S.M. The CXC chemokine MIP-2 stimulates neutrophil mobilization from the rat bone marrow in a CD49d-dependent manner. Blood 2005, 105, 2543–2548. [Google Scholar] [CrossRef] [Green Version]
- Rankin, S.M. The bone marrow: a site of neutrophil clearance. J. Leuk. Biol. 2010, 88, 241–251. [Google Scholar] [CrossRef]
- Dominguez, P.M.; Ardavin, C. Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol. Rev. 2010, 234, 90–104. [Google Scholar] [CrossRef]
- Serbina, N.V.; Salazar-Mather, T.P.; Biron, C.A.; Kuziel, W.A.; Pamer, E.G. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity 2003, 19, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Bosschaerts, T.; Guilliams, M.; Stijlemans, B.; Morias, Y.; Engel, D.; Tacke, F.; Hérin, M.; De Baetselier, P.; Beschin, A. Tip-DC development during parasitic infection is regulated by IL-10 and requires CCL2/CCR2, IFN-gamma and MyD88 signaling. PLoS Pathog. 2010, 6, e1001045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalbandian, G.; Paharkova-Vatchkova, V.; Mao, A.; Nale, S.; Kovats, S. The selective estrogen receptor modulators, tamoxifen and raloxifene, impair dendritic cell differentiation and activation. J. Immunol. 2005, 175, 2666–2675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smani, Y.; Docobo-Pérez, F.; López-Rojas, R.; Domínguez-Herrera, J.; Ibáñez-Martínez, J.; Pachón, J. Platelet-activating factor receptor initiates contact of Acinetobacter baumannii expressing phosphorylcholine with host cells. J. Biol. Chem. 2012, 287, 26901–26910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra-Millán, R.; Guerrero-Gómez, D.; Ayerbe-Algaba, R.; Pachón-Ibáñez, M.E.; Miranda-Vizuete, A.; Pachón, J.; Smani, Y. Inracellular trafficking and persistence of Acinetobacter baumannii requires transcription factor EB. mSphere 2018, 3, e00106-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomita, T.; Kanegasaki, S. Enhanced phagocytic response of macrophages to bacteria by physical impact caused by bacterial motility or centrifugation. Infect. Immun. 1982, 38, 865–870. [Google Scholar] [CrossRef] [Green Version]
- Mittal, R.; Lisi, C.V.; Kumari, H.; Grati, M.; Blackwelder, P.; Yan, D.; Jain, C.; Mathee, K.; Weckwerth, P.H. Otopathogenic Pseudomonas aeruginosa enters and survives inside macrophages. Front. Microbiol. 2016, 7, 1828. [Google Scholar] [CrossRef] [Green Version]
- Garai, P.; Berry, L.; Moussouni, M.; Bleves, S.; Blanc-Potard, A.B. Killing from the inside: Intracellular role of T3SS in the fate of Pseudomonas aeruginosa within macrophages revealed by mgtC and oprF mutants. PLoS Pathog. 2019, 15, e1007812. [Google Scholar] [CrossRef] [Green Version]
- Corbun, J.; Frank, D.W. Macrophages and epithelial cells respond differently to the Pseudomonas aeruginosa type III secretion system. Infect. Immun. 1999, 67, 3151–3154. [Google Scholar]
- Gil-Marqués, M.L.; Pachón-Ibáñez, M.E.; Pachón, J.; Smani, Y. Effect of hypoxia on the pathogenesis of Acinetobacter baumannii and Pseudomonas aeruginosa in vitro and in murine experimental models of infection. Infect. Immun. 2018, 86, e00543-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baumann, P.; Doudoroff, M.; Stanier, M.R. A 377 study of the Moraxella grouP. II. Oxidative negative species (genus Acinetobacter). J. Bacteriol. 1968, 95, 1520–1541. [Google Scholar] [CrossRef] [Green Version]
- Holloway, I.W. Genetic recombination in Pseudomonas aeruginosa. J. Gen. Microbial. 1955, 13, 572–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Type Culture Collection (ATCC). Escherichia coli (Migula) Castellani and Chalmers (ATCC® 25922™); FDA strain Seattle [DSM 1103, NCIB 12210]; ATCC: Manassas, VA, USA, 1946. [Google Scholar]
- Peña, C.; Suarez, C.; Gonzalo, M.; Murillas, J.; Almirante, B.; Pomar, V.; Aguilar, M.; Granados, A.; Calbo, E.; Rodríguez-Baño, J.; et al. Propsective muticenter study of the impact of carbapenem resistance on mortality in Pseudomonas aeruginosa bloodstream infections. Antimicrob. Agents Chemother. 2012, 56, 1265–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Algaba, R.A.; Álvarez-Marín, R.; Praena, J.; Smani, Y. Escherichia coli causing meningitis in an adult: A case report and experimental characterization of its virulence. Enferm. Infecc. Microbiol. Clin. 2019, 37, 418–419. [Google Scholar] [CrossRef] [PubMed]
- Yanat, B.; Machuca, J.; Yahia, R.D.; Touati, A.; Pascual, Á.; Rodríguez-Martínez, J.M. First report of the plasmid-mediated colistin resistance gene mcr-1 in a clinical Escherichia coli isolate in Algeria. Int. J. Antimicrob. Agents 2016, 48, 760–761. [Google Scholar] [CrossRef]
- Del Toro, R.; Chèvre, R.; Rodríguez, C.; Ordóñez, A.; Martínez-González, J.; Andrés, V.; Méndez-Ferrer, S. Nestin+ cells direct inflammatoy cell migration in atherosclerosis. Nat. Commun. 2016, 7, 12706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, G.; Song, L.; Tian, C.; Wang, Y.; Miao, F.; Zheng, J.; Lu, C.; Alsadun, S.; Graves, D.T. FOXO1 regulates bacteria-induced neutrophil activity. Front. Immunol. 2017, 8, 1088. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Treatment | N | Bacterial Load (log CFU/g or mL ± SEM) | 3-Days Survival (%) | ||
|---|---|---|---|---|---|---|
| Spleen | Lung | Blood | ||||
| A. baumannii ATCC 17978 | CTL | 6 | 9.51 ± 0.17 | 9.77 ± 0.17 | 6.14 ± 0.94 | 0 |
| TAM | 6 | 2.87 ± 1.21 a | 2.61 ± 1.07 a | 0.61 ± 0.61 a | 100 a | |
| P. aeruginosa PAO1 | CTL | 5 | 8.91 ± 0.15 | 9.24 ± 0.17 | 6.71 ± 0.27 | 0 |
| TAM | 6 | 5.33 ± 1.08 b | 4.14 ± 1.50 b | 1.26 ± 1.26 b | 66.7 b | |
| E. coli ATCC 25922 | CTL | 6 | 8.71 ± 0.05 | 8.88 ± 0.16 | 8.18 ± 0.37 | 0 |
| TAM | 6 | 5.01 ± 1.20 c | 4.72 ± 1.08 c | 3.87 ± 0.99 c | 83.3c | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miró-Canturri, A.; Ayerbe-Algaba, R.; del Toro, R.; Mejías, M.E.-J.; Pachón, J.; Smani, Y. Potential Tamoxifen Repurposing to Combat Infections by Multidrug-Resistant Gram-Negative Bacilli. Pharmaceuticals 2021, 14, 507. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060507
Miró-Canturri A, Ayerbe-Algaba R, del Toro R, Mejías ME-J, Pachón J, Smani Y. Potential Tamoxifen Repurposing to Combat Infections by Multidrug-Resistant Gram-Negative Bacilli. Pharmaceuticals. 2021; 14(6):507. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060507
Chicago/Turabian StyleMiró-Canturri, Andrea, Rafael Ayerbe-Algaba, Raquel del Toro, Manuel Enrique-Jiménez Mejías, Jerónimo Pachón, and Younes Smani. 2021. "Potential Tamoxifen Repurposing to Combat Infections by Multidrug-Resistant Gram-Negative Bacilli" Pharmaceuticals 14, no. 6: 507. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060507