
Phytosterols and Novel Triterpenes Recovered from Industrial Fermentation Coproducts Exert In Vitro Anti-Inflammatory Activity in Macrophages

 , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
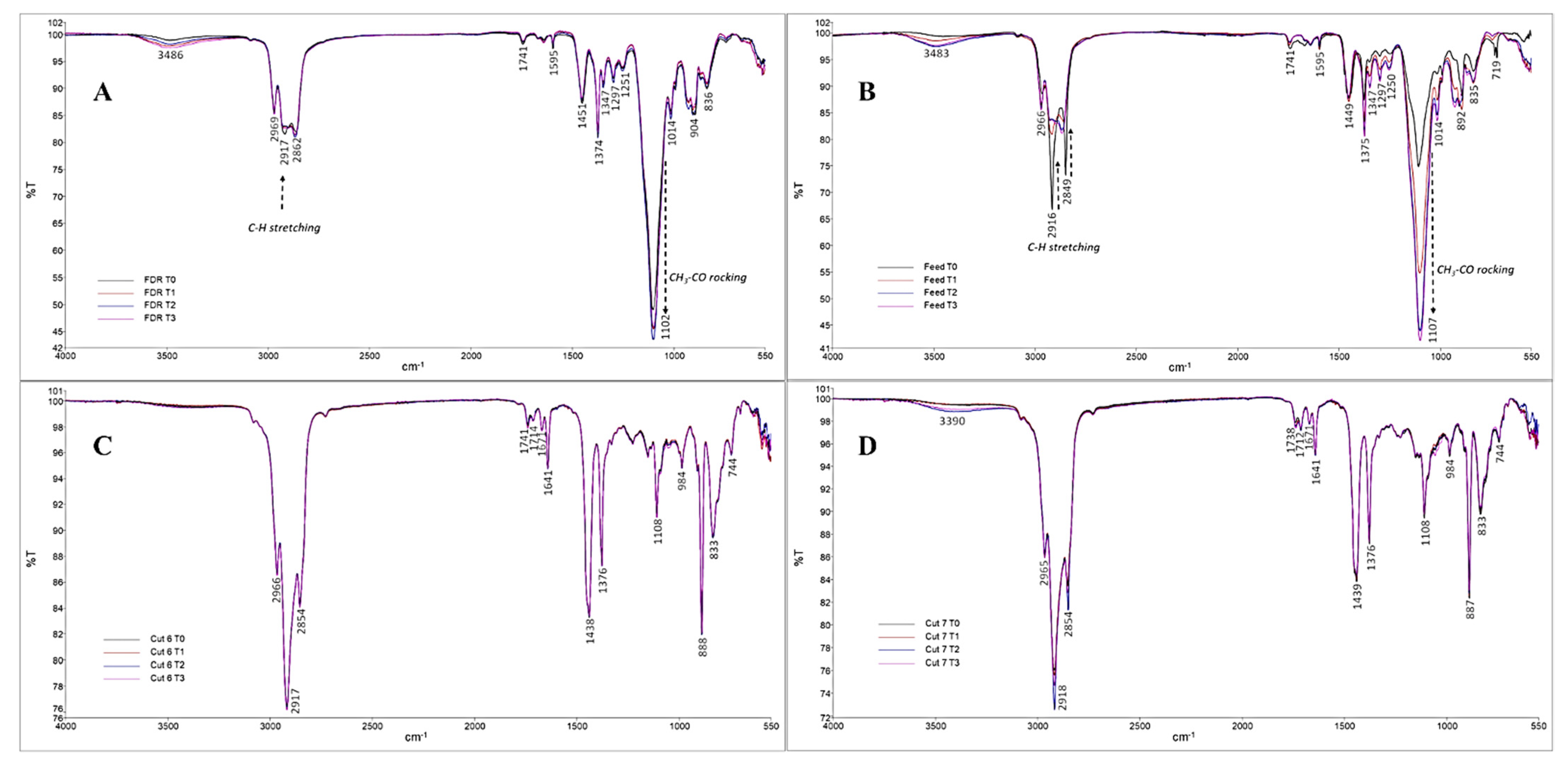
2.1. Composition and Presence of Potentially Bioactive Lipids in β-Farnesene Distillation Residue (FDR) Samples
2.2. Recovery of Bioactive Compounds by Molecular Distillation
2.3. Stability of the Distillates Enriched in Bioactive Lipids
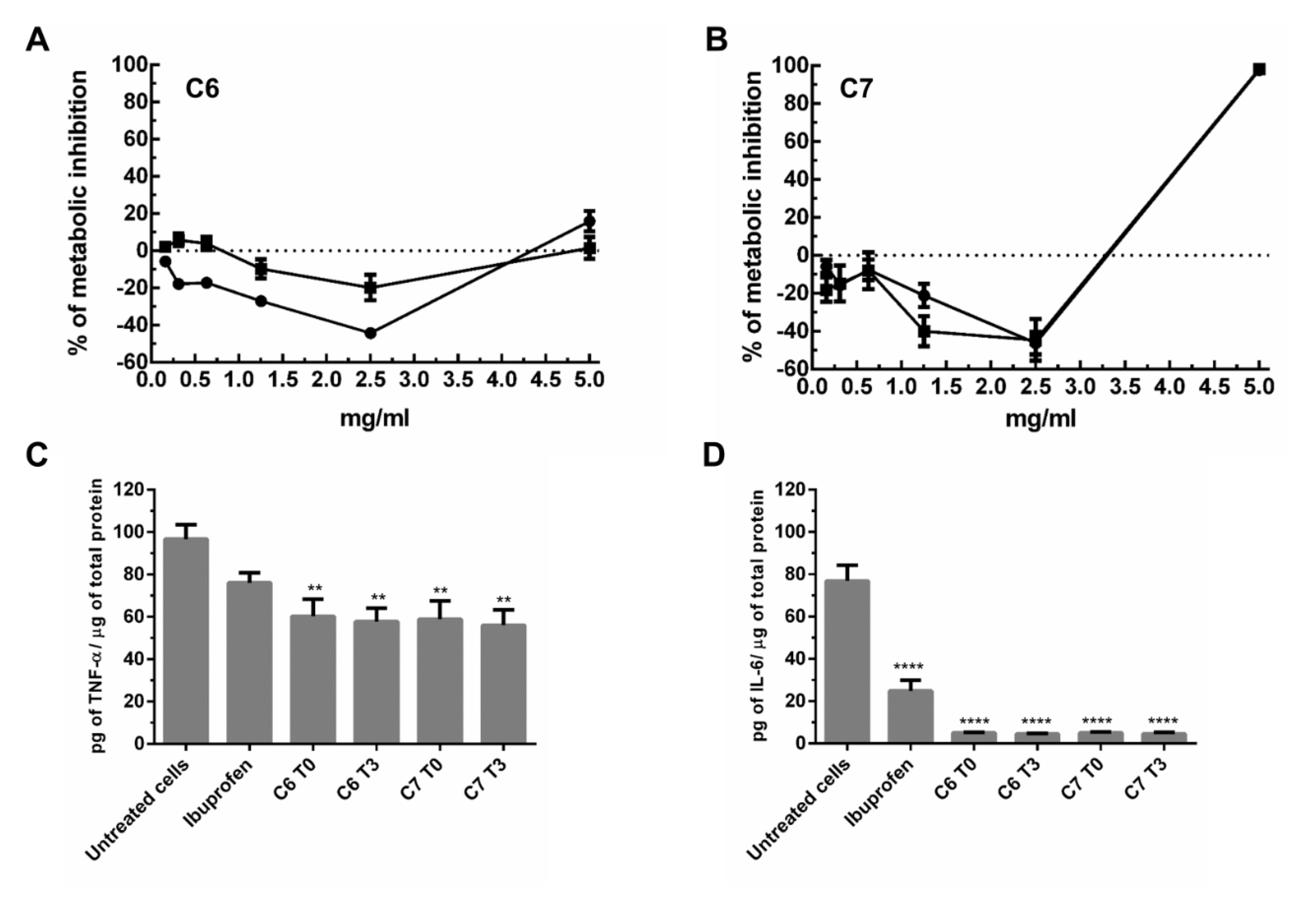
2.4. Bioactivity Studies of C6 and C7 Samples
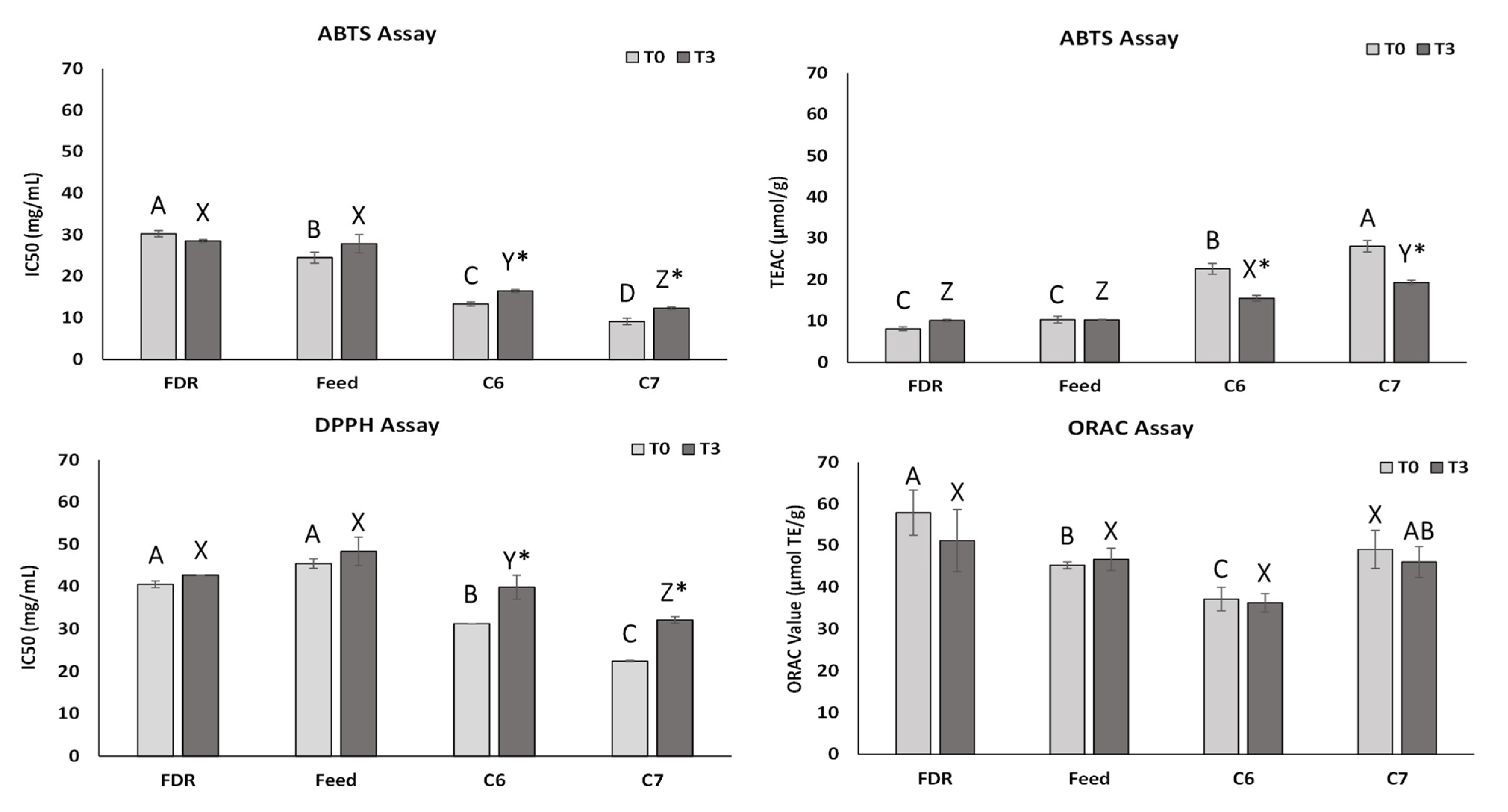
2.5. Changes in Antioxidant Activity of the Assayed Samples during the Stability Test
2.6. Crystallization, Melting, and Decomposition Temperature
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Distillation
3.3. Accelerated Stability Test
3.4. HPLC-ELSD Analyses
3.5. GC-MS Analyses
3.6. Fourier Transform Infrared Spectroscopy with Attenuated Total Reflectance (FTIR-ATR)
3.7. Cell Culture
3.8. Cytotoxicity Assays
3.9. Immune Response
3.10. Antioxidant Activity
3.11. Differential Scanning Calorimetry (DSC)
3.12. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- United Nations. World Population Prospects 2019; United Nations: New York, NY, USA, 2019; ISBN 9789211483161. [Google Scholar]
- Bell, J.; Paula, L.; Dodd, T.; Németh, S.; Nanou, C.; Mega, V.; Campos, P. EU ambition to build the world’s leading bioeconomy—Uncertain times demand innovative and sustainable solutions. N. Biotechnol. 2018, 40, 25–30. [Google Scholar] [CrossRef]
- Pimentel, D.; Burgess, M. World human population problems. In Encyclopedia of the Anthropocene; Elsevier: Amsterdam, The Netherlands, 2017; Volume 1–5, pp. 313–317. ISBN 9780128096659. [Google Scholar]
- Willett, W.; Rockström, J.; Loken, B.; Springmann, M.; Lang, T.; Vermeulen, S.; Garnett, T.; Tilman, D.; DeClerck, F.; Wood, A.; et al. Food in the Anthropocene: The EAT–Lancet Commission on healthy diets from sustainable food systems. Lancet 2019, 393, 447–492. [Google Scholar] [CrossRef]
- Geissdoerfer, M.; Savaget, P.; Bocken, N.M.P.; Hultink, E.J. The Circular Economy—A new sustainability paradigm? J. Clean. Prod. 2017, 143, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Hanson, R. The Great Filter. Available online: http://mason.gmu.edu/~rhanson/greatfilter.html (accessed on 21 March 2021).
- D’Amato, D.; Droste, N.; Allen, B.; Kettunen, M.; Lähtinen, K.; Korhonen, J.; Leskinen, P.; Matthies, B.D.; Toppinen, A. Green, circular, bio economy: A comparative analysis of sustainability avenues. J. Clean. Prod. 2017, 168, 716–734. [Google Scholar] [CrossRef]
- Lee, J.K.; Patel, S.K.S.; Sung, B.H.; Kalia, V.C. Biomolecules from municipal and food industry wastes: An overview. Bioresour. Technol. 2020, 298, 122346. [Google Scholar] [CrossRef]
- Mello, B.L.B.; Fernandes, A.M.; de Oliveira, T.S.; Leonel, F.P.; Glória, L.S.; Silva, R.S.T. Feed intake, digestibility, and energy contents in growing bull fed pineapple crop waste silage in different planes of nutrition. Trop. Anim. Health Prod. 2021, 53, 188. [Google Scholar] [CrossRef]
- Klunklin, W.; Jantanasakulwong, K.; Phimolsiripol, Y.; Leksawasdi, N.; Seesuriyachan, P.; Chaiyaso, T.; Insomphun, C.; Phongthai, S.; Jantrawut, P.; Sommano, S.R.; et al. Synthesis, characterization, and application of carboxymethyl cellulose from asparagus stalk end. Polymers 2021, 13, 81. [Google Scholar] [CrossRef]
- Piccirillo, C.; Rocha, C.; Tobaldi, D.M.; Pullar, R.C.; Labrincha, J.A.; Ferreira, M.O.; Castro, P.M.L.; Pintado, M.M.E. A hydroxyapatite-Fe2O3 based material of natural origin as an active sunscreen filter. J. Mater. Chem. B 2014, 2, 5999–6009. [Google Scholar] [CrossRef] [PubMed]
- OECD-FAO Agricultural Outlook 2015–2024; OECD: Paris, France, 2015.
- Roukas, T.; Kotzekidou, P. Rotary biofilm reactor: A new tool for long-term bioethanol production from non-sterilized beet molasses by Saccharomyces cerevisiae in repeated-batch fermentation. J. Clean. Prod. 2020, 257, 120519. [Google Scholar] [CrossRef]
- Bušić, A.; Mardetko, N.; Kundas, S.; Morzak, G.; Belskaya, H.; Šantek, M.I.; Komes, D.; Novak, S.; Šantek, B. Bioethanol production from renewable raw materials and its separation and purification: A review. Food Technol. Biotechnol. 2018, 56, 289–311. [Google Scholar] [CrossRef] [PubMed]
- Zabed, H.; Sahu, J.N.; Suely, A.; Boyce, A.N.; Faruq, G. Bioethanol production from renewable sources: Current perspectives and technological progress. Renew. Sustain. Energy Rev. 2017, 71, 475–501. [Google Scholar] [CrossRef]
- Jambo, S.A.; Abdulla, R.; Mohd Azhar, S.H.; Marbawi, H.; Gansau, J.A.; Ravindra, P. A review on third generation bioethanol feedstock. Renew. Sustain. Energy Rev. 2016, 65, 756–769. [Google Scholar] [CrossRef]
- FAOSTAT. FAOSTAT: Statistical Database. Available online: http://www.fao.org/faostat/en/#home (accessed on 1 September 2020).
- Fadhil, A.B.; Al-Tikrity, E.T.B.; Albadree, M.A. Biodiesel production from mixed non-edible oils, castor seed oil and waste fish oil. Fuel 2017, 210, 721–728. [Google Scholar] [CrossRef]
- Nautiyal, P.; Subramanian, K.A.; Dastidar, M.G. Experimental investigation on performance and emission characteristics of a compression ignition engine fueled with biodiesel from waste tallow. Clean Technol. Environ. Policy 2017, 19, 1667–1677. [Google Scholar] [CrossRef]
- Xu, P.; Vansiri, A.; Bhan, N.; Koffas, M.A.G. EPathBrick: A synthetic biology platform for engineering metabolic pathways in E. coli. ACS Synth. Biol. 2012, 1, 256–266. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Y.; Fu, L.; Guo, E.; Wang, B.; Dai, L.; Si, T. Accelerating strain engineering in biofuel research via build and test automation of synthetic biology. Curr. Opin. Biotechnol. 2021, 67, 88–98. [Google Scholar] [CrossRef]
- Zhang, Y.; Nielsen, J.; Liu, Z. Engineering yeast metabolism for production of terpenoids for use as perfume ingredients, pharmaceuticals and biofuels. FEMS Yeast Res. 2017, 17, 1–11. [Google Scholar] [CrossRef]
- Paddon, C.J.; Westfall, P.J.; Pitera, D.J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M.D.; Tai, A.; Main, A.; Eng, D.; et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Meadows, A.L.; Hawkins, K.M.; Tsegaye, Y.; Antipov, E.; Kim, Y.; Raetz, L.; Dahl, R.H.; Tai, A.; Mahatdejkul-Meadows, T.; Xu, L.; et al. Rewriting yeast central carbon metabolism for industrial isoprenoid production. Nature 2016, 537, 694–697. [Google Scholar] [CrossRef] [PubMed]
- Ejsing, C.S.; Sampaio, J.L.; Surendranath, V.; Duchoslav, E.; Ekroos, K.; Klemm, R.W.; Simons, K.; Shevchenko, A. Global analysis of the yeast lipidome by quantitative shotgun mass spectrometry. Proc. Natl. Acad. Sci. USA 2009, 106, 2136–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Zhao, S.; Zhu, C.; Gao, Q.; Bai, J.; Si, J.; Chen, Y. Ergosterol ameliorates renal inflammatory responses in mice model of diabetic nephropathy. Biomed. Pharmacother. 2020, 128, 110252. [Google Scholar] [CrossRef]
- Nallathamby, N.; Guan-Serm, L.; Vidyadaran, S.; Malek, S.N.A.; Raman, J.; Sabaratnam, V. Ergosterol of Cordyceps militaris Attenuates LPS Induced Inflammation in BV2 Microglia Cells. Nat. Prod. Commun. 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Asikin, Y.; Takahashi, M.; Hirose, N.; Hou, D.X.; Takara, K.; Wada, K. Wax, policosanol, and long-chain aldehydes of different sugarcane (Saccharum officinarum L.) cultivars. Eur. J. Lipid Sci. Technol. 2012, 114, 583–591. [Google Scholar] [CrossRef]
- del Río, J.C.; Marques, G.; Lino, A.G.; Lima, C.F.; Colodette, J.L.; Gutiérrez, A. Lipophilic phytoSSSSSSchemicals from sugarcane bagasse and straw. Ind. Crops Prod. 2015, 77, 992–1000. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Lin, Q.; Li, X.; Nie, Y.; Wang, L.; Shi, L.; Xu, W.; Hu, T.; Guo, T.; Luo, F. Octacosanol Attenuates Inflammation in Both RAW264.7 Macrophages and a Mouse Model of Colitis. J. Agric. Food Chem. 2017, 65, 3647–3658. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, L.L.; Rodríguez-Alcalá, L.M. Cholesterol, inflammation, and phospholipids: COVID-9 share traits with cardiovascular disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, T.; Miyazawa, T. Separation and determination of glycolipids from edible plant sources by high-performance liquid chromatography and evaporative light-scattering detection. Lipids 1999, 34, 1231–1237. [Google Scholar] [CrossRef] [PubMed]
- Fouquet, T.N.J. The Kendrick analysis for polymer mass spectrometry. J. Mass Spectrom. 2019, 54, 933–947. [Google Scholar] [CrossRef] [PubMed]
- Yoo, T.; Henning, S.K. Synthesis and characterization of farnesene-based polymers. Rubber Chem. Technol. 2017, 90, 308–324. [Google Scholar] [CrossRef]
- Sahu, P.; Bhowmick, A.K. Redox Emulsion Polymerization of Terpenes: Mapping the Effect of the System, Structure, and Reactivity. Ind. Eng. Chem. Res. 2019, 58, 20946–20960. [Google Scholar] [CrossRef]
- Fisher, K.; Woolard, F.X. Farnesene Dimers and/or Farnesane Dimers and Compositions Thereof. U.S. Patent 7,592,295, 22 September 2009. [Google Scholar]
- Reddy, L.H.; Couvreur, P. Squalene: A natural triterpene for use in disease management and therapy. Adv. Drug Deliv. Rev. 2009, 61, 1412–1426. [Google Scholar] [CrossRef] [PubMed]
- Tegenge, M.A.; Von Tungeln, L.S.; Anderson, S.A.; Mitkus, R.J.; Vanlandingham, M.M.; Forshee, R.A.; Beland, F.A. Comparative pharmacokinetic and biodistribution study of two distinct squalene-containing oil-in-water emulsion adjuvants in H5N1 influenza vaccines. Regul. Toxicol. Pharmacol. 2019, 108, 104436. [Google Scholar] [CrossRef]
- Dormont, F.; Brusini, R.; Cailleau, C.; Reynaud, F.; Reynaud, F.; Peramo, A.; Gendron, A.; Mougin, J.; Gaudin, F.; Gaudin, F.; et al. Squalene-based multidrug nanoparticles for improved mitigation of uncontrolled inflammation in rodents. Sci. Adv. 2020, 6, eaaz5466. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Lepetre-Mouelhi, S.; Gautier, A.; Mura, S.; Cailleau, C.; Coudore, F.; Hamon, M.; Couvreur, P. A new painkiller nanomedicine to bypass the blood-brain barrier and the use of morphine. Sci. Adv. 2019, 5, eaau5148. [Google Scholar] [CrossRef] [Green Version]
- Sharma, R.; Matsuzaka, T.; Kaushik, M.K.; Sugasawa, T. Octacosanol and policosanol prevent high-fat diet-induced obesity and metabolic disorders by activating brown adipose tissue and improving liver metabolism. Sci. Rep. 2019, 9, 5169. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Scott, S.D.; Pekas, E.J.; Lee, J.; Park, S. Improvement of Lipids and Reduction of Oxidative Stress with Octacosanol after Taekwondo Training. Int. J. Sports Physiol. Perform. 2019, 14, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, M.K.; Aritake, K.; Takeuchi, A.; Yanagisawa, M.; Urade, Y. Octacosanol restores stress-affected sleep in mice by alleviating stress. Sci. Rep. 2017, 7, 8892. [Google Scholar] [CrossRef] [Green Version]
- European Comission COMMISSION REGULATION (EU) No 432/2012 of 16 May 2012 establishing a list of permitted health claims made on foods, other than those referring to the reduction of disease risk and to children’s development and health. Off. J. Eur. Union 2012, L136, 1–40. Available online: http://eur-lex.europa.eu/pri/en/oj/dat/2003/l_285/l_28520031101en00330037.pdf (accessed on 1 January 2021).
- Attard, T.M.; McElroy, C.R.; Rezende, C.A.; Polikarpov, I.; Clark, J.H.; Hunt, A.J. Sugarcane waste as a valuable source of lipophilic molecules. Ind. Crops Prod. 2015, 76, 95–103. [Google Scholar] [CrossRef]
- Ishaka, A.; Imam, M.U.; Mahamud, R.; Zuki, A.B.Z.; Maznah, I. Characterization of rice bran wax policosanol and its nanoemulsion formulation. Int. J. Nanomedicine 2014, 9, 2261–2269. [Google Scholar] [CrossRef] [Green Version]
- Canizares, D.; Angers, P.; Ratti, C. Selective wax recovery from flax and wheat straw. Ind. Crops Prod. 2019, 141, 111700. [Google Scholar] [CrossRef]
- Rahmania, H.; Kato, S.; Sawada, K.; Hayashi, C.; Hashimoto, H.; Nakajima, S.; Otoki, Y.; Ito, J.; Nakagawa, K. Revealing the thermal oxidation stability and its mechanism of rice bran oil. Sci. Rep. 2020, 10, 14091. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Luo, Z.; Zeng, F.; Liu, S.; Khan, Z.U. Effect of water, metallic ions, fatty acid and temperature on oxidative stability of 1-octacosanol from sugarcane rind. Food Chem. 2015, 182, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Ogihara, Y.; Ohashi, H.; Akita, T.; Honma, T.; Oji, H.; Haruta, M. Base-free direct oxidation of 1-octanol to octanoic acid and its octyl ester over supported gold catalysts. ChemSusChem 2012, 5, 2243–2248. [Google Scholar] [CrossRef] [PubMed]
- Corberán, V.C.; Gómez-Avilés, A.; Martínez-González, S.; Ivanova, S.; Domínguez, M.I.; González-Pérez, M.E. Heterogeneous selective oxidation of fatty alcohols: Oxidation of 1-tetradecanol as a model substrate. Catal. Today 2014, 238, 49–53. [Google Scholar] [CrossRef]
- Ekinci, N.; Şeker, M.; Aydin, F.; Gündoğdu, M.A. Possible chemical mechanism and determination of inhibitory effects of 1-MCP on superficial scald of the Granny Smith apple variety. Turk. J. Agric. For. 2016, 40, 38–44. [Google Scholar] [CrossRef]
- Gachumi, G.; Poudel, A.; Wasan, K.M.; El-Aneed, A. Analytical strategies to analyze the oxidation products of phytosterols, and formulation-based approaches to reduce their generation. Pharmaceutics 2021, 13, 268. [Google Scholar] [CrossRef]
- Lengyel, J.; Rimarčík, J.; Vagánek, A.; Fedor, J.; Lukeš, V.; Klein, E. Oxidation of sterols: Energetics of C-H and O-H bond cleavage. Food Chem. 2012, 133, 1435–1440. [Google Scholar] [CrossRef]
- Choe, E.; Min, D.B. Mechanisms and Factors for Edible Oil Oxidation. Compr. Rev. Food Sci. Food Saf. 2006, 5, 169–186. [Google Scholar] [CrossRef]
- Khor, Y.P.; Shin Hew, K.; Abas, F.; Lai, O.M.; Cheong, L.Z.; Nehdi, I.A.; Sbihi, H.M.; Gewik, M.M.; Tan, C.P. Oxidation and Polymerization of Triacylglycerols: In-Depth Investigations towards the Impact of Heating Profiles. Foods 2019, 8, 475. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Alcalá, L.M.; García-Martínez, M.C.; Cachón, F.; Marmesat, S.; Alonso, L.; Márquez-Ruiz, G.; Fontecha, J. Changes in the lipid composition of powdered infant formulas during long-term storage. J. Agric. Food Chem. 2007, 55, 6533–6538. [Google Scholar] [CrossRef] [Green Version]
- del Carmen García-Martínez, M.; Rodríguez-Alcalá, L.M.; Marmesat, S.; Alonso, L.; Fontecha, J.; Márquez-Ruiz, G. Lipid stability in powdered infant formula stored at ambient temperatures. Int. J. Food Sci. Technol. 2010, 45, 2337–2344. [Google Scholar] [CrossRef]
- de Almeida, D.T.; Viana, T.V.; Costa, M.M.; Silva, C.D.S.; Feitosa, S. Effects of different storage conditions on the oxidative stability of crude and refined palm oil, olein and stearin (Elaeis guineensis). Food Sci. Technol. 2019, 39, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Toci, A.T.; Neto, V.J.M.F.; Torres, A.G.; Farah, A. Changes in triacylglycerols and free fatty acids composition during storage of roasted coffee. LWT—Food Sci. Technol. 2013, 50, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Chung, H.J.; Lim, S.T. Effect of various heat treatments on rancidity and some bioactive compounds of rice bran. J. Cereal Sci. 2014, 60, 243–248. [Google Scholar] [CrossRef]
- Aldini, R.; Micucci, M.; Cevenini, M.; Fato, R.; Bergamini, C.; Nanni, C.; Cont, M.; Camborata, C.; Spinozzi, S.; Montagnani, M.; et al. Antiinflammatory Effect of Phytosterols in Experimental Murine Colitis Model: Prevention, Induction, Remission Study. PLoS ONE 2014, 9, e108112. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Zhang, F.; Shen, M.; Jia, S.; Xie, J. Phytosterols suppress phagocytosis and inhibit inflammatory mediators via ERK pathway on LPS-triggered inflammatory responses in RAW264.7 macrophages and the correlation with their structure. Foods 2019, 8, 582. [Google Scholar] [CrossRef] [Green Version]
- CPMP/ICH. ICH Topic Q1 A Stability Testing Guidelines: Stability Testing of New Drug Substances and Products. Int. Conf. Harmon. 2003, 4, 1–14. [Google Scholar]
- Greenspan, L. Humidity fixed points of binary saturated aqueous solutions. J. Res. Natl. Bur. Stand. Sect. A Phys. Chem 1977, 81A, 89–96. [Google Scholar] [CrossRef]
- Abreu, S.; Solgadi, A.; Chaminade, P. Optimization of normal phase chromatographic conditions for lipid analysis and comparison of associated detection techniques. J. Chromatogr. A 2017, 1514, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Homan, R.; Anderson, M.K. Rapid separation and quantitation of combined neutral and polar lipid classes by high-performance liquid chromatography and evaporative light- scattering mass detection. J. Chromatogr. B Biomed. Appl. 1998, 708, 21–26. [Google Scholar] [CrossRef]
- George, S. Infrared and Raman Characteristic Group Frequencies Contents, 3rd ed.; John Wiley & Sons, Inc.: London, UK, 2001; ISBN 0471852988. [Google Scholar]
- Bobo-García, G.; Davidov-Pardo, G.; Arroqui, C.; Vírseda, P.; Marín-Arroyo, M.R.; Navarro, M. Intra-laboratory validation of microplate methods for total phenolic content and antioxidant activity on polyphenolic extracts, and comparison with conventional spectrophotometric methods. J. Sci. Food Agric. 2015, 95, 204–209. [Google Scholar] [CrossRef]
- Oliveira, S. Evaluation of antiradical assays used in determining the antioxidant capacity of pure compounds and plant extracts. Quim. Nov. 2014, 37, 497–503. [Google Scholar] [CrossRef]
- Sanchez-Moreno, C. Review: Methods used to evaluate the free radical scavenging activity in foods and biological systems. Food Sci. Technol. Int. 2002, 8, 121–137. [Google Scholar] [CrossRef]
- Benteldjoune, M.; Boudiar, T.; Bakhouche, A.; Contreras, M.; Lozano-sánchez, J.; Bensouici, C.; Kabouche, Z.; Segura-carretero, A.; Contreras, M.; Lozano-sánchez, J.; et al. Antioxidant activity and characterization of flavonoids and phenolic acids of Ammoides atlantica by RP—UHPLC—ESI—QTOF—MS. Nat. Prod. Res. 2019, 1–5. [Google Scholar] [CrossRef]
- Dávalos, A.; Gómez-Cordovés, C.; Bartolomé, B. Extending Applicability of the Oxygen Radical Absorbance Capacity (ORAC−Fluorescein) Assay. J. Agric. Food Chem. 2004, 52, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Yu, L. (Ed.) Wheat Antioxidants; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; ISBN 9780470228333. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| FDR | T0 | T1 | T2 | T3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Mean | SD | Mean | SD | Mean | SD | Mean | SD | ||||
| Hydrocarbons | B 43.29 | ± | 0.17 | B 44.23 | ± | 1.61 | A 52.82 | ± | 0.51 | A 54.79 | ± | 0.02 |
| Wax esters | AB 0.22 | ± | 0.05 | A 0.23 | ± | 0.01 | B 0.17 | ± | 0.01 | C 0.14 | ± | 0.02 |
| Triglycerides | A 4.21 | ± | 0.03 | A 4.32 | ± | 0.10 | B 3.01 | ± | 0.25 | B 2.59 | ± | 0.11 |
| Fatty alcohols | A 2.00 | ± | 0.10 | A 2.18 | ± | 0.09 | B 1.49 | ± | 0.17 | B 1.29 | ± | 0.19 |
| Phytosterols | A 0.78 | ± | 0.01 | A 0.74 | ± | 0.02 | B 0.53 | ± | 0.03 | C 0.36 | ± | 0.07 |
| Diglycerides | A 2.62 | ± | 0.12 | A 2.51 | ± | 0.02 | B 1.67 | ± | 0.13 | B 1.61 | ± | 0.02 |
| Free fatty acids | B 0.33 | ± | 0.03 | A 0.48 | ± | 0.05 | B 0.31 | ± | 0.02 | B 0.34 | ± | 0.03 |
| Monoglycerides | C0.04 | ± | 0.01 | B 0.10 | ± | 0.02 | A 0.17 | ± | 0.01 | A 0.16 | ± | 0.01 |
| Polymer | A 46.51 | ± | 0.22 | A 45.20 | ± | 1.67 | B 39.84 | ± | 0.52 | B 38.70 | ± | 0.46 |
| Feed | T0 | T1 | T2 | T3 | ||||||||
| Compound | Mean | SD | Mean | SD | Mean | SD | Mean | SD | ||||
| Hydrocarbons | B 39.54 | ± | 0.92 | B 39.71 | ± | 0.91 | A 52.60 | ± | 0.62 | A 49.46 | ± | 2.34 |
| Wax esters | A 0.11 | ± | 0.01 | A 0.11 | ± | 0.02 | B 0.07 | ± | 0.01 | C 0.06 | ± | 0.01 |
| Triglycerides | A 4.34 | ± | 0.15 | A 4.56 | ± | 0.16 | B 2.72 | ± | 0.06 | C 2.05 | ± | 0.15 |
| Fatty alcohols | A 1.75 | ± | 0.03 | A 1.75 | ± | 0.05 | B 0.98 | ± | 0.01 | B 0.98 | ± | 0.06 |
| Phytosterols | A 0.73 | ± | 0.02 | A 0.74 | ± | 0.03 | B 0.44 | ± | 0.02 | C 0.37 | ± | 0.03 |
| Diglycerides | A 2.08 | ± | 0.05 | A 1.95 | ± | 0.20 | B 1.22 | ± | 0.05 | C 0.90 | ± | 0.22 |
| Free fatty acids | 0.30 | ± | 0.04 | 0.31 | ± | 0.07 | 0.20 | ± | 0.02 | 0.22 | ± | 0.01 |
| Monoglycerides | C 0.04 | ± | 0.01 | A 0.10 | ± | 0.01 | B 0.06 | ± | 0.01 | A 0.09 | ± | 0.01 |
| Polymer | A 51.11 | ± | 0.79 | A 50.78 | ± | 0.85 | A 41.69 | ± | 0.66 | B 45.88 | ± | 2.37 |
| FDR | T0 | T1 | T2 | T3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Mean | SD | Mean | SD | Mean | SD | Mean | SD | ||||
| β-Farnesene (PR 15) | A 133.48 | ± | 2.81 | B 124.35 | ± | 5.56 | C 107.19 | ± | 1.11 | D 97.15 | ± | 3.1 |
| Farnesol (Z,E) (PR 15:2c2t6) | A 16.26 | ± | 0.38 | B 14.95 | ± | 0.38 | B 15.10 | ± | 0.92 | C 13.02 | ± | 0.48 |
| Farnesol (E,E) (PR 15:2t2t6) | 1.36 | ± | 0.04 | 1.15 | ± | 0.08 | 1.35 | ± | 0.14 | 1.07 | ± | 0.11 |
| Sesterterpene (PR 25) | 13.5 | ± | 0.28 | 12.86 | ± | 0.60 | 11.68 | ± | 1.36 | 10.53 | ± | 0.76 |
| Triterpene I (PR 30) | D 2.18 | ± | 0.01 | C 2.47 | ± | 0.04 | A 3.05 | ± | 0.18 | B 2.62 | ± | 0.30 |
| Triterpene III (PR 30) | B 2.36 | ± | 0.05 | A 3.01 | ± | 0.09 | A 3.35 | ± | 0.16 | A 3.09 | ± | 0.24 |
| Triterpene IV (PR 30) | C 42.22 | ± | 0.48 | A 52.96 | ± | 2.15 | B 46.86 | ± | 1.28 | B 44.96 | ± | 0.95 |
| Triterpene VI (PR 30) | 1.77 | ± | 0.05 | 2.12 | ± | 0.02 | 1.30 | ± | 0.25 | 1.63 | ± | 0.19 |
| Triterpene VIII (PR 30) | C 24.03 | ± | 0.38 | A 30.41 | ± | 1.11 | B 26.21 | ± | 0.80 | B 25.86 | ± | 0.94 |
| Σ TERPENES | A 237.16 | ± | 4.46 | A 244.28 | ± | 9.00 | B 216.09 | ± | 5.86 | C 199.93 | ± | 5.06 |
| 1-Octacosanol (FOH 28:0) | 1.45 | ± | 0.31 | 1.13 | ± | 0.08 | 1.09 | ± | 0.11 | 0.92 | ± | 0.12 |
| Σ FOH | A 1.45 | ± | 0.31 | B 1.13 | ± | 0.08 | B 1.09 | ± | 0.11 | B 0.92 | ± | 0.12 |
| Ergosterol (ST 28:3;O) | B 3.19 | ± | 0.14 | A 3.72 | ± | 0.09 | B 3.07 | ± | 0.26 | C 1.70 | ± | 0.22 |
| Campesterol (ST 28:1;O) | 0.72 | ± | 0.05 | 0.86 | ± | 0.01 | 0.94 | ± | 0.07 | 0.77 | ± | 0.09 |
| Stigmasterol (ST 29:2;O) | 0.92 | ± | 0.04 | 1.03 | ± | 0.02 | 1.07 | ± | 0.12 | 0.98 | ± | 0.11 |
| β-Sitosterol (ST 29:1;O) | 1.29 | ± | 0.05 | 1.51 | ± | 0.05 | 1.60 | ± | 0.15 | 1.47 | ± | 0.15 |
| Σ ST | A 6.11 | ± | 0.28 | A 7.11 | ± | 0.14 | A 6.68 | ± | 0.59 | B 4.92 | ± | 0.57 |
| Feed | T0 | T1 | T2 | T3 | ||||||||
| Compound | Mean | SD | Mean | SD | Mean | SD | Mean | SD | ||||
| β-Farnesene (PR 15) | B 116.09 | ± | 1.52 | A 119.95 | ± | 2.5 | C 101.27 | ± | 0.7 | D 66.30 | ± | 1.46 |
| Farnesol (Z,E) (PR 15:2c2t6) | A 15.67 | ± | 0.17 | A 16.47 | ± | 0.21 | A 16.74 | ± | 0.15 | B 14.74 | ± | 0.63 |
| Farnesol (E,E) (PR 15:2t2t6) | 1.27 | ± | 0.03 | 1.33 | ± | 0.02 | 1.49 | ± | 0.02 | 1.24 | ± | 0.03 |
| Sesterterpene (PR 25) | 14.83 | ± | 0.14 | 15.14 | ± | 0.37 | 13.56 | ± | 0.03 | 13.04 | ± | 0.80 |
| Triterpene I (PR 30) | C 2.08 | ± | 0.14 | C 2.67 | ± | 0.12 | A 3.08 | ± | 0.12 | B 2.98 | ± | 0.20 |
| Triterpene III (PR 30) | B 2.61 | ± | 0.12 | A 2.84 | ± | 0.34 | B 3.27 | ± | 0.09 | B 3.16 | ± | 0.11 |
| Triterpene IV (PR 30) | B 45.69 | ± | 0.46 | A 50.11 | ± | 0.45 | C 44.94 | ± | 0.63 | C 43.95 | ± | 1.45 |
| Triterpene VI (PR 30) | A 2.02 | ± | 0.06 | A 2.44 | ± | 0.08 | C 1.57 | ± | 0.06 | B 1.82 | ± | 0.16 |
| Triterpene VIII (PR 30) | B 25.99 | ± | 0.26 | A 28.64 | ± | 0.36 | C 25.05 | ± | 0.33 | C 24.84 | ± | 0.86 |
| Σ TERPENES | A 226.26 | ± | 0.13 | A 239.59 | ± | 1.39 | B 210.97 | ± | 1.75 | C 172.08 | ± | 5.46 |
| 1-Octacosanol (FOH 28:0) | 0.25 | ± | 0.11 | 0.23 | ± | 0.01 | 0.24 | ± | 0.02 | 0.21 | ± | 0.02 |
| Σ FOH | 0.25 | ± | 0.11 | 0.23 | ± | 0.01 | 0.24 | ± | 0.02 | 0.21 | ± | 0.02 |
| Ergosterol (ST 28:3;O) | B 3.54 | ± | 0.12 | A 3.70 | ± | 0.17 | B 3.00 | ± | 0.14 | C 1.79 | ± | 0.16 |
| Campesterol (ST 28:1;O) | 0.71 | ± | 0.01 | 0.78 | ± | 0.05 | 0.88 | ± | 0.03 | 0.83 | ± | 0.06 |
| Stigmasterol (ST 29:2;O) | 0.90 | ± | 0.03 | 1.13 | ± | 0.02 | 1.22 | ± | 0.06 | 1.23 | ± | 0.14 |
| β-Sitosterol (ST 29:1;O) | 1.36 | ± | 0.08 | 1.69 | ± | 0.1 | 1.79 | ± | 0.08 | 1.76 | ± | 0.14 |
| Σ ST | A 6.52 | ± | 0.24 | A 7.31 | ± | 0.29 | A 6.89 | ± | 0.17 | B 5.62 | ± | 0.49 |
| C6 | T0 | T1 | T2 | T3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Mean | SD | Mean | SD | Mean | SD | Mean | SD | ||||
| Sesterterpene (PR 25) | A 3.40 | ± | 0.20 | A 3.38 | ± | 0.24 | B 2.28 | ± | 0.05 | B 2.15 | ± | 0.21 |
| Triterpene I (PR 30) | B 22.86 | ± | 2.93 | A 29.15 | ± | 0.60 | B 26.23 | ± | 0.76 | B 24.35 | ± | 0.26 |
| Triterpene II (PR 30) | C 3.32 | ± | 0.27 | B 4.57 | ± | 0.28 | A 6.38 | ± | 0.54 | D 3.90 | ± | 0.22 |
| Triterpene III (PR 30) | B 33.36 | ± | 3.74 | A 41.27 | ± | 0.36 | B 36.79 | ± | 1.29 | B 34.74 | ± | 0.08 |
| Triterpene IV (PR 30) | B 438.12 | ± | 19.4 | A 504.66 | ± | 0.44 | C 404.35 | ± | 8.84 | C 411.04 | ± | 6.10 |
| Triterpene V (PR 30) | C 5.82 | ± | 0.25 | C 5.74 | ± | 0.13 | A 14.26 | ± | 1.23 | B 7.61 | ± | 1.25 |
| Triterpene VII (PR 30) | A 7.42 | ± | 0.51 | A 7.90 | ± | 0.51 | A 8.81 | ± | 1.53 | B 6.50 | ± | 0.03 |
| Triterpene VIII (PR 30) | B 242.75 | ± | 13.2 | A 282.45 | ± | 3.14 | D 184.12 | ± | 2.83 | C 220.45 | ± | 8.02 |
| Σ TERPENES | B 757.05 | ± | 40.5 | A 879.12 | ± | 3.7 | C 680.93 | ± | 17 | C 710.74 | ± | 13.1 |
| 1-Octacosanol (FOH 28:0) | A 1.61 | ± | 0.18 | B 1.49 | ± | 0.06 | C <LOD | na | C <LOD | na | ||
| Σ FOH | A 1.61 | ± | 0.18 | B 1.49 | ± | 0.06 | C <LOD | na | C <LOD | na | ||
| Ergosterol (ST 28:3;O) | B 5.91 | ± | 0.98 | A 9.93 | ± | 0.28 | C 2.20 | ± | 0.35 | C 2.51 | ± | 0.21 |
| Campesterol (ST 28:1;O) | C 1.64 | ± | 0.01 | A 2.70 | ± | 0.1 | B 2.01 | ± | 0.33 | D 1.12 | ± | 0.06 |
| Stigmasterol (ST 29:2;O) | C 1.67 | ± | 0.06 | A 2.81 | ± | 0.04 | B 2.30 | ± | 0.01 | D 1.37 | ± | 0.14 |
| β-Sitosterol (ST 29:1;O) | B 2.18 | ± | 0.25 | A 3.08 | ± | 0.21 | C 1.67 | ± | 0.13 | C 1.61 | ± | 0.04 |
| Σ ST | B 11.40 | ± | 1.27 | A 18.53 | ± | 0.53 | C 8.18 | ± | 0.82 | D 6.61 | ± | 0.37 |
| C7 | T0 | T1 | T2 | T3 | ||||||||
| Compound | Mean | SD | Mean | SD | Mean | SD | Mean | SD | ||||
| Triterpene I (PR 30) | C 9.80 | ± | 1.19 | A 11.48 | ± | 0.2 | C 9.67 | ± | 0.54 | B 10.66 | ± | 0.52 |
| Triterpene II (PR 30) | C 6.65 | ± | 0.91 | A 8.15 | ± | 0.27 | C 6.82 | ± | 0.2 | B 6.95 | ± | 0.26 |
| Triterpene III (PR 30) | D 20.30 | ± | 2.13 | A 23.66 | ± | 0.32 | C 21.08 | ± | 0.34 | B 21.45 | ± | 0.87 |
| Triterpene IV (PR 30) | A 352.42 | ± | 23.4 | A 383.56 | ± | 1.47 | C 309.84 | ± | 3.85 | B 328.42 | ± | 5.28 |
| Triterpene V (PR 30) | 9.41 | ± | 0.90 | 9.61 | ± | 0.25 | 9.95 | ± | 0.91 | 9.76 | ± | 0.34 |
| Triterpene VII (PR 30) | 5.26 | ± | 0.69 | 5.57 | ± | 0.35 | 4.83 | ± | 0.62 | 3.88 | ± | 0.20 |
| Triterpene VIII (PR 30) | B 230.35 | ± | 13.1 | A 252.10 | ± | 1.64 | D 195.89 | ± | 6.00 | C 201.61 | ± | 5.36 |
| Σ TERPENES | B 634.20 | ± | 42.3 | A 694.14 | ± | 3.74 | D 558.09 | ± | 9.2 | C 582.73 | ± | 9.08 |
| 1-Octacosanol (FOH 28:0) | A 3.88 | ± | 0.36 | B 3.42 | ± | 0.03 | C <LOD | na | C <LOD | na | ||
| Σ FOH | A 3.88 | ± | 0.36 | B 3.42 | ± | 0.03 | C <LOD | na | C <LOD | na | ||
| Ergosterol (ST 28:3;O) | B 15.93 | ± | 1.82 | A 18.95 | ± | 0.28 | C 11.21 | ± | 0.82 | D 5.88 | ± | 0.25 |
| Campesterol (ST 28:1;O) | B 4.20 | ± | 0.39 | A 5.11 | ± | 0.27 | C 3.91 | ± | 0.27 | D 2.83 | ± | 0.07 |
| Stigmasterol (ST 29:2;O) | B 4.63 | ± | 0.45 | A 5.68 | ± | 0.04 | C 3.85 | ± | 0.33 | D 3.12 | ± | 0.15 |
| β-Sitosterol (ST 29:1;O) | B 6.08 | ± | 0.79 | A 7.04 | ± | 0.29 | C 4.50 | ± | 0.05 | C 3.61 | ± | 0.16 |
| Σ ST | B 30.84 | ± | 3.45 | A 36.77 | ± | 0.64 | C 23.47 | ± | 1.33 | D 15.44 | ± | 0.38 |
| C6 | T0 | T1 | T2 | T3 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Compound | Mean | SD | Mean | SD | Mean | SD | Mean | SD | ||||
| Hydrocarbons | C 90.93 | ± | 0.64 | B 91.42 | ± | 0.70 | A 92.68 | ± | 0.23 | A 93.50 | ± | 0.16 |
| Wax esters | B 0.21 | ± | 0.01 | A 0.42 | ± | 0.02 | B 0.24 | ± | 0.12 | B 0.37 | ± | 0.01 |
| Triglycerides | A 0.98 | ± | 0.10 | A 0.86 | ± | 0.09 | B 0.73 | ± | 0.03 | C 0.52 | ± | 0.05 |
| Fatty alcohols | A 3.11 | ± | 0.24 | A 3.03 | ± | 0.28 | B 2.60 | ± | 0.12 | B 2.47 | ± | 0.14 |
| Phytosterols | A 1.31 | ± | 0.10 | A 1.25 | ± | 0.09 | B 1.13 | ± | 0.01 | B 1.02 | ± | 0.03 |
| Diglycerides | A 3.01 | ± | 0.08 | B 2.42 | ± | 0.24 | B 2.41 | ± | 0.05 | C 2.01 | ± | 0.02 |
| Free fatty acids | B 0.46 | ± | 0.11 | A 0.60 | ± | 0.09 | C 0.21 | ± | 0.01 | D 0.11 | ± | 0.01 |
| C7 | T0 | T1 | T2 | T3 | ||||||||
| Compound | Mean | SD | Mean | SD | Mean | SD | Mean | SD | ||||
| Hydrocarbons | C 85.81 | ± | 0.31 | A 87.52 | ± | 0.59 | B 87.79 | ± | 0.25 | C 85.77 | ± | 0.33 |
| Wax esters | B 0.41 | ± | 0.01 | A 0.52 | ± | 0.04 | C 0.29 | ± | 0.01 | B 0.46 | ± | 0.01 |
| Triglycerides | A 1.29 | ± | 0.15 | A 0.98 | ± | 0.05 | B 0.80 | ± | 0.03 | B 0.78 | ± | 0.01 |
| Fatty alcohols | A 3.59 | ± | 0.59 | A 3.39 | ± | 0.03 | B 2.47 | ± | 0.10 | B 2.34 | ± | 0.07 |
| Phytosterols | A 3.40 | ± | 0.08 | B 3.30 | ± | 0.06 | B 3.06 | ± | 0.10 | C 2.89 | ± | 0.07 |
| Diglycerides | 4.68 | ± | 1.09 | 4.35 | ± | 0.32 | 5.42 | ± | 0.08 | 5.76 | ± | 0.21 |
| Free fatty acids | B 0.82 | ± | 0.04 | A 1.11 | ± | 0.13 | B 0.46 | ± | 0.01 | C 0.55 | ± | 0.02 |
| Temperature (°C) (|ΔH| (J/g)) | |||
|---|---|---|---|
| FDR | Crystallization | Melting | Decomposition |
| T0 | 40.9 (4.7) | 44.1(6.4) | 400.5 (553.9) |
| T1 | na | na | 401.5 (317.2) |
| T2 | na | na | 408.1 (536.5) |
| T3 | na | na | 403.9 (324.1) |
| Temperature (°C) (|ΔH| (J/g)) | |||
| Feed | Crystallization | Melting | Decomposition |
| T0 | 29.6 (1.6) | 39.4 (4.3) | 400.7 (373.5) |
| T1 | na | na | 399.2 (348.4) |
| T2 | na | na | 399.4 (409.7) |
| T3 | na | na | 399.2 (295.7) |
| Temperature (°C) (|ΔH| (J/g)) | |||
| C6 | Crystallization | Melting | Decomposition |
| T0 | 28.6 (2.6) | 41.5 (2.7) | 365.3 (215.5) |
| T1 | 28.3 (2.2) | 39.3 (2.2) | 383.9 (180.8) |
| T2 | 27.1 (2.2) | 37.9 (2.3) | 374.5 (172.9) |
| T3 | 26.7 (1.8) | 35.4 (1.7) | 373.5 (137.2) |
| Temperature (°C) (|ΔH| (J/g)) | |||
| C7 | Crystallization | Melting | Decomposition |
| T0 | 37.2 (5.3) | 46.4 (5.4) | 370.9 (297.3) |
| T1 | 39.3 (4.4) | 47.4 (4.7) | 377.4 (117.9) |
| T2 | 38.2 (3.7) | 47.6 (5.4) | 371.6 (158.2) |
| T3 | 35.8 (4.1) | 44.2 (3.4) | 371.8 (166.3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teixeira, F.S.; Vidigal, S.S.M.P.; Pimentel, L.L.; Costa, P.T.; Tavares-Valente, D.; Azevedo-Silva, J.; Pintado, M.E.; Fernandes, J.C.; Rodríguez-Alcalá, L.M. Phytosterols and Novel Triterpenes Recovered from Industrial Fermentation Coproducts Exert In Vitro Anti-Inflammatory Activity in Macrophages. Pharmaceuticals 2021, 14, 583. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060583
Teixeira FS, Vidigal SSMP, Pimentel LL, Costa PT, Tavares-Valente D, Azevedo-Silva J, Pintado ME, Fernandes JC, Rodríguez-Alcalá LM. Phytosterols and Novel Triterpenes Recovered from Industrial Fermentation Coproducts Exert In Vitro Anti-Inflammatory Activity in Macrophages. Pharmaceuticals. 2021; 14(6):583. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060583
Chicago/Turabian StyleTeixeira, Francisca S., Susana S. M. P. Vidigal, Lígia L. Pimentel, Paula T. Costa, Diana Tavares-Valente, João Azevedo-Silva, Manuela E. Pintado, João C. Fernandes, and Luís M. Rodríguez-Alcalá. 2021. "Phytosterols and Novel Triterpenes Recovered from Industrial Fermentation Coproducts Exert In Vitro Anti-Inflammatory Activity in Macrophages" Pharmaceuticals 14, no. 6: 583. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14060583