Do Small Molecules Activate the TrkB Receptor in the Same Manner as BDNF? Limitations of Published TrkB Low Molecular Agonists and Screening for Novel TrkB Orthosteric Agonists

 , add
Show full author list
, add
Show full author list
Abstract
:1. Introduction
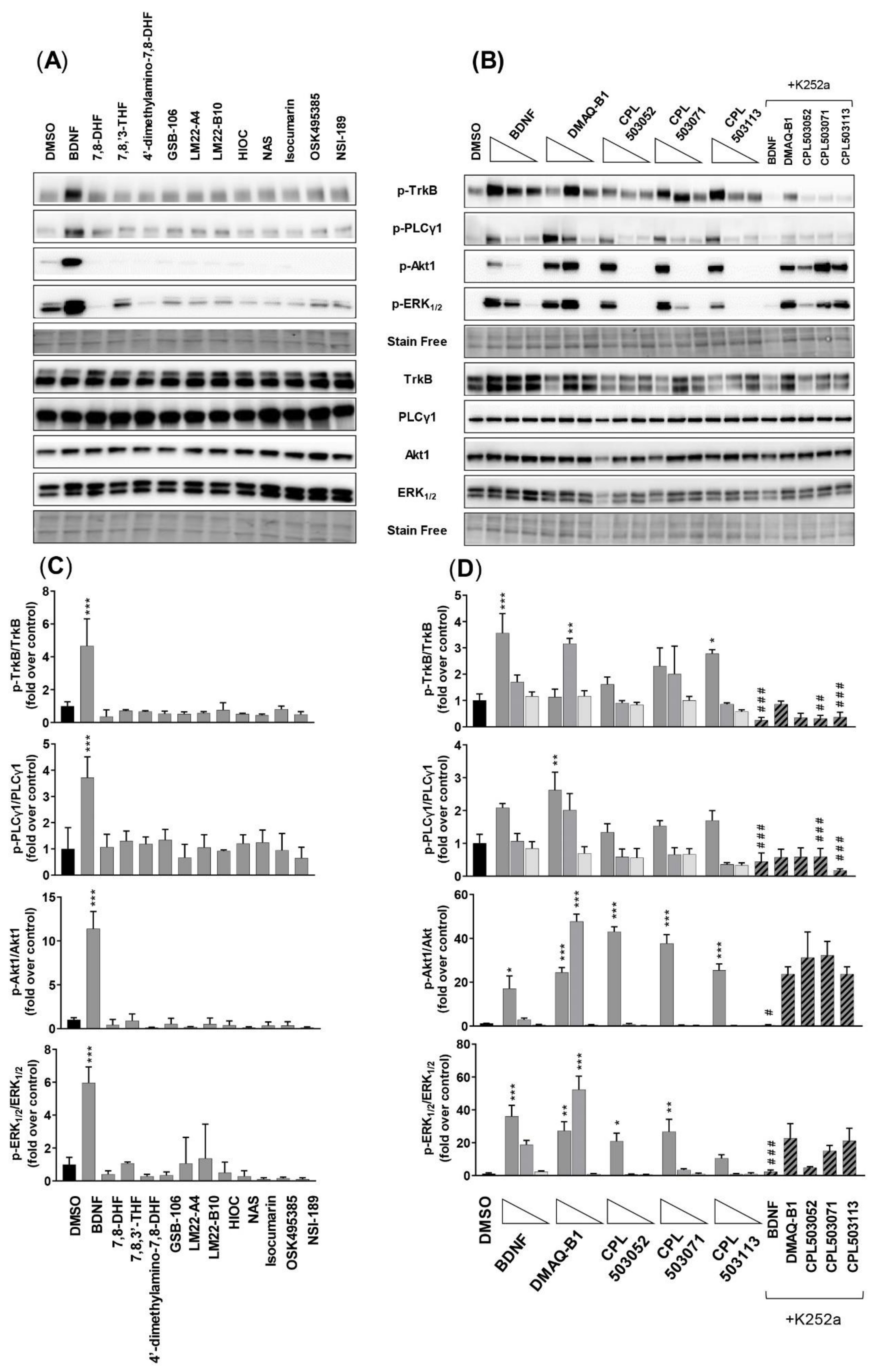
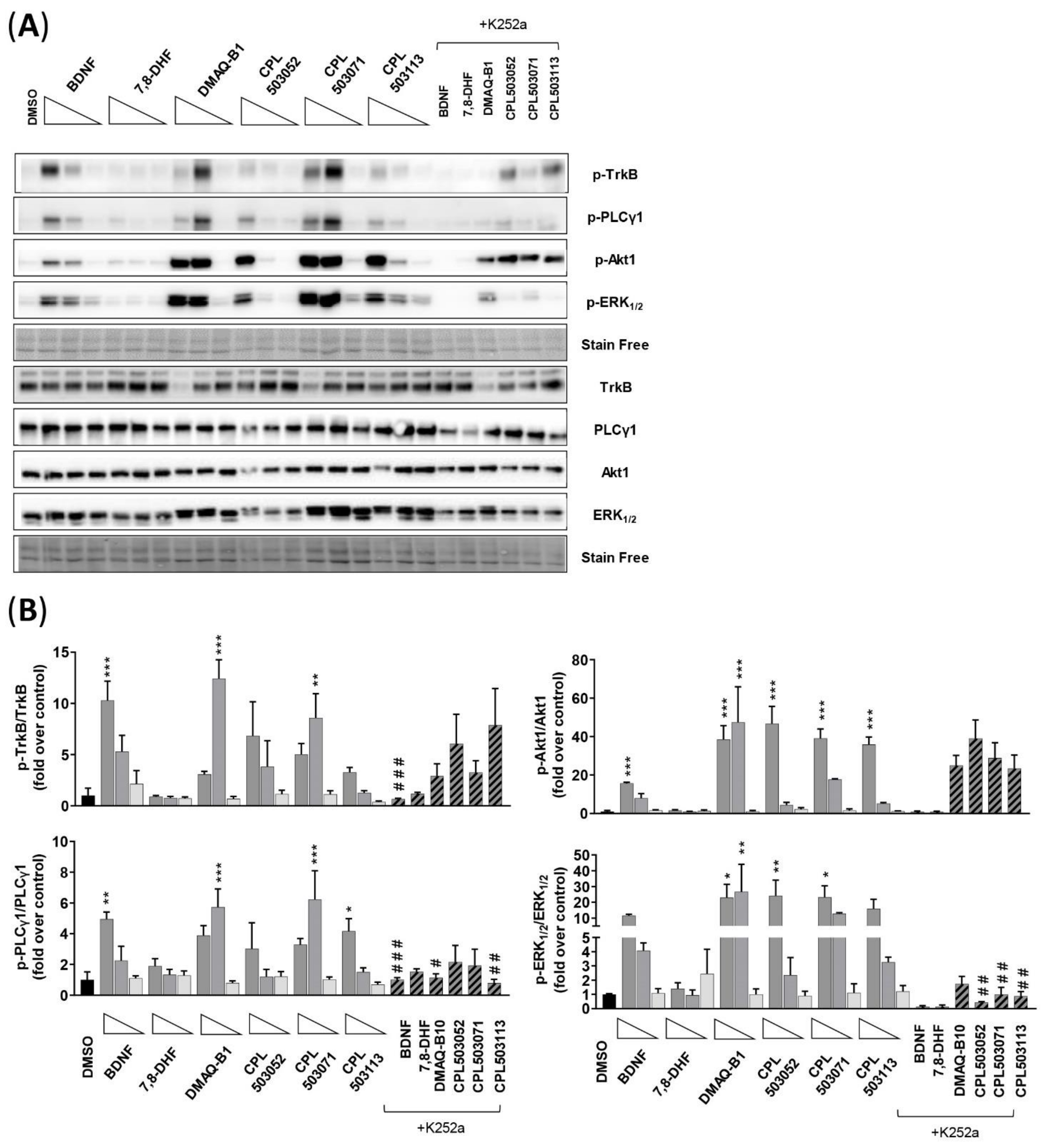
2. Results
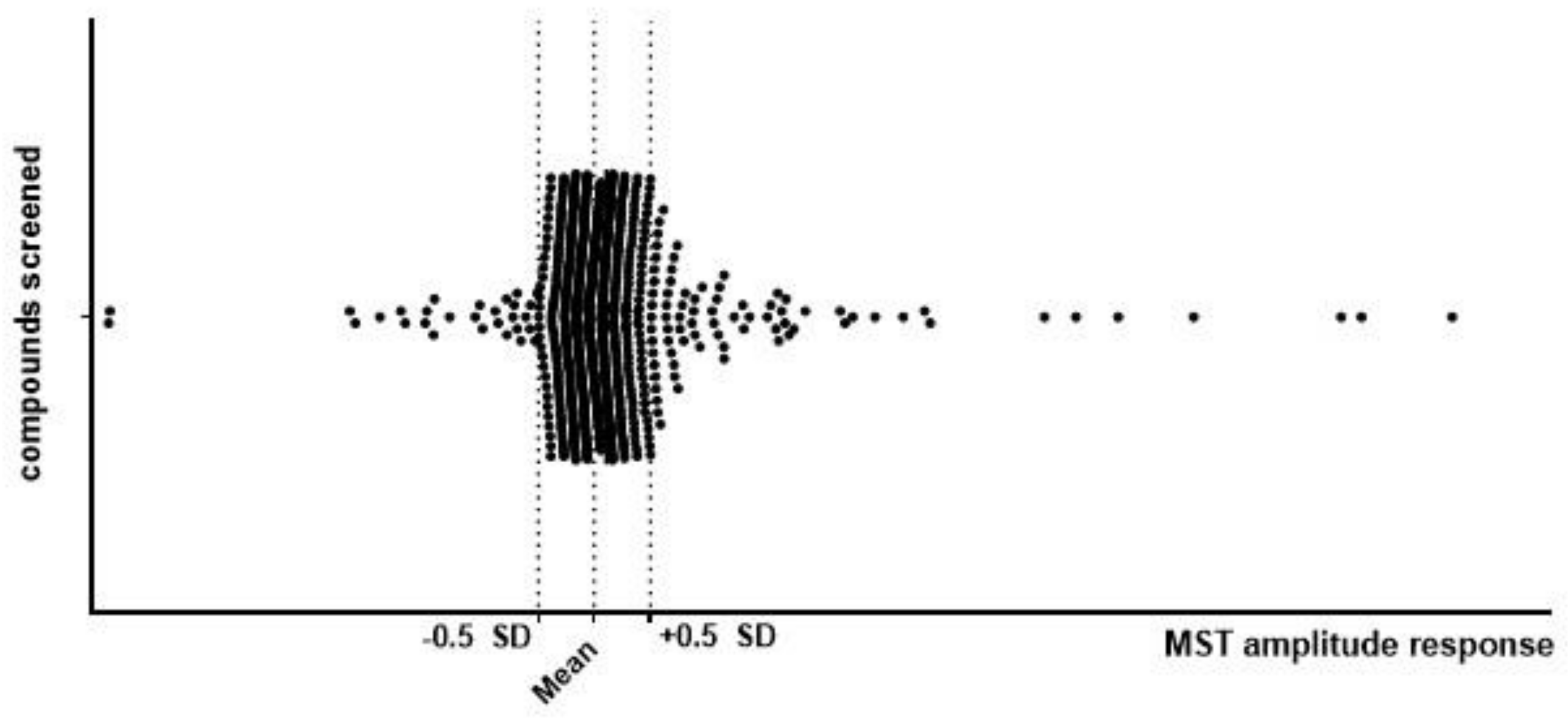
2.1. Primary Screening
2.1.1. Identification of Molecules Binding to TrkB Using MicroScale Thermophoresis
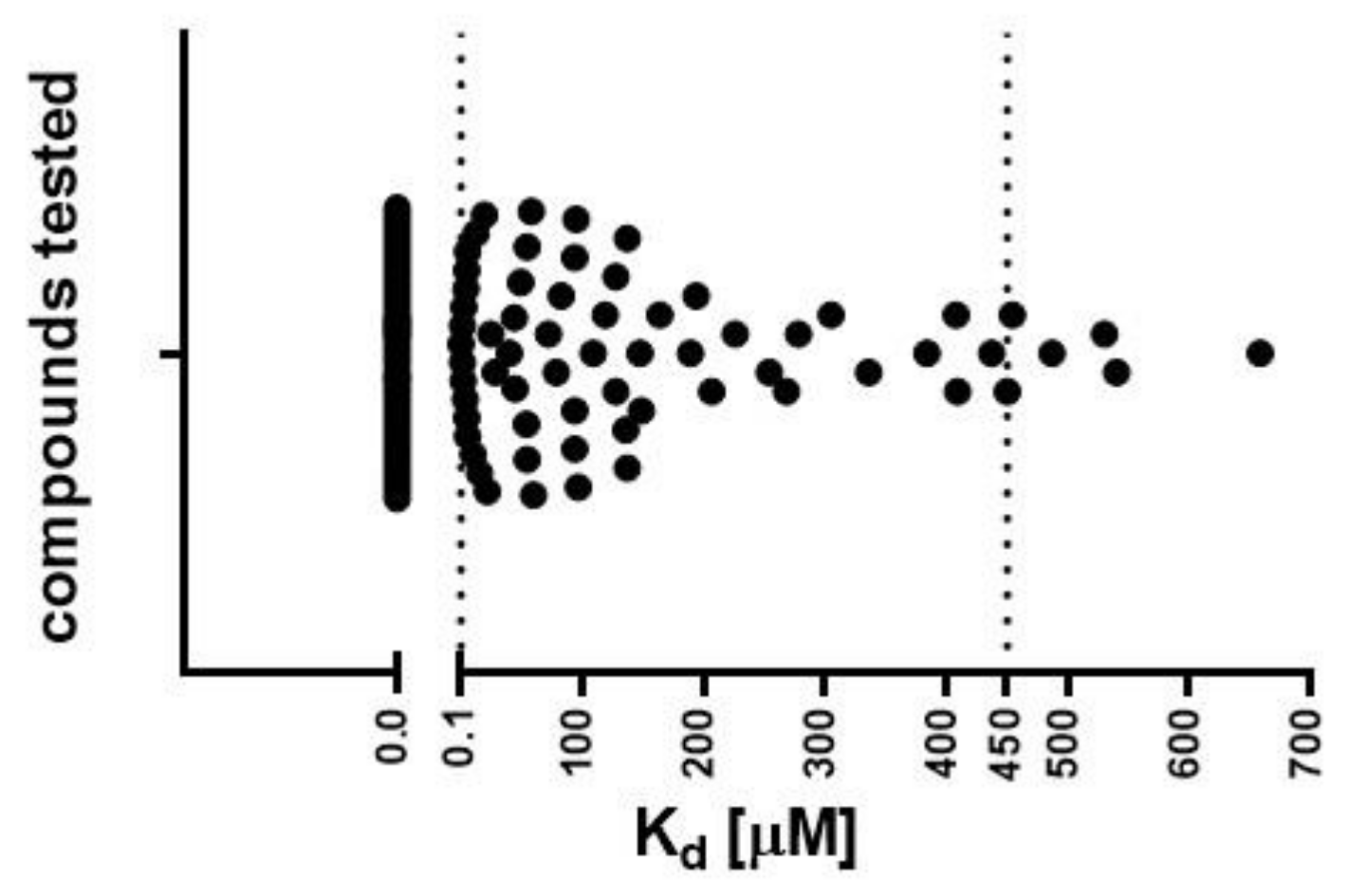
2.1.2. Determination of Dissociation Constant (Kd) by MST
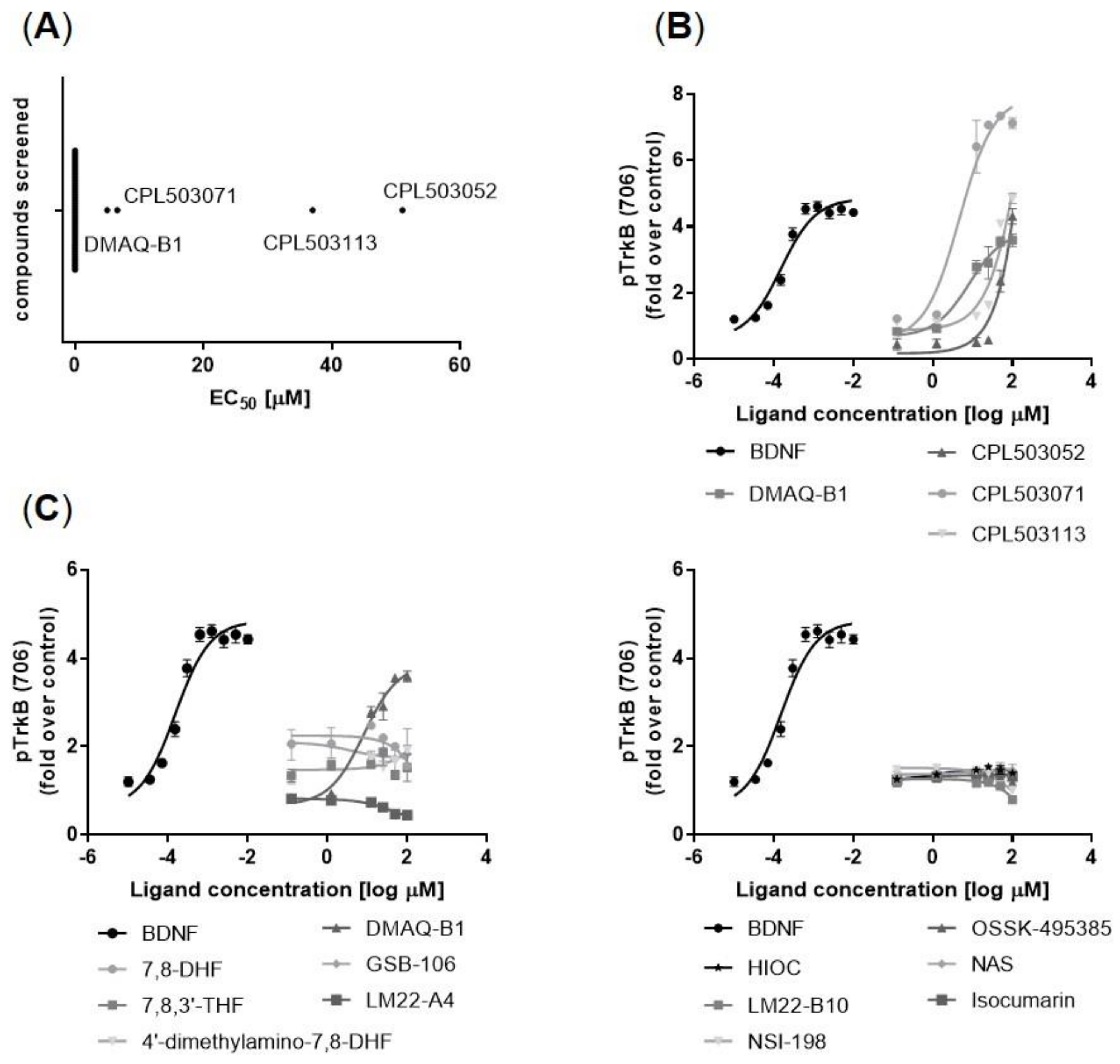
2.2. Secondary Screening
TrkB Receptor Activation in a Functional Model
2.3. Evaluation of Compounds’ Activity on TrkB Downstream Signaling
2.3.1. SN56 T48 Cell Line
2.3.2. SH-SY5Y Cell Line
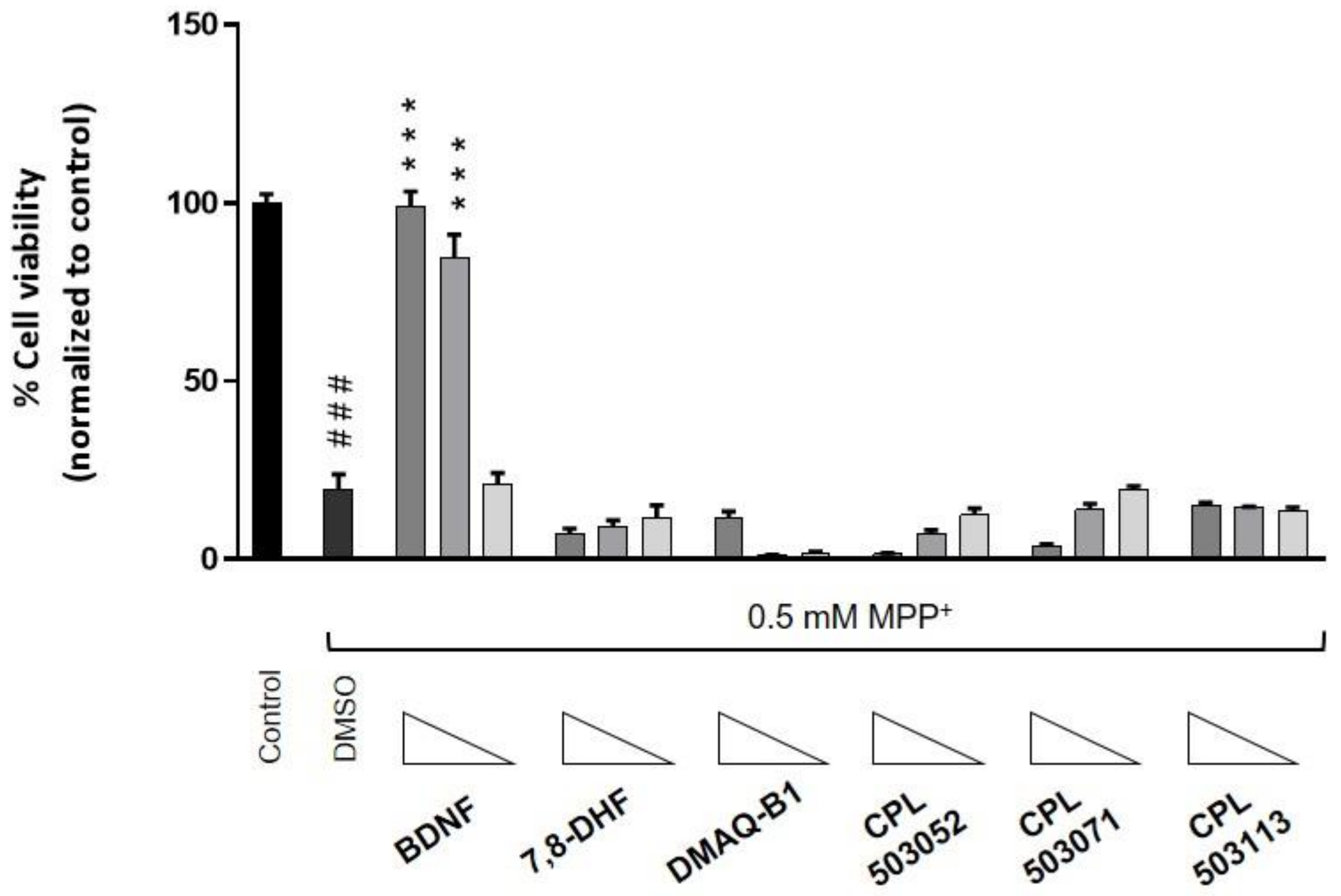
2.4. Evaluation of the Cytoprotective Effect of TrkB Agonists
2.5. Physicochemical and ADME Characterization
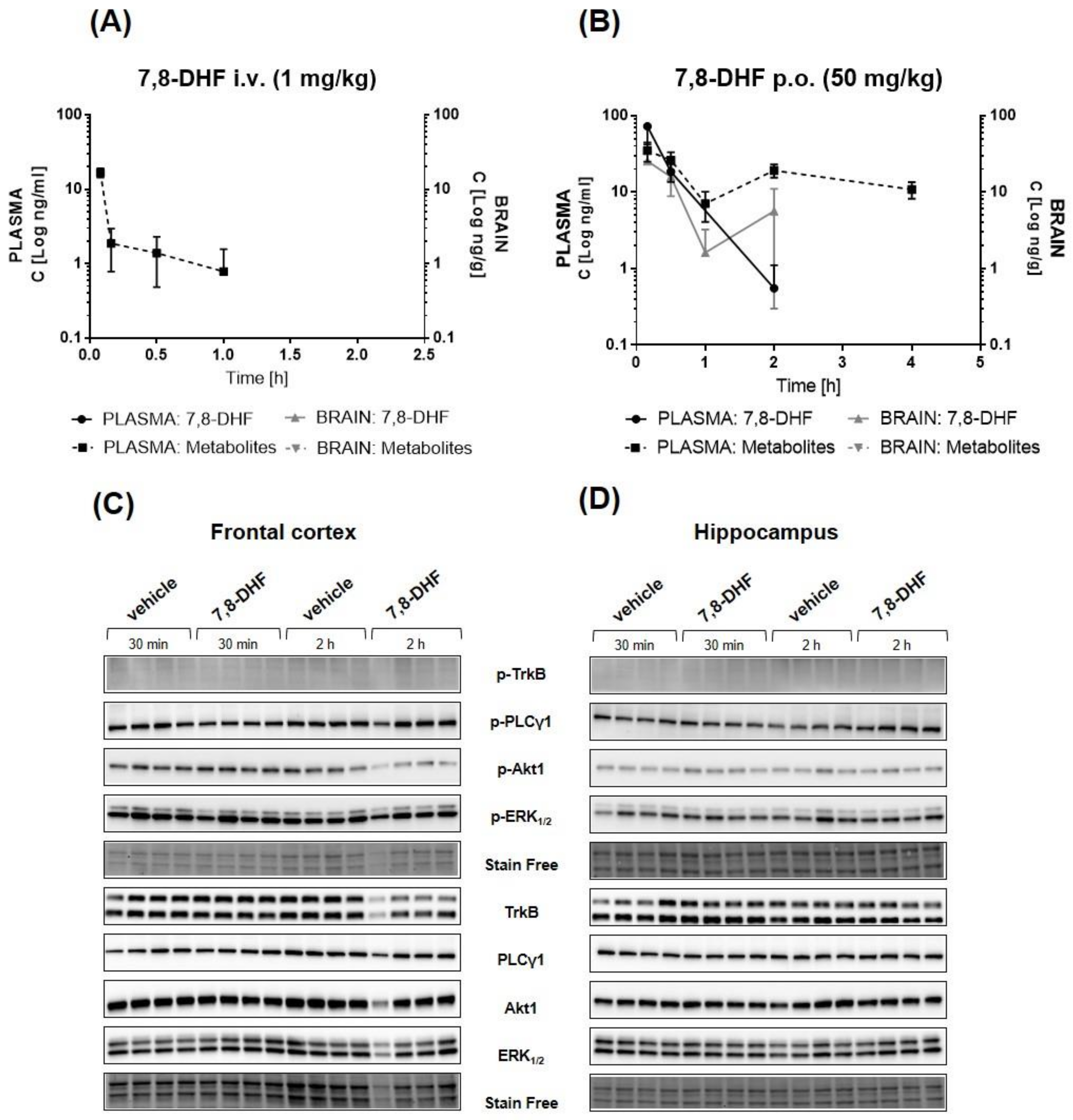
2.6. Pharmacokinetic/Pharmacodynamic (PK/PD) Profiling of 7,8-DHF
2.7. Selectivity of 7,8-DHF in a Panel of Various Molecular Targets
3. Discussion
4. Materials and Methods
4.1. Molecules Tested for Trkb Agonism
4.2. Primary Screening and Analysis of Compound Interactions with exTrkb
4.2.1. Preparation of exTrkB Protein
4.2.2. Single-Point and Binding Affinity Screening with Microscale Thermophoresis (MST)
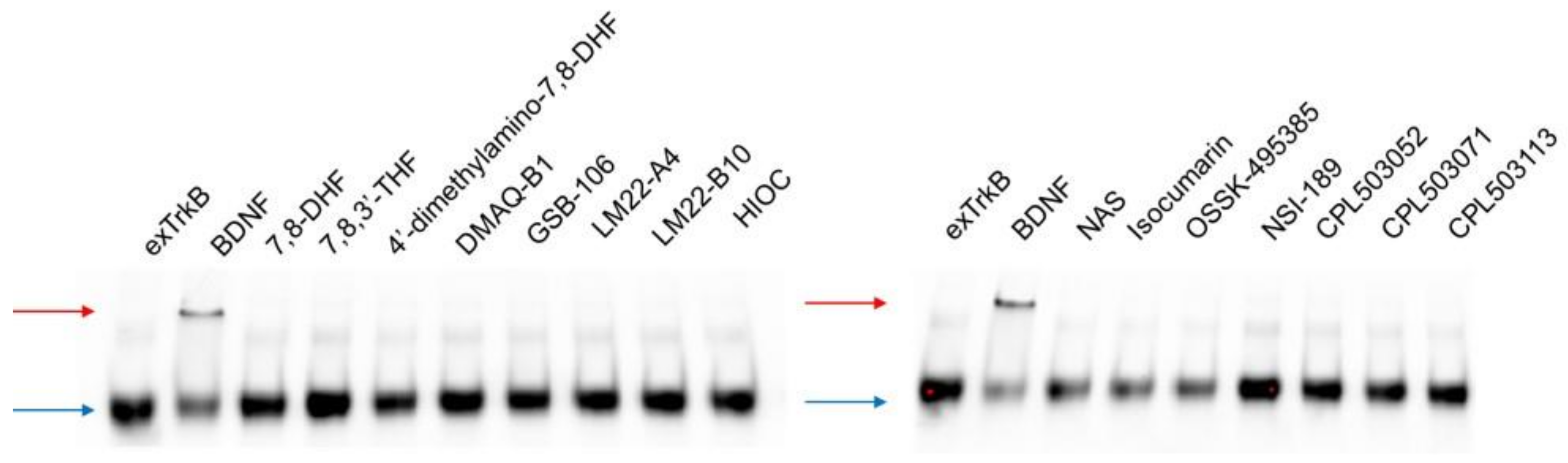
4.2.3. Ligand-Induced exTrkB Dimerization Assay
4.3. In Vitro Functional Experiments
4.3.1. Cell Lines and Materials
4.3.2. Enzyme-Linked Fixed Cell Immunoassay (ELFI) Method for Allosteric and Orthosteric Examination
4.3.3. Downstream Signaling Analysis
4.3.4. Evaluation of Neuroprotective Effect of TrkB Agonists
4.4. Physiochemical Characterization and ADME Profiling
4.4.1. Chemical Stability
4.4.2. Parallel Artificial Membrane Permeability Assay (PAMPA)
4.4.3. Caco-2 Permeability
4.4.4. Metabolic Stability
4.4.5. In Vivo
4.4.6. Binding Study
4.4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, E.J.; Reichardt, L.F. Neurotrophins: Roles in neuronal development and function. Annu. Rev. Neurosci. 2001, 24, 677–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Hyman, C.; Hofer, M.; Barde, Y.A.; Juhasz, M.; Yancopoulos, G.D.; Squinto, S.P.; Lindsay, R.M. BDNF Is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 1991, 350, 230–232. [Google Scholar] [CrossRef]
- Hetman, M.; Kanning, K.; Cavanaugh, J.E.; Xia, Z. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J. Biol. Chem. 1999, 274, 22569–22580. [Google Scholar] [CrossRef] [Green Version]
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF Function and intracellular signaling in neurons. Histol. Histopathol. 2010, 25, 237–258. [Google Scholar]
- Zahavi, E.E.; Steinberg, N.; Altman, T.; Chein, M.; Joshi, Y.; Gradus-Pery, T.; Perlson, E. The receptor tyrosine kinase TrkB signals without dimerization at the plasma membrane. Sci. Signal. 2018, 11, eaao4006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Chen, Z.Y. The role of BDNF in depression on the basis of its location in the neural circuitry. Acta Pharmacol. Sin. 2010, 32, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Dwivedi, Y. Brain-derived neurotrophic factor: Role in depression and suicide. Neuropsychiatr. Dis. Treatment. 2009, 5, 433–449. [Google Scholar] [CrossRef] [Green Version]
- Monteggia, L.M.; Björkholm, C. BDNF—A key transducer of antidepressant effects. Neuropharmacology 2017, 102, 72–79. [Google Scholar]
- Ferguson, J.M. SSRI antidepressant medications: Adverse effects and tolerability. Prim. Care Companion J. Clin. Psychiatry 2001, 3, 22–27. [Google Scholar] [CrossRef]
- Fava, M. Diagnosis and definition of treatment-resistant depression. Biol. Psychiatry 2003, 53, 649–659. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Yepes, M.; Shepherd, K.R.; Miller, G.W.; Liu, Y.; Wilson, W.D.; Xiao, G.; Blanchi, B.; Sun, Y.E.; et al. Selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. USA 2010, 107, 2687–2692. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chan, C.B.; Jang, S.W.; Pradoldej, S.; Huang, J.; He, K.; Phun, L.H.; France, S.; Xiao, G.; Jia, Y.; et al. Synthetic 7,8-dihydroxyflavone derivative promotes neurogenesis and exhibits potent antidepressant effect. J. Med. Chem. 2010, 53, 8274–8286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.A.; Wang, Y.H.; Tung, C.S.; Yeh, C.B.; Liu, Y.P. 7, 8-Dihydroxyflavone, a tropomyosin-kinase related receptor b agonist, produces fast-onset antidepressant-like effects in rats exposed to chronic mild stress. Psychiatry Investig. 2016, 13, 531–540. [Google Scholar] [CrossRef]
- Zhang, M.W.; Zhang, S.F.; Li, Z.H.; Han, F. 7,8-Dihydroxyflavone reverses the depressive symptoms in mouse chronic mild stress. Neurosci. Lett. 2016, 635, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.; Xie, S.; Tan, X.; Chen, Y.; Ren, Q.; Botchway, B.O.A.; Hu, S.; Ma, Y.; Hu, Z.; Fang, M. Optimized integration of fluoxetine and 7, 8-dihydroxyflavone as an efficient therapy for reversing depressive-like behavior in mice during the perimenopausal period. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 101, 109939. [Google Scholar] [CrossRef]
- Wilkie, N.; Wingrove, P.B.; Bilsland, J.G.; Young, L.; Harper, S.J.; Hefti, F.; Ellis, S.; Pollack, S.J. The non-peptidyl fungal metabolite L-783,281 activates Trk neurotrophin receptors. J. Neurochem. 2001, 78, 1135–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massa, S.M.; Yang, T.; Xie, Y.; Shi, J.; Bilgen, M.; Joyce, J.N.; Nehama, D.; Rajadas, J.; Longo, F.M. Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J. Clin. Investig. 2010, 120, 1774–1785. [Google Scholar] [CrossRef] [Green Version]
- Gudasheva, T.A.; Logvinov, I.O.; Antipova, T.A.; Seredenin, S.B. Brain-derived neurotrophic factor loop 4 dipeptide mimetic GSB-106 activates TrkB, Erk, and Akt and promotes neuronal survival in vitro. Dokl. Biochem. Biophys. 2013, 451, 212–214. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Pradoldej, S.; Tosini, G.; Chang, Q.; Iuvone, P.M.; Ye, K. N-Acetylserotonin activates TrkB receptor in a circadian rhythm. Proc. Natl. Acad. Sci. USA 2010, 107, 3876–3881. [Google Scholar] [CrossRef] [Green Version]
- Sudarshan, K.; Boda, A.K.; Dogra, S.; Bose, I.; Yadav, P.N.; Aidhen, I.S. Discovery of an isocoumarin analogue that modulates neuronal functions via neurotrophin receptor TrkB. Bioorg. Med. Chem. Lett. 2019, 29, 585–590. [Google Scholar] [CrossRef]
- Liu, Y.; Johe, K.; Sun, J.; Hao, X.; Wang, Y.; Bi, X.; Baudry, M. Enhancement of Synaptic plasticity and reversal of impairments in motor and cognitive functions in a mouse model of angelman syndrome by a small neurogenic molecule, NSI-189. Neuropharmacology 2019, 144, 337–344. [Google Scholar] [CrossRef]
- Shen, J.; Ghai, K.; Sompol, P.; Liu, X.; Cao, X.; Iuvone, P.M.; Ye, K. N-Acetyl serotonin derivatives as potent neuroprotectants for retinas. Proc. Natl. Acad. Sci. USA 2012, 109, 3540–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, M.; Takatori, A.; Nakamura, Y.; Suganami, A.; Hoshino, T.; Tamura, Y.; Nakagawara, A. Effects of novel small compounds targeting TrkB on neuronal cell survival and depression-like behavior. Neurochem. Int. 2016, 97, 42–48. [Google Scholar] [CrossRef]
- Todd, D.; Gowers, I.; Dowler, S.J.; Wall, M.D.; McAllister, G.; Fischer, D.F.; Dijkstra, S.; Fratantoni, S.A.; Van De Bospoort, R.; Veenman-Koepke, J.; et al. A monoclonal antibody TrkB receptor agonist as a potential therapeutic for Huntington’s disease. PLoS ONE 2014, 9, e87923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boltaev, U.; Meyer, Y.; Tolibzoda, F.; Jacques, T.; Gassaway, M.; Xu, Q.; Wagner, F.; Zhang, Y.L.; Palmer, M.; Holson, E.; et al. Multiplex quantitative assays indicate a need for re-evaluating reported small-molecule TrkB agonists. Sci. Signal. 2017, 10, eaal1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalevich, J.; Langford, D. Considerations for the use of SH-SY5Y neuroblastoma cells in neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [PubMed] [Green Version]
- Colucci-D’amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic factor Bdnf, physiological functions and therapeutic potential in depression, neurodegeneration and brain cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qi, Q.; Xiao, G.; Li, J.; Luo, H.R.; Ye, K. O-Methylated metabolite of 7,8-dihydroxyflavone activates TrkB receptor and displays antidepressant activity. Pharmacology 2013, 91, 185–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowes, J.; Brown, A.J.; Hamon, J.; Jarolimek, W.; Sridhar, A.; Waldron, G.; Whitebread, S. Reducing Safety-related drug attrition: The use of in vitro pharmacological profiling. Nat. Rev. Drug Discov. 2012, 11, 909–922. [Google Scholar] [CrossRef]
- Reichardt, L.F. Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1545–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumagalli, F.; Racagni, G.; Riva, M.A. The Expanding Role of BDNF: A therapeutic target for Alzheimer’s disease? Pharm. J. 2006, 6, 8–15. [Google Scholar] [CrossRef] [Green Version]
- Casarotto, P.; Girych, M.; Fred, S.; Kovaleva, V.; Moliner, R.; Enkavi, G.; Biojone, C.; Cannarozzo, C.; Sahu, M.P.; Kaurinkoski, K.; et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell 2021, 184, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Emili, M.; Guidi, S.; Uguagliati, B.; Giacomini, A.; Bartesaghi, R.; Stagni, F. Treatment with the flavonoid 7,8-dihydroxyflavone: A promising strategy for a constellation of body and brain disorders. Crit. Rev. Food Sci. Nutr. 2020, 1–38, in press. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Obiany, O.; Chan, C.B.; Huang, J.; Xue, S.; Yang, J.J.; Zeng, F.; Goodman, M.; Ye, K. Biochemical and Biophysical Investigation of the Brain-Derived Neurotrophic Factor Mimetic 7,8-Dihydroxyflavone in the Binding and Activation of the Trkb Receptor. J. Biol. Chem. 2014, 289, 27571–27584. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Chua, K.W.; Chua, C.C.; Yu, H.; Pei, A.; Chua, B.H.L.; Hamdy, R.C.; Xu, X.; Liu, C.F. Antioxidant activity of 7,8-dihydroxyflavone provides neuroprotection against glutamate-induced toxicity. Neurosci. Lett. 2011, 499, 181–185. [Google Scholar] [CrossRef]
- Han, X.; Zhu, S.; Wang, B.; Chen, L.; Li, R.; Yao, W.; Qu, Z. Antioxidant action of 7,8-dihydroxyflavone protects pc12 cells against 6-hydroxydopamine-induced cytotoxicity. Neurochem. Int. 2014, 64, 18–23. [Google Scholar] [CrossRef]
- Song, Y.; Han, X. 7,8-dihydroxyflavone protects pc12 cells against mpp+-induced cytotoxicity by heme oxygenase-1. Int. J. Sci. 2019, 8, 55–59. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.W.; Liu, X.; Chan, C.B.; France, S.A.; Sayeed, I.; Tang, W.; Lin, X.; Xiao, G.; Andero, R.; Chang, Q. Deoxygedunin, a natural product with potent neurotrophic activity in mice. PLoS ONE 2010, 5, e11528. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wang, Z.; Zhang, Z.; Liu, X.; Kang, S.S.; Zhang, Y.; Ye, K. The prodrug of 7,8-dihydroxyflavone development and therapeutic efficacy for treating alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, 578–583. [Google Scholar] [CrossRef] [Green Version]
- El Atifi-Borel, M.; Buggia-Prevot, V.; Platet, N.; Benabid, A.L.; Berger, F.; Sgambato-Faure, V. De novo and long-term l-dopa induce both common and distinct striatal gene profiles in the hemiparkinsonian rat. Neurobiol. Dis. 2009, 34, 340–350. [Google Scholar] [CrossRef]
- Luscher, B.; Shen, Q.; Sahir, N. The GABAergic deficit hypothesis of major depressive disorder. Mol. Psychiatry 2010, 16, 383–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago, R.M.; Barbiero, J.; Martynhak, B.J.; Boschen, S.L.; Da Silva, L.M.; Werner, M.F.P.; Da Cunha, C.; Andreatini, R.; Lima, M.M.S.; Vital, M.A.B.F. Antidepressant-like effect of celecoxib piroxicam in rat models of depression. J. Neural Transm. 2014, 121, 671–682. [Google Scholar] [CrossRef]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Akhondzadeh, S.; Jafari, S.; Raisi, F.; Nasehi, A.A.; Ghoreishi, A.; Salehi, B.; Mohebbi-Rasa, S.; Raznahan, M.; Kamalipour, A. Clinical trial of adjunctive celecoxib treatment in patients with major depression: A double blind and placebo controlled trial. Depress. Anxiety 2009, 26, 607–611. [Google Scholar] [CrossRef]
- Abbasi, S.H.; Hosseini, F.; Modabbernia, A.; Ashrafi, M.; Akhondzadeh, S. Effect of celecoxib add-on treatment on symptoms and serum IL-6 concentrations in patients with major depressive disorder: Randomized double-blind placebo-controlled study. J. Affect. Disord. 2012, 141, 308–314. [Google Scholar] [CrossRef]
- Arai, K.; Shimizu, S.; Taguchi, Y.; Yamamoto, Y. Metabolic products of Aspergillus terreus. VII. Astechrome: An iron-containing metabolite of the strain IFO 6123. Chem. Pharm. Bull. 1981, 29, 1510–1517. [Google Scholar] [CrossRef] [Green Version]
- Pirrung, M.C.; Park, K.; Li, Z. Synthesis of 3-indolyl-2,5-dihydroxybenzoquinones. Org. Lett. 2001, 3, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Presgraves, S.P.; Borwege, S.; Millan, M.J.; Joyce, J.N. Involvement of dopamine D 2/D 3 receptors and BDNF in the neuroprotective effects of S32504 and pramipexole against 1-Methyl-4-Phenylpyridinium in terminally differentiated SH-SY5Y Cells. Exp. Neurol. 2004, 190, 157–170. [Google Scholar] [CrossRef]
- Sugano, K.; Okazaki, A.; Sugimoto, S.; Tavornvipas, S.; Omura, A.; Mano, T. Solubility and dissolution profile assessment in drug discovery. Drug Metab. Pharmacokinet. 2007, 22, 225–254. [Google Scholar] [CrossRef] [PubMed]
- Guha, R.; Dexheimer, T.S.; Kestranek, A.N.; Jadhav, A.; Chervenak, A.M.; Ford, M.G.; Simeonov, A.; Roth, G.P.; Thomas, C.J. Exploratory analysis of kinetic solubility measurements of a small molecule library. Bioorg. Med. Chem. 2011, 19, 4127–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredlund, L.; Winiwarter, S.; Hilgendorf, C. In vitro intrinsic permeability: A transporter-independent measure of Caco-2 cell permeability in drug design and development. Mol. Pharm. 2017, 14, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Gunerka, P.; Gala, K.; Banach, M.; Dominowski, J.; Hucz-Kalitowska, J.; Mulewski, K.; Hajnal, A.; Mikus, E.G.; Smuga, D.; Zagozda, M.; et al. Preclinical characterization of CPL302-253, a selective inhibitor of PI3Kδ, as the candidate for the inhalatory treatment and prevention of asthma. PLoS ONE 2020, 15, e0236159. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Kd [μM] | EC50 [μM] | PAM Activity | TrkB Dimerization |
|---|---|---|---|---|
| BDNF | 0.093 | 0.004 | NO | YES |
| 7,8-DHF | 1.3 | 0 | NO | NO |
| OSK495385 | 2.32 | 0 | NO | NO |
| DMAQ-B1 | 5.6 | 5 | NO | NO |
| LM22-B10 | 83 | 0 | NO | NO |
| 7,8,3′-THF | 0 | 0 | NO | NO |
| 4′-dimethylamino-7,8-DHF | 0 | 0 | NO | NO |
| GSB-106 | 0 | 0 | NO | NO |
| LM22-A4 | 0 | 0 | NO | NO |
| HIOC | 0 | 0 | NO | NO |
| NAS | 0 | 0 | NO | NO |
| Isocumarin | 0 | 0 | NO | NO |
| NSI-189 | 0 | 0 | NO | NO |
| CPL503071 | 25 | 6.6 | NO | NO |
| CPL503113 | 44.2 | 37 | NO | NO |
| CPL503052 | 2.6 | 51 | NO | NO |
| Compound | Kinetic Solubility [μM] | Chemical Stability [% of Degradation] | PAMPA [10−6·cm/s] | Papp,AB [10−6·cm/s] | Papp,BA [10−6·cm/s] | Clint HLM [μL·min−1 ·mgprotein−1] | Clint MLM [μL·min−1 ·mgprotein−1] |
|---|---|---|---|---|---|---|---|
| 7,8,3′-THF | 168 | Stable | 1.1 | 3.00 | 11.14 | 49.4 | 26.6 |
| 4′-dimethylamino-7,8-DHF | 46.6 | 1.31 | 15.5 | 15.61 | 9.03 | 175.7 | 365.1 |
| DMAQ-B1 | 500 | 12.76 | 6.93 | 0.00 | 0.00 | ** | ** |
| GSB-106 | 152.7 | Stable | 0.85 | ** | ** | ** | ** |
| LM22-A4 | 397 | Stable | 0.01 | 0.00 | 0.12 | 3.7 | 0.0 |
| LM22-B10 | <1 | 4.18 | 2.36 | 6.47 | 4.97 | 29 | 4.8 |
| HIOC | 437 | Stable | 1.24 | 1.11 | 2.29 | 0.0 | 2.1 |
| NAS | 404 | Stable | <0.01 | 8.86 | 8.00 | 0.3 | 1.5 |
| Isocumarin | 5 | Stable | 0.22 | 1.60 | 2.18 | 780.7 | 407.4 |
| OSSK-495385 | 500 | Stable | 0.03 | 0.61 | 10.86 | 10 | 3.3 |
| NSI-189 | 500 | 0.65 | 8.91 | 28.23 | 13.22 | 466.6 | 1316.7 |
| CPL503052 | 21 | 89.84 | * | * | * | * | * |
| CPL503071 | 67,8 | 38.67 | * | * | * | * | * |
| CPL503113 | 195 | 12.43 | * | * | * | * | * |
| 7,8-DHF p.o. (50 mg/kg) | 7,8-DHF i.v. (1 mg/kg) | ||||||
|---|---|---|---|---|---|---|---|
| AUC | Cmax | Tmax [h] | AUC | Cmax | Tmax [h] | ||
| PLASMA | 7,8-DHF | 20.9 | 72.42 ng/mL | 0.16 | N/D | N/D | N/D |
| 7H8M-flavone 8H7M-flavone | 61.8 | 34.68 ng/mL | 0.16 | 2.2 | 16.72 ng/mL | 0.08 | |
| BRAIN | 7,8-DHF | 5.2 | 6.35 ng/g | 0.16 | N/D | N/D | N/D |
| 7H8M-flavone 8H7M-flavone | N/D | N/D | N/D | N/D | N/D | N/D | |
| Target | Assay Type | Family | % Inhibition of Control Specific Binding |
|---|---|---|---|
| A1 | Agonist | Receptor | 99 |
| Xanthine oxidase/ superoxide O2-scavenging | Non-Kinase Enzyme | 91.5 | |
| MT3 (ML2) | Agonist | Receptor | 86 |
| COX2 | Non-Kinase Enzyme | 81.6 | |
| A2B | Antagonist | Receptor | 78.3 |
| A2A | Agonist | Receptor | 74.9 |
| MMP-9 | Non-Kinase Enzyme | 72 | |
| BZD | Agonist | Ion Channel | 70.6 |
| A3 | Agonist | Receptor | 68.2 |
| MMP-2 | Non-Kinase Enzyme | 56.5 | |
| Lyn A kinase | Kinase | 52.6 | |
| 5-HT2B | Agonist | Receptor | 50.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pankiewicz, P.; Szybiński, M.; Kisielewska, K.; Gołębiowski, F.; Krzemiński, P.; Rutkowska-Włodarczyk, I.; Moszczyński-Pętkowski, R.; Gurba-Bryśkiewicz, L.; Delis, M.; Mulewski, K.; et al. Do Small Molecules Activate the TrkB Receptor in the Same Manner as BDNF? Limitations of Published TrkB Low Molecular Agonists and Screening for Novel TrkB Orthosteric Agonists. Pharmaceuticals 2021, 14, 704. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14080704
Pankiewicz P, Szybiński M, Kisielewska K, Gołębiowski F, Krzemiński P, Rutkowska-Włodarczyk I, Moszczyński-Pętkowski R, Gurba-Bryśkiewicz L, Delis M, Mulewski K, et al. Do Small Molecules Activate the TrkB Receptor in the Same Manner as BDNF? Limitations of Published TrkB Low Molecular Agonists and Screening for Novel TrkB Orthosteric Agonists. Pharmaceuticals. 2021; 14(8):704. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14080704
Chicago/Turabian StylePankiewicz, Piotr, Marcin Szybiński, Katarzyna Kisielewska, Filip Gołębiowski, Patryk Krzemiński, Izabela Rutkowska-Włodarczyk, Rafał Moszczyński-Pętkowski, Lidia Gurba-Bryśkiewicz, Monika Delis, Krzysztof Mulewski, and et al. 2021. "Do Small Molecules Activate the TrkB Receptor in the Same Manner as BDNF? Limitations of Published TrkB Low Molecular Agonists and Screening for Novel TrkB Orthosteric Agonists" Pharmaceuticals 14, no. 8: 704. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14080704