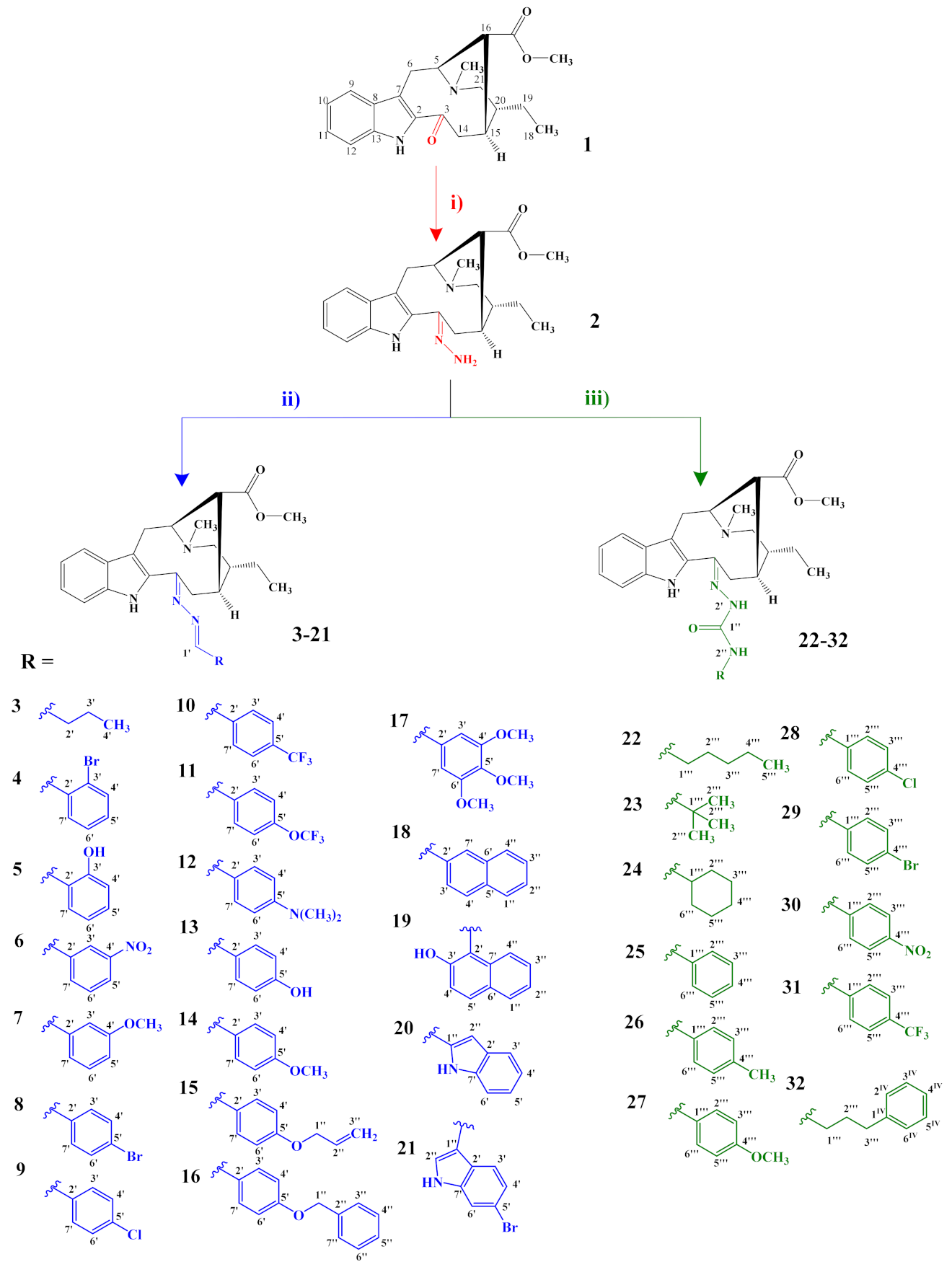
3.2.1. General Preparation of Dregamine Azines 3–21
Compound (2) (15–32 mg, 1 equiv.) was dissolved in ethanol (EtOH, 2 mL) with suitable aldehyde (5–6 equiv.), and a catalytic amount of acetic acid was added. The mixture was stirred, at room temperature and under nitrogen atmosphere, for 2 h. After evaporating the solvent, under vacuum at 40 °C, the residue was submitted to flash chromatography and further purified by preparative TLC.
3-[(butylidene)hydrazineylidene]dregamine (3). Obtained from reaction of compound 2 (20 mg, 0.054 mmol, 1 equiv.) with butyraldehyde (29 µL, 0.33 mmol, 6 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 6 mg (0.014 mmol, yield 26%) of a yellow oil. 1H NMR (300 MHz, MeOD) δ 7.86 (1H, t, J = 5.4 Hz, H-1′), 7.60 (1H, bd, J = 8.0 Hz, H-9), 7.33 (1H, bd, J = 8.1 Hz, H-12), 7.20 (1H, m, H-11), 7.05 (1H, m, H-10), 3.94 (1H, m, H-5), 3.63 (1H, dd, J = 13.1, 6.1 Hz, H-14a), 3.15 (1H, dd, J = 14.7, 10.3 Hz, H-6a), 2.72–2.64* (5H, H-6b, H-21b, –COOMe), 2.62 (1H, m, H-15), 2.60–2.49* (5H, H-16, H-21a, N-Me), 2.47–2.31* (3H, H-14b, H-2′), 1.81 (1H, m, H-20), 1.68 (2H, m, H-3′), 1.43–1.34* (2H, m, H-19), 1.09–1.01* (6H, H-18, H-4′) ppm; 13C NMR (75 MHz, MeOD) δ 172.0 (–COOMe), 162.8 (C-1′), 160.0 (C-3), 138.0 (C-13), 133.6 (C-2), 130.5 (C-8), 125.4 (C-11), 120.2 (C-10), 120.0 (C-9), 117.5 (C-7), 112.2 (C-12), 58.9 (C-5), 50.9 (–COOMe), 50.0–48.0* (C-16, C-21), 44.1 (C-20), 42.2 (N-Me), 36.0 (C-2′), 31.8 (C-15), 25.4 (C-14), 24.3 (C-19), 21.2 (C-3′), 20.7 (C-6), 14.2 (C-4′), 11.5 (C-18) ppm. * signals overlapped. ESIMS m/z 423 (M + H)+.
3-[(2″-bromobenzylidene)hydrazineylidene]dregamine (4). Obtained from reaction of compound 2 (32 mg, 0.087 mmol, 1 equiv.) with 2-bromobenzaldehyde (60 µL, 0.52 mmol, 6 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 98.5:1.5) and preparative TLC (CH2Cl2/MeOH, 49:1) to afford 13 mg (0.023 mmol, yield 28%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD) δ 8.91 (1H, s, H-1′), 8.12 (1H, dd, J = 7.6, 1.8 Hz, H-4′), 7.63–7.55* (2H, m, H-9, H-7′), 7.39 (1H, bd, J = 7.9 Hz, H-12), 7.36–7.28* (2H, H-5′, H-6′), 7.23 (1H, m, H-11), 7.07 (1H, dd, J = 7.9, 7.1 Hz, H-10), 3.90 (1H, m, H-5), 3.79 (1H, dd, J = 13.2, 6.5 Hz, H-14a), 3.15 (1H, dd, J = 14.9, 8.2 Hz, H-6a), 2.85 (1H, dd, J = 15.1, 10.0 Hz, H-6b), 2.73–2.63* (5H, H-15, H-21b, –COOMe), 2.51–2.42* (5H, H-16, H-21a, N-Me), 2.19 (1H, t, J = 13.1, H-14b), 1.78 (1H, m, H-20), 1.38 (2H, m, H-19), 1.04 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD) δ 171.9 (–COOMe), 162.9 (C-3), 158.3 (C-1′), 138.3 (C-13), 134.9 (C-2′), 134.4 (C-7′), 133.7 (C-2), 133.1 (C-6′), 130.4 (C-8), 129.4 (C-4′), 128.7 (C-5′), 126.2 (C-3′), 125.9 (C-11), 120.4 (C-10), 120.2 (C-9), 118.4 (C-7), 112.5 (C-12), 58.7 (C-5), 51.0 (–COOMe), 50.0–48.0* (C-16, C-21), 43.9 (C-20), 42.0 (N-Me), 31.6 (C-15), 25.5 (C-14), 24.2 (C-19), 21.3 (C-6), 11.4 (C-18) ppm. * signals overlapped. ESIMS m/z 537 (M + H)+.
3-[(2″-hydroxybenzylidene)hydrazineylidene]dregamine (5). Obtained from reaction of compound 2 (27 mg, 0.073 mmol, 1 equiv.) with 2-hydroxybenzaldehyde (46 µL, 0.44 mmol, 6 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 25 mg (0.053 mmol, yield 72%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD/CDCl3) δ 8.63 (1H, s, H-1′), 7.62 (1H, bd, J = 8.0 Hz, H-9), 7.38 (1H, bd, J = 8.1 Hz, H-12), 7.35–7.25* (2H, H-5′, H-11), 7.21 (1H, dd, J = 7.4, 1.4 Hz, H-7′), 7.12 (1H, t, J = 7.2 Hz, H-10), 6.99 (1H, d, J = 8.2 Hz, H-4′), 6.87 (1H, t, J = 7.4 Hz, H-6′), 3.88 (1H, m, H-5), 3.40 (1H, m, H-14a), 3.17 (1H, dd, J = 14.8, 8.2 Hz, H-6a), 2.80 (1H, dd, J = 14.6, 10.2 Hz, H-6b), 2.72–2.61* (5H, H-15, H-21b, –COOMe), 2.48–2.37* (4H, H-16, N-Me), 2.29–2.10* (2H, H-14b, H-21a), 1.76 (1H, m, H-20), 1.46 (1H, m, H-19a), 1.31 (1H, m, H-19b), 1.04 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD/CDCl3) δ 171.4 (–COOMe), 163.4 (C-3′), 161.1 (C-3), 159.7 (C-1′), 137.0 (C-13), 132.9 (C-5′), 132.6 (C-7′), 132.5 (C-2), 129.5 (C-8), 125.7 (C-11), 120.1 (C-10), 120.0 (C-9), 119.6 (C-6′), 118.7 (C-7), 118.3 (C-2′), 116.8 (C-4′), 111.7 (C-12), 57.6 (C-5), 50.7 (–COOMe), 50.0–48.0* (C-16, C-21), 43.6 (C-20), 42.0 (N-Me), 31.5 (C-15), 25.2 (C-14), 23.9 (C-19), 20.6 (C-6), 11.8 (C-18) ppm. * signals overlapped. ESIMS m/z 473 (M + H)+.
3-[(3″-nitrobenzylidene)hydrazineylidene]dregamine (6). Obtained from reaction of compound 2 (28 mg, 0.076 mmol, 1 equiv.) with 3-nitrobenzaldehyde (69 mg, 0.46 mmol, 6 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 10 mg (0.019 mmol, yield 26%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD/CDCl3) δ 8.73 (1H, t, J = 1.6 Hz, H-3′), 8.68 (1H, s, H-1′), 8.28 (1H, dd, J = 8.2, 1.4 Hz, H-5′), 8.11 (1H, bd, J = 7.7 Hz, H-9), 7.70–7.64* (2H, H-6′, H-7′), 7.40 (1H, bd, J = 8.2 Hz, H-12), 7.28 (1H, t, J = 7.4 Hz, H-11), 7.12 (1H, t, J = 7.4 Hz, H-10), 4.08 (1H, m, H-5), 3.99 (1H, dd, J = 13.1, 6.3 Hz, H-14a), 3.38 (1H, m, H-6a), 3.15 (1H, dd, J = 14.5, 10.3 Hz, H-6b), 2.75–2.87* (3H, H-15, H-21a, H-21b), 2.57–2.70* (7H, N-Me, –COOMe, H-16), 2.40 (1H, t, J = 13.0 Hz, H-14b), 1.92 (1H, m, H-20), 1.52 (2H, m, H-19), 1.16 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD/CDCl3) δ 171.0 (–COOMe), 162.9 (C-3), 157.2 (C-1′), 149.2 (C-4′), 137.3 (C-13), 137.3 (C-2′), 134.8 (C-7′), 132.9 (C-2), 130.5 (C-6′), 129.5 (C-8), 125.8 (C-11), 125.4 (C-3′), 122.6 (C-5′), 120.2 (C-10), 119.7 (C-9), 117.6 (C-7), 111.8 (C-12), 58.2 (C-5), 50.9 (–COOMe), 50.0–48.0* (C-16, C-21), 42.9 (C-20), 41.8 (N-Me), 30.5 (C-15), 24.9 (C-14), 23.6 (C-19), 21.0 (C-6), 11.1 (C-18) ppm. * signals overlapped. ESIMS m/z 502 (M + H)+.
3-[(3″-methoxybenzylidene)hydrazineylidene]dregamine (7). Obtained from reaction of compound 2 (23 mg, 0.062 mmol, 1 equiv.) with 3-methoxybenzaldehyde (51 mg, 0.37 mmol, 6 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 9 mg (0.019 mmol, yield 30%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD) δ 8.50 (1H, s, H-1′), 7.56 (1H, bd, J = 8.0 Hz, H-9), 7.42 (1H, m, H-6′), 7.37 (1H, bd, J = 8.2 Hz, H-12), 7.34–7.28* (2H, H-3′, H-5′), 7.21 (1H, ddd, J = 8.1, 7.0, 1.0 Hz, H-11), 7.05 (1H, ddd, J = 8.0, 7.1, 1.0 Hz, H-10), 7.00 (1H, m, H-7′), 3.87–3.76* (5H, H-5, H-14a, 4′-OCH3), 3.13 (1H, dd, J = 14.9, 8.2 Hz, H-6a), 2.90 (1H, dd, J = 14.7, 10.1 Hz, H-6b), 2.72–2.63* (4H, H-21b, –COOMe), 2.61 (1H, m, H-15), 2.47–2.29* (5H, H-16, H-21a, N-Me), 2.22 (1H, t, J = 12.8 Hz, H-14b), 1.75 (1H, m, H-20), 1.37 (2H, m, H-19), 1.04 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD) δ 172.5 (–COOMe), 162.5 (C-4′), 161.4 (C-3), 159.7 (C-1′), 138.2 (C-13), 137.8 (C-2′), 133.8 (C-2), 130.7 (C-6′), 130.5 (C-8), 125.6 (C-11), 122.7 (C-7′), 120.3 (C-10), 120.2 (C-9), 118.8 (C-5′), 118.3 (C-7), 112.7 (C-3′), 112.3 (C-12), 58.6 (C-5), 55.7 (O–CH3), 50.8 (–COOMe), 50.0–48.0* (C-16, C-21), 44.2 (C-20), 42.3 (N-Me), 31.7 (C-15), 25.5 (C-14), 24.3 (C-19), 21.3 (C-6), 11.5 (C-18) ppm. * signals overlapped. ESIMS m/z 487 (M + H)+.
3-[(4″-bromobenzylidene)hydrazineylidene]dregamine (8). Obtained from reaction of compound 2 (23 mg, 0.062 mmol, 1 equiv.) with 4-bromobenzaldehyde (58 mg, 0.31 mmol, 5 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 8 mg (0.015 mmol, yield 24%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD/CDCl3) δ 8.50 (1H, s, H-1′), 7.70 (2H, d, J = 8.6 Hz, H-3′, H-7′), 7.61–7.53* (3H, H-9, H-4′, H-6′), 7.37 (1H, bd, J = 8.2 Hz, H-12), 7.23 (1H, ddd, J = 8.1, 6.9, 1.0 Hz, H-11), 7.07 (1H, ddd, J = 8.0, 6.9, 0.9 Hz, H-10), 3.93–3.80* (2H, H-5, H-14a), 3.20 (1H, dd, J = 14.8, 8.2 Hz, H-6a), 3.01 (1H, dd, J = 14.6, 10.0 Hz, H-6b), 2.75–2.61* (5H, H-15, H-21b, –COOMe), 2.53 (1H, m, H-16), 2.48–2.37* (5H, H-14b, H-21a, N-Me), 1.80 (1H, m, H-20), 1.39 (2H, m, H-19), 1.05 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD/CDCl3) δ 172.3 (–COOMe), 162.5 (C-3), 158.2 (C-1′), 138.0 (C-13), 135.3 (C-2′), 133.5 (C-2), 132.9 (C-4′, C-6′), 130.5 (C-3′, C-7′), 130.3 (C-8), 125.7 (C-11), 125.7 (C-5′), 120.2 (C-10), 120.0 (C-9), 118.7 (C-7), 112.1 (C-12), 58.4 (C-5), 50.8 (–COOMe), 50.0–48.0* (C-16, C-21), 44.1 (C-20), 42.3 (N-Me), 31.6 (C-15), 25.4 (C-14), 24.2 (C-19), 21.2 (C-6), 11.4 (C-18) ppm. * Signals overlapped. ESIMS m/z 537 (M + H)+.
3-[(4″-chlorobenzylidene)hydrazineylidene]dregamine (9). Obtained from reaction of compound 2 (23 mg, 0.062 mmol, 1 equiv.) with 4-chlorobenzaldehyde (53 mg, 0.37 mmol, 6 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 49:1) and preparative TLC (CH2Cl2/MeOH, 49:1) to afford 11 mg (0.022 mmol, yield 36%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD) δ 8.50 (1H, s, H-1′), 7.75 (2H, d, J = 8.4 Hz, H-3′, H-7′), 7.59 (1H, bd, J = 7.9 Hz, H-9), 7.44–7.38* (3H, H-12, H-4′, H-6′), 7.25 (1H, t, J = 7.4 Hz, H-11), 7.09 (1H, t, J = 7.5 Hz, H-10), 3.89–3.73* (2H, H-5, H-14a), 3.10 (1H, m, H-6a), 2.83 (1H, m, H-6b), 2.71–2.58* (5H, H-15, H-21b, –COOMe), 2.44–2.32* (5H, H-16, H-21a, N-Me), 2.17 (1H, m, H-14b), 1.75 (1H, m, H-20), 1.36 (2H, m, H-19), 1.04 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD) δ 172.3 (–COOMe), 162.8 (C-3), 158.3 (C-1′), 138.2 (C-13), 137.5 (C-5′), 135.2 (C-2′), 133.8 (C-2), 130.5 (C-4′, C-6′), 130.5 (C-8), 130.0 (C-3′, C-7′), 125.7 (C-11), 120.3 (C-10), 120.2 (C-9), 118.7 (C-7), 112.3 (C-12), 58.5 (C-5), 50.9 (–COOMe), 50.0–48.0* (C-16, C-21), 44.1 (C-20), 42.1 (N-Me), 31.7 (C-15), 25.4 (C-14), 24.3 (C-19), 21.2 (C-6), 11.4 (C-18) ppm. * Signals overlapped. ESIMS m/z 491 (M + H)+.
3-[(4″-(trifluoromethyl)benzylidene)hydrazineylidene]dregamine (10). Obtained from reaction of compound 2 (15 mg, 0.041 mmol, 1 equiv.) with 4-trifluoromethylbenzaldehyde (35 mg, 0.37 mmol, 5 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 6 mg (0.011 mmol, yield 28%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD) δ 8.60 (1H, s, H-1′), 7.97 (2H, d, J = 8.1 Hz, H-3′, H-7′), 7.71 (2H, d, J = 8.3 Hz, H-4′, H-6′), 7.62 (1H, bd, J = 8.1 Hz, H-9), 7.39 (1H, bd, J = 8.2 Hz, H-12), 7.25 (1H, t, J = 7.3 Hz, H-11), 7.08 (1H, t, J = 7.5 Hz, H-10), 3.94 (1H, m, H-5), 3.85 (1H, dd, J = 13.3, 6.4 Hz, H-14a), 3.25 (1H, dd, J = 15.1, 8.5 Hz, H-6a), 3.02 (1H, dd, J = 14.7, 10.2 Hz, H-6b), 2.75–2.63* (5H, H-15, H-21b, –COOMe), 2.61–2.46* (5H, H-16, H-21a, N-Me), 2.35 (1H, t, J = 13.2 Hz, H-14b), 1.81 (1H, m, H-20), 1.41 (2H, m, H-19), 1.06 (3H, t, J = 7.3 Hz, H-18); 13C NMR (75 MHz, MeOD) δ 172.1 (–COOMe), 163.0 (C-3), 157.9 (C-1′), 140.1 (C-2′), 138.3 (C-13), 133.7 (C-2), 130.5 (C-8), 129.5 (C-3′, C-7′), 126.7 (C-4′), 126.6 (C-6′), 125.9 (C-11), 120.4 (C-10), 120.3 (C-9), 118.7 (C-7), 112.4 (C-12), 58.7 (C-5), 50.9 (–COOMe), 50.0–48.0* (C-16, C-21), 44.0 (C-20), 42.1 (N-Me), 31.7 (C-15), 25.6 (C-14), 24.2 (C-19), 21.4 (C-6), 11.3 (C-18) ppm. * Signals overlapped. ESIMS m/z 525 (M + H)+.
3-[(4″-(trifluoromethoxy)benzylidene)hydrazineylidene]dregamine (11). Obtained from reaction of compound 2 (15 mg, 0.041 mmol, 1 equiv.) with 4-trifluoromethoxybenzaldehyde (29 µL, 0.20 mmol, 5 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 15 mg (0.028 mmol, yield 68%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD) δ 8.55 (1H, s, H-1′), 7.87 (2H, d, J = 8.6, 2.3 Hz, H-4′, H-6′), 7.58 (1H, bd, J = 8.1 Hz, H-9), 7.40 (1H, bd, J = 8.2 Hz, H-12), 7.31 (2H, d, J = 8.3 Hz, H-3′, H-7′), 7.24 (1H, ddd, J = 8.1, 7.1, 1.0 Hz, H-11), 7.08 (1H, ddd, J = 7.9, 7.0, 0.9 Hz, H-10), 3.88–3.73* (2H, H-5, H-14a), 3.10 (1H, dd, J = 14.9, 8.2 Hz, H-6a), 2.80 (1H, dd, J = 14.6, 10.2 Hz, H-6b), 2.71–2.57* (5H, H-15, H-21b, –COOMe), 2.39–2.29* (5H, H-16, H-21a, N-Me), 2.15 (1H, t, J = 12.7 Hz, H-14b), 1.73 (1H, m, H-20), 1.37 (2H, m, H-19), 1.04 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD) δ 172.3 (–COOMe), 163.0 (C-3), 158.0 (C-1′), 151.8 (C-5′), 138.2 (C-13), 135.4 (C-2′), 133.7 (C-2), 130.8 (C-3′, C-7′), 130.5 (C-8), 125.7 (C-11), 122.1 (C-4′, C-6′), 120.3 (C-10), 120.3 (C-9), 118.8 (C-7), 112.4 (C-12), 58.5 (C-5), 50.9 (–COOMe), 50.0–48.0* (C-16, C-21), 44.1 (C-20), 42.1 (N-Me), 31.7 (C-15), 25.4 (C-14), 24.3 (C-19), 21.2 (C-6), 11.4 (C-18) ppm. * Signals overlapped. ESIMS m/z 541 (M + H)+.
3-[(4″-(dimethylamino)benzylidene)hydrazineylidene]dregamine (12). Obtained from reaction of compound 2 (20 mg, 0.054 mmol, 1 equiv.) with 4-dimethylaminobenzaldehyde (25 µL, 0.27 mmol, 5 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 7 mg (0.013 mmol, yield 26%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD) δ 8.45 (1H, s, H-1′), 7.67 (2H, d, J = 8.9 Hz, H-3′, H-7′), 7.57 (1H, bd, J = 8.0 Hz, H-9), 7.37 (1H, bd, J = 8.1 Hz, H-12), 7.21 (1H, ddd, J = 8.1, 7.0, 1.0 Hz, H-11), 7.05 (1H, ddd, J = 8.0, 6.9, 0.9 Hz, H-10), 6.75 (2H, d, J = 8.9 Hz, H-4′, H-6′), 3.99–3.88* (2H, H-5, H-14a), 3.20 (1H, dd, J = 14.9, 8.3 Hz, H-6a), 3.03 (6H, s, 5′-N-Me), 2.97 (1H, dd, J = 13.7, 9.4 Hz, H-6b), 2.74–2.62* (5H, H-15, H-21a, –COOMe), 2.60–2.45* (5H, H-16, H-21b, N-Me), 2.21 (1H, t, J = 12.8 Hz, H-14b), 1.80 (1H, m, H-20), 1.39 (2H, m, H-19), 1.08 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD) δ 172.0 (–COOMe), 160.5 (C-5′), 160.5 (C-3), 153.8 (C-1′), 138.0 (C-13), 134.3 (C-2), 130.9 (C-3′, C-7′), 130.6 (C-8), 125.3 (C-11), 123.9 (C-2′), 120.2 (C-10), 120.0 (C-9), 116.9 (C-7), 112.8 (C-4′, C-6′), 112.2 (C-12), 58.9 (C-5), 50.9 (–COOMe), 50.0–48.0* (C-16, C-21), 43.9 (C-20), 42.0 (N-Me), 40.3 (5′-N-Me), 31.5 (C-15), 25.1 (C-14), 24.2 (C-19), 21.3 (C-6), 11.4 (C-18) ppm. * Signals overlapped. ESIMS m/z 500 (M + H)+.
3-[(4″-hydroxybenzylidene)hydrazineylidene]dregamine (13). Obtained from reaction of compound 2 (29 mg, 0.078 mmol, 1 equiv.) with 4-hydroxybenzaldehyde (36 µL, 0.47 mmol, 6 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 6 mg (0.013 mmol, yield 16%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD) δ 8.44 (1H, s, H-1′), 7.67 (2H, d, J = 8.6 Hz, H-3′, H-7′), 7.59 (1H, bd, J = 8.0 Hz, H-9), 7.37 (1H, bd, J = 8.1 Hz, H-12), 7.21 (1H, ddd, J = 8.1, 7.0, 0.9 Hz, H-11), 7.06 (1H, ddd, J = 8.0, 7.0, 0.9 Hz, H-10), 6.84 (2H, d, J = 8.6 Hz, H-4′, H-6′), 3.97–3.84* (2H, H-5, H-14a), 3.19 (1H, dd, J = 14.4, 6.1 Hz, H-6a), 2.98 (1H, dd, J = 14.6, 10.3 Hz, H-6b), 2.76–2.63* (5H, H-15, H-21b, –COOMe), 2.56–2.42* (5H, H-16, N-Me, H-21a), 2.24 (1H, m, H-14b), 1.79 (1H, m, H-20), 1.38 (2H, m, H-19), 1.06 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD) δ 172.1 (–COOMe), 161.5 (C-5′), 161.2 (C-3), 159.7 (C-1′), 138.1 (C-13), 134.1 (C-2), 131.2 (C-3′, C-7′), 130.5 (C-8), 127.9 (C-2′), 125.5 (C-11), 120.3 (C-10), 120.1 (C-9), 117.5 (C-7), 116.6 (C-4′, C-6′), 112.2 (C-12), 58.8 (C-5), 50.9 (–COOMe), 50.0–48.0* (C-16, C-21), 44.0 (C-20), 42.1 (N-Me), 31.6 (C-15), 25.3 (C-14), 24.2 (C-19), 21.3 (C-6), 11.3 (C-18) ppm. * Signals overlapped. ESIMS m/z 473 (M + H)+.
3-[(4″-methoxybenzylidene)hydrazineylidene]dregamine (14). Obtained from reaction of compound 2 (25 mg, 0.068 mmol, 1 equiv.) with 4-methoxybenzaldehyde (50 µL, 0.41 mmol, 6 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 5 mg (0.009 mmol, yield 15%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD) δ 8.49 (1H, s, H-1′), 7.75 (2H, d, J = 8.8 Hz, H-3′, H-7′), 7.58 (1H, bd, J = 8.0 Hz, H-9), 7.37 (1H, bd, J = 8.2 Hz, H-12), 7.21 (1H, ddd, J = 8.1, 7.0, 0.9 Hz, H-11), 7.06 (1H, ddd, J = 8.0, 7.0, 1.0 Hz, H-10), 6.96 (2H, d, J = 8.8 Hz, H-4′, H-6′), 3.94–3.83* (5H, H-5, H-14a, 5′-OCH3), 3.20 (1H, dd, J = 15.3, 9.0 Hz, H-6a), 3.00 (1H, dd, J = 15.2, 10.4 Hz, H-6b), 2.74–2.63* (5H, H-15, H-21b, –COOMe), 2.54 (1H, m, H-16), 2.49–2.38* (4H, H-21a, N-Me), 2.28 (1H, t, J = 12.7 Hz, H-14b), 1.78 (1H, m, H-20), 1.39 (2H, m, H-19), 1.06 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD) δ 172.2 (–COOMe), 163.4 (C-5′), 161.6 (C-3), 159.3 (C-1′), 138.1 (C-13), 134.0 (C-2), 130.9 (C-3′, C-7′), 130.5 (C-8), 129.1 (C-2′), 125.5 (C-11), 120.2 (C-10), 120.1 (C-9), 117.9 (C-7), 115.2 (C-4′, C-6′), 112.2 (C-12), 58.8 (C-5), 55.9 (–OCH3), 50.9 (–COOMe), 50.0–48.0* (C-16, C-21), 44.1 (C-20), 42.2 (N-Me), 31.7 (C-15), 25.4 (C-14), 24.3 (C-19), 21.3 (C-6), 11.4 (C-18) ppm. * Signals overlapped. ESIMS m/z 487 (M + H)+.
3-[(4″-(allyloxy)benzylidene)hydrazineylidene]dregamine (15). Obtained from reaction of compound 2 (20 mg, 0.054 mmol, 1 equiv.) with 4-allyloxybenzaldehyde (39 µL, 0.27 mmol, 5 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 13 mg (0.025 mmol, yield 47%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD) δ 8.47 (1H, s, H-1′), 7.73 (2H, d, J = 8.7 Hz, H-3′, H-7′), 7.55 (1H, bd, J = 8.0 Hz, H-9), 7.37 (1H, bd, J = 8.1 Hz, H-12), 7.20 (1H, t, J = 7.5 Hz, H-11), 7.04 (1H, t, J = 7.5 Hz, H-10), 6.95 (2H, d, J = 8.7 Hz, H-4′, H-6′), 6.09 (1H, ddt, J = 17.1, 10.4, 5.2 Hz, H-2″), 5.44 (1H, dd, J = 17.3, 1.6 Hz, H-3″a), 5.29 (1H, dd, J = 10.6, 1.3 Hz, H-3″b), 4.59 (2H, dt, J = 5.1, 1.3 Hz, H-1″), 3.87–3.77* (2H, H-5, H-14a), 3.08 (1H, dd, J = 14.9, 8.2 Hz, H-6a), 2.81 (1H, dd, J = 14.6, 10.3 Hz, H-6b), 2.68–2.58* (5H, H-15, H-21b, –COOMe), 2.40–2.31* (5H, H-16, H-21a, N-Me), 2.12 (1H, t, J = 12.8 Hz, H-14b), 1.71 (1H, m, H-20), 1.35 (2H, m, H-19), 1.03 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD) δ 172.3 (–COOMe), 162.2 (C-5′), 161.8 (C-3), 159.4 (C-1′), 138.1 (C-13), 134.5 (C-2″), 134.0 (C-2), 130.9 (C-3′, C-7′), 130.5 (C-8), 129.2 (C-2′), 125.5 (C-11), 120.2 (C-10), 120.1 (C-9), 118.1 (C-3″), 117.8 (C-7), 115.9 (C-4′, C-6′), 112.3 (C-12), 69.8 (C-1″), 58.6 (C-5), 50.9 (–COOMe), 50.0–48.0* (C-16, C-21), 44.1 (C-20), 42.2 (N-Me), 31.6 (C-15), 25.3 (C-14), 24.3 (C-19), 21.2 (C-6), 11.4 (C-18) ppm. * Signals overlapped. ESIMS m/z 513 (M + H)+.
3-[(4″-(benzyloxy)benzylidene)hydrazineylidene]dregamine (16). Obtained from reaction of compound 2 (20 mg, 0.054 mmol, 1 equiv.) with 4-(benzyloxy)benzaldehyde (51 µL, 0.27 mmol, 5 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 16 mg (0.028 mmol, yield 52%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD) δ 8.47 (1H, s, H-1′), 7.72 (2H, d, J = 8.8, 2.3 Hz, H-3′, H-7′), 7.55 (1H, bd, J = 8.0 Hz, H-9), 7.45–7.30* (6H, H-12, H-3″, H-4″, H-5″, H-6″, H-7″), 7.21 (1H, ddd, J = 8.1, 7.0, 0.9 Hz, H-11), 7.06 (1H, ddd, J = 8.0, 6.9, 0.9 Hz, H-10), 6.98 (2H, d, J = 8.8, 2.4 Hz, H-4′, H-6′), 5.06 (2H, s, H-1″), 3.87–3.75* (2H, H-5, H-14a), 3.06 (1H, dd, J = 14.9, 8.2 Hz, H-6a), 2.77 (1H, dd, J = 14.6, 10.2 Hz, H-6b), 2.69–2.56* (5H, H-15, H-21b, –COOMe), 2.41–2.28* (5H, H-16, H-21a, N-Me), 2.07 (1H, t, J = 12.7 Hz, H-14b), 1.72 (1H, m, H-20), 1.33 (2H, m, H-19), 1.02 (3H, t, J = 7.2 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD) δ 172.3 (–COOMe), 162.4 (C-5′), 161.8 (C-3), 159.4 (C-1′), 138.2 (C-2″), 138.1 (C-13), 134.0 (C-2), 131.0 (C-3′, C-7′), 130.5 (C-8), 129.5 (C-4″, C-6″), 129.3 (C-5″), 129.0 (C-2′), 128.6 (C-3″, C-7″), 125.5 (C-11), 120.3 (C-10), 120.1 (C-9), 118.1 (C-7), 116.1 (C-4′, C-6′), 112.3 (C-12), 71.0 (C-1″), 58.6 (C-5), 50.9 (–COOMe), 50.0–48.0* (C-16, C-21), 44.1 (C-20), 42.1 (N-Me), 31.6 (C-15), 25.3 (C-14), 24.3 (C-19), 21.2 (C-6), 11.4 (C-18) ppm. * Signals overlapped. ESIMS m/z 563 (M + H)+.
3-[(3″,4″,5″-trimethoxybenzylidene)hydrazineylidene]dregamine (17). Obtained from reaction of compound 2 (36 mg, 0.098 mmol, 1 equiv.) with 3,4,5-trimethoxybenzaldehyde (60 µL, 0.49 mmol, 5 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 8 mg (0.015 mmol, yield 15%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD) δ 8.48 (1H, s, H-1′), 7.58 (1H, bd, J = 7.9 Hz, H-9), 7.38 (1H, bd, J = 8.2 Hz, H-12), 7.22 (1H, t, J = 7.6 Hz, H-11), 7.17–7.13* (2H, H-3′, H-7′), 7.06 (1H, t, J = 7.6 Hz, H-10), 3.91–3.81* (11H, 4′-OCH3, 5′-OCH3, 6′-OCH3, H-5, H-14a), 3.18 (1H, dd, J = 14.9, 8.1 Hz, H-6a), 2.96 (1H, dd, J = 14.8, 10.4 Hz, H-6b), 2.79–2.59* (5H, –COOMe, H-15, H-21b), 2.51, (1H, m, H-16), 2.44 (3H, s, N-Me), 2.40–2.22* (2H, H-14b, H-21a), 1.78 (1H, m, H-20), 1.36 (2H, m, H-19), 1.08 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD) δ 172.5 (–COOMe), 162.5 (C-3), 159.8 (C-1′), 154.8 (C-4′, C-6′), 141.5 (C-5′), 138.2 (C-13), 133.9 (C-2), 132.2 (C-2′), 130.6 (C-8), 125.6 (C-11), 120.3 (C-10), 120.2 (C-9), 118.7 (C-7), 112.2 (C-12), 106.5 (C-3′, C-7′), 61.2 (5′-OCH3), 58.6 (C-5), 56.6 (4′-OCH3, 6′-OCH3), 50.8 (–COOMe), 50.0–48.0* (C-16, C-21), 44.1 (C-20), 42.3 (N-Me), 31.5 (C-15), 25.4 (C-14), 24.3 (C-19), 21.3 (C-6), 11.6 (C-18) ppm. * Signals overlapped. ESIMS m/z 547 (M + H)+.
3-[(2″-naphthalenylmethylene)hydrazineylidene]dregamine (18). Obtained from reaction of compound 2 (15 mg, 0.041 mmol, 1 equiv.) with 2-naphthaldehyde (27 µL, 0.20 mmol, 5 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 13 mg (0.026 mmol, yield 63%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD/CDCl3) δ 8.46 (1H, s, H-1′), 7.87 (1H, dd, J = 8.4, 1.4 Hz, H-3′), 7.79 (1H, s, H-7′), 7.67–7.54* (3H, H-9, H-1″, H-4″), 7.41 (1H, d, J = 8.2 Hz, H-4′), 7.37–7.25* (2H, H-2″, H-3″), 7.21 (1H, bd, J = 8.1 Hz, H-12), 7.07 (1H, t, J = 7.7 Hz, H-11), 6.91 (1H, t, J = 7.7 Hz, H-10), 3.80–3.70* (2H, H-5, H-14a), 3.01 (1H, dd, J = 14.8, 8.2 Hz, H-6a), 2.72 (1H, dd, J = 14.7, 10.3 Hz, H-6b), 2.62–2.45* (5H, H-15, H-21a, –COOMe), 2.34–2.23* (5H, H-16, H-21a, N-Me), 2.02 (1H, m, H-14b), 1.63 (1H, m, H-20), 1.23 (2H, m, H-19), 0.91 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD/CDCl3) δ 171.7 (-COOMe), 161.9 (C-3), 159.7 (C-1′), 137.67 (C-13), 135.6 (C-5′), 134.2 (C-6′), 133.5 (C-2), 133.5 (C-2′), 131.3 (C-7′), 130.0 (C-8), 129.3 (C-4″), 129.1 (C-2″), 128.5 (C-3″), 128.0 (C-4′), 127.3 (C-1″), 125.6 (C-11), 124.2 (C-3′), 120.2 (C-10), 119.8 (C-9), 117.7 (C-7), 112.0 (C-12), 58.3 (C-5), 50.8 (–COOMe), 50.0–48.0* (C-16, C-21), 43.5 (C-20), 42.0 (N-Me), 31.1 (C-15), 25.1 (C-14), 23.9 (C-19), 21.1 (C-6), 11.3 (C-18) ppm. *Signals overlapped. ESIMS m/z 507 (M + H)+.
3-[((2″-hydroxy-1″naphthalenyl)methylene)hydrazineylidene]dregamine (19). Obtained from reaction of compound 2 (15 mg, 0.041 mmol, 1 equiv.) with 2-hydroxy-1-naphthaldehyde (30 µL, 0.20 mmol, 5 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) and preparative TLC (CH2Cl2/MeOH, 97:3) to afford 12 mg (0.023 mmol, yield 56%) of an amorphous light orange powder. 1H NMR (300 MHz, MeOD/CDCl3) δ 9.55 (1H, s, H-1′), 8.00 (1H, d, J = 8.4 Hz, H-4″), 7.66–7.55* (3H, H-9, H-5′, H-1″), 7.48–7.37* (2H, H-12, H-3″), 7.34–7.25* (2H, H-11, H-2″), 7.13 (1H, m, H-10), 6.99 (1H, d, J = 9.0 Hz, H-4′), 3.88 (1H, m, H-5), 3.41 (1H, m, H-14a), 3.14 (1H, dd, J = 14.9, 8.2 Hz, H-6a), 2.76–2.63* (6H, H-6b, H-15, H-21b, -COOMe), 2.47–2.33* (4H, H-16, N-Me), 2.23–2.09* (2H, H-14b, H-21a), 1.75 (1H, m, H-20), 1.53 (1H, m, H-19a), 1.36 (1H, m, H-19b), 1.07 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD/CDCl3) δ 171.6 (-COOMe), 161.3 (C-3″), 160.5 (C-3), 160.3 (C-1′), 137.7 (C-13), 134.7 (C-1′’), 133.3 (C-6′), 133.2 (C-2), 129.9 (C-8), 129.6 (C-5′), 128.9 (C-7′), 128.2 (C-3′’), 125.8 (C-11), 124.2 (C-2″), 120.7 (C-4″), 120.4 (C-10), 119.9 (C-9), 119.1 (C-4′), 118.2 (C-7), 112.1 (C-12), 109.5 (C-2′), 58.1 (C-5), 50.9 (–COOMe), 50.0–48.0* (C-16, C-21), 43.9 (C-20), 42.0 (N-Me), 31.8 (C-15), 25.6 (C-14), 24.3 (C-19), 20.9 (C-6), 12.0 (C-18) ppm. * Signals overlapped. ESIMS m/z 523 (M + H)+.
3-[((1H-indol-2-yl)methylene)hydrazineylidene]dregamine (20). Obtained from reaction of compound 2 (30 mg, 0.081 mmol, 1 equiv.) with indole-2-carboxaldehyde (37 µL, 0.41 mmol, 5 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 96.5:3.5) and preparative TLC (CH2Cl2/MeOH, 96.5:3.5) to afford 10 mg (0.020 mmol, yield 25%) of an amorphous yellow powder. 1H NMR (300 MHz, MeOD) δ 8.50 (1H, s, H-1′), 7.63–7.54* (2H, H-9, H-3′), 7.44–7.34* (2H, H-12, H-6′), 7.25–7.17* (2H, H-11, H-4′), 7.09–7.02* (2H, H-10, H-5′), 6.84 (1H, s, H-2″), 3.93–3.79* (2H, H-5, H-14a), 3.21 (1H, dd, J = 14.8, 8.1 Hz, H-6a), 3.00 (1H, dd, J = 14.5, 10.3 Hz, H-6b), 2.70–2.64* (5H, H-15, H-21b, –COOMe), 2.50–2.43* (5H, H-16, H-21a, N-Me), 2.28 (1H, t, J = 12.7 Hz, H-14b), 1.75 (1H, m, H-20), 1.50 (1H, m, H-19a), 1.31 (1H, m, H-19b), 0.99 (3H, t, J = 7.4 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD) δ 172.4 (–COOMe), 161.1 (C-3), 150.7 (C-1′), 139.5 (C-7′), 138.1 (C-13), 135.2 (C-2), 134.0 (C-2′), 130.6 (C-8), 129.7 (C-1″), 125.57 (C-11), 125.1 (C-4′), 122.3 (C-5′), 120.9 (C-3′), 120.2 (C-10), 120.1 (C-9), 118.2 (C-7), 112.6 (C-6′), 112.2 (C-12), 109.2 (C-2″), 58.7 (C-5), 50.9 (–COOMe), 50.0–48.0* (C-16, C-21), 44.4 (C-20), 42.3 (N-Me), 32.4 (C-15), 25.6 (C-14), 24.5 (C-19), 21.2 (C-6), 11.7 (C-18) ppm. * Signals overlapped. ESIMS m/z 496 (M + H)+.
3-[((6-bromo-1H-indol-3-yl)methylene)hydrazineylidene]dregamine (21). Obtained from reaction of compound 2 (37.5 mg, 0.102 mmol, 1 equiv.) with 6-bromoindole-3-carboxaldehyde (71 µL, 0.51 mmol, 5 equiv.). After stirring the mixture for 2 h at room temperature, the reaction product was sequentially purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 24:1) and preparative TLC (CH2Cl2/MeOH, 24:1) to afford 20 mg (0.035 mmol, yield 34%) of an amorphous orange powder. 1H NMR (300 MHz, MeOD) δ 8.74 (1H, s, H-1′), 8.16 (1H, d, J = 8.4 Hz, H-3′), 7.59–7.51* (3H, H-9, H-6′, H-2″), 7.40 (1H, bd, J = 8.1 Hz, H-12), 7.23 (1H, ddd, J = 8.1, 7.0, 0.9 Hz, H-11), 7.13–7.02* (2H, H-10, H-4′), 4.10–3.98* (2H, H-5, H-14a), 3.11 (1H, dd, J = 14.8, 8.3 Hz, H-6a), 2.75–2.64* (6H, H-6b, H-15, H-16, –COOMe), 2.60 (1H, m, H-21b), 2.53 (3H, s, N-Me), 2.45 (1H, m, H-21a), 2.00 (1H, m, H-14b), 1.81 (1H, m, H-20), 1.46 (1H, m, H-19a), 1.39 (1H, m, H-19b), 1.04 (1H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, MeOD) δ 171.2 (–COOMe), 159.5 (C-3), 156.8 (C-1′), 139.7 (C-7′), 137.9 (C-13), 134.7 (C-2), 133.2 (C-2″), 130.4 (C-8), 125.4 (C-11), 125.2 (C-2′, C-3′), 124.7 (C-4′), 120.4 (C-10), 120.0 (C-9), 117.3 (C-7), 115.6 (C-6′), 115.3 (C-5′), 114.9 (C-1″), 112.2 (C-12), 59.0 (C-5), 51.1 (–COOMe), 50.0–48.0* (C-16, C-21), 43.6 (C-20), 41.5 (N-Me), 31.6 (C-15), 25.1 (C-14), 24.3 (C-19), 21.3 (C-6), 12.0 (C-18) ppm. * Signals overlapped. ESIMS m/z 576 (M + H)+.
3.2.2. General Preparation of Dregamine Semicarbazones 22–32
Compound 2 (50 mg, 1 equiv.) and the suitable isocyanate (1.2–1.5 equiv.) were dissolved in tetrahydrofuran (THF, 2 mL) at room temperature. The mixture was stirred at 70 °C for 1 h under nitrogen atmosphere. After evaporating the solvent under vacuum, at 40 °C, the obtained residue was purified by flash chromatography, followed by preparative TLC.
3-[(2″-(pentylcarbamoyl)hydrazineylidene]dregamine (22). Obtained from reaction of compound 2 (50 mg, 0.136 mmol, 1 equiv.) with pentyl isocyanate (19 µL, 0.18 mmol, 1.3 equiv.). The mixture was stirred for 1 h at 70 °C and purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 24:1) followed by preparative TLC (CH2Cl2/MeOH, 24:1) to afford 5 mg (0.010 mmol, yield 8%) of an amorphous light yellow powder. 1H NMR (300 MHz, CDCl3) δ 8.94 (1H, s, NH), 8.04 (1H, s, NH-2′), 7.57 (1H, bd, J = 7.9 Hz, H-9), 7.27 (1H, b, J = 7.8 Hz, H-12), 7.21 (1H, ddd, J = 8.0, 6.8, 1.1 Hz, H-11), 7.09 (1H, ddd, J = 8.0, 6.9, 1.2 Hz, H-10), 6.27 (1H, t, J = 5.9 Hz, NH-2″), 3.96 (1H, m, H-5), 3.40 (2H, m, H-1‴), 3.25 (1H, dd, J = 15.0, 8.2 Hz, H-6a), 3.11 (1H, dd, J = 14.8, 10.1 Hz, H-6b), 2.83–2.69* (3H, H-14a, H-15, H-16), 2.62 (3H, s, –COOMe), 2.57 (3H, s, N-Me), 2.61–2.53* (3H, H-14b, H-21), 1.83 (1H, m, H-20), 1.64 (2H, m, H-2‴), 1.50–1.27* (6H, H-19, H-3‴, H-4‴), 1.01 (3H, t, J = 7.4 Hz, H-18), 0.92 (3H, t, J = 7.0 Hz, H-5‴) ppm; 13C NMR (75 MHz, CDCl3) δ 171.0 (–COOMe), 156.6 (C-1″), 143.6 (C-3), 135.9 (C-13), 132.5 (C-8), 129.7 (C-2), 124.4 (C-11), 119.7 (C-10), 119.0 (C-9), 115.3 (C-7), 110.6 (C-12), 58.1 (C-5), 50.3 (–COOMe), 49.0 (C-16), 48.4 (C-21), 43.6 (C-20), 42.6 (N-Me), 40.2 (C-1‴), 31.3 (C-15), 30.2 (C-3‴), 29.3 (C-2‴), 24.0 (C-19), 23.3 (C-14), 22.6 (C-4‴), 20.0 (C-6), 14.2 (C-5‴), 12.2 (C-18) ppm. * Signals overlapped. ESIMS m/z 482 (M + H)+.
3-[(2″-(tert-butylcarbamoyl)hydrazineylidene]dregamine (23). Obtained from reaction of compound 2 (50 mg, 0.136 mmol, 1 equiv.) with tert-butyl isocyanate (23 µL, 0.20 mmol, 1.5 equiv.). The mixture was stirred for 1 h at 70 °C and purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 19:1) followed by preparative TLC (CH2Cl2/MeOH, 19:1) to afford 8 mg (0.018 mmol, yield 13%) of an amorphous light yellow powder. 1H NMR (300 MHz, CDCl3) δ 8.69 (1H, s, NH), 7.58 (1H, s, NH-2′), 7.56 (1H, bd, J = 7.8 Hz, H-9), 7.27 (1H, d, J = 7.9 Hz, H-12), 7.20 (1H, ddd, J = 8.1, 6.8, 1.1 Hz, H-11), 7.08 (1H, ddd, J = 8.0, 6.8, 1.2 Hz, H-10), 5.97 (1H, s, NH-2″), 3.97 (1H, m, H-5), 3.26 (1H, dd, J = 15.0, 8.3 Hz, H-6a), 3.16 (1H, dd, J = 14.9, 10.0 Hz, H-6b), 2.81–2.66* (5H, H-14a, H-15, H-16, H-21), 2.63 (1H, m, H-14b), 2.61 (3H, s, –COOMe), 2.58 (3H, s, N-Me), 1.88 (1H, m, H-20), 1.47* (9H, s, H-2‴), 1.38 (2H, m, H-19), 1.03 (3H, t, J = 7.4 Hz, H-18) ppm; 13C NMR (75 MHz, CDCl3) δ 171.1 (–COOMe), 155.1 (C-1″), 143.1 (C-3), 135.8 (C-13), 132.3 (C-8), 129.7 (C-2), 124.4 (C-11), 119.7 (C-10), 119.0 (C-9), 115.3 (C-7), 110.6 (C-12), 58.1 (C-5), 50.7 (C-1‴), 50.3 (–COOMe), 48.9 (C-16), 48.6 (C-21), 43.5 (C-20), 42.6 (N-Me), 31.1 (C-15), 29.4 (C-2‴), 23.9 (C-19), 23.3 (C-14), 20.1 (C-6), 12.1 (C-18) ppm. * Signals overlapped. ESIMS m/z 468 (M + H)+.
3-[(2″-(cyclohexylcarbamoyl)hydrazineylidene]dregamine (24). Obtained from reaction of compound 2 (50 mg, 0.136 mmol, 1 equiv.) with cyclohexyl isocyanate (23 µL, 0.18 mmol, 1.3 equiv.). The mixture was stirred for 1 h at 70 °C and purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 19:1) followed by preparative TLC (CH2Cl2/MeOH, 19:1) to afford 17 mg (0.033 mmol, yield 25%) of an amorphous light yellow powder. 1H NMR (300 MHz, CDCl3) δ 8.99 (1H, s, NH), 8.23 (1H, s, NH-2′), 7.56 (1H, bd, J = 8.0 Hz, H-9), 7.26 (1H, bd, J = 7.9 Hz, H-12), 7.19 (1H, ddd, J = 8.0, 6.9, 1.0 Hz, H-11), 7.07 (1H, ddd, J = 8.0, 6.9, 1.2 Hz, H-10), 6.15 (1H, d, J = 8.3 Hz, NH-2″), 3.95 (1H, m, H-5), 3.80 (1H, m, H-1‴), 3.24 (1H, dd, J = 15.0, 8.2 Hz, H-6a), 3.13 (1H, dd, J = 14.7, 10.1 Hz, H-6b), 2.89–2.70* (3H, H-14a, H-15, H-16), 2.61 (3H, s, –COOMe), 2.60–2.53* (2H, H-14b, H-21), 2.57 (3H, s, N-Me), 2.06 (2H, m, H-2‴), 1.88–1.73* (3H, H-20, H-3‴), 1.65 (1H, m, H-4‴a), 1.51 (1H, m, H-19a), 1.39–1.14* (6H, H-19b, H-6‴, H-5‴, H-4‴b), 1.01 (3H, t, J = 7.4 Hz, H-18) ppm; 13C NMR (75 MHz, CDCl3) δ 171.0 (–COOMe), 155.9 (C-1″), 143.8 (C-3), 135.9 (C-13), 132.5 (C-8), 129.6 (C-2), 124.3 (C-11), 119.5 (C-10), 118.9 (C-9), 115.1 (C-7), 110.6 (C-12), 58.0 (C-5), 50.2 (–COOMe), 49.1 (C-1‴), 49.0 (C-16), 48.4 (C-21), 43.7 (C-20), 42.6 (N-Me), 33.9 (C-2‴), 33.9 (C-6‴), 31.4 (C-15), 25.7 (C-4‴), 25.4 (C-3‴, C-5‴), 23.9 (C-19), 23.5 (C-14), 19.9 (C-6), 12.2 (C-18) ppm. * Signals overlapped. ESIMS m/z 494 (M + H)+.
3-[(2″-(phenylcarbamoyl)hydrazineylidene]dregamine (25). Obtained from reaction of compound 2 (50 mg, 0.136 mmol, 1 equiv.) with phenyl isocyanate (19 µL, 0.18 mmol, 1.3 equiv.). The mixture was stirred for 1 h at 70 °C and purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 24:1) followed by preparative TLC (CH2Cl2/MeOH, 19:1) to afford 20 mg (0.041 mmol, yield 30%) of an amorphous yellow powder. 1H NMR (300 MHz, CDCl3) δ 9.43 (1H, s, NH), 8.82 (1H, s, NH-2′), 8.30 (1H, s, NH-2″), 7.61–7.50* (3H, H-9, H-2‴), 7.31–7.17* (4H, H-11, H-12, H-3‴), 7.14–6.99* (2H, H-10, H-4‴), 3.93 (1H, m, H-5), 3.22 (1H, dd, J = 14.8, 8.1 Hz, H-6a), 3.06 (1H, dd, J = 14.5, 10.2 Hz, H-6b), 2.90–2.66* (3H, H-14a, H-15, H-16), 2.61 (3H, s, –COOMe), 2.54 (3H, s, N-Me), 2.52–2.44* (3H, H-14b, H-21), 1.74 (1H, m, H-20), 1.30 (2H, m, H-19), 0.88 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, CDCl3) δ 171.4 (–COOMe)), 154.6 (C-1″), 146.2 (C-3), 138.2 (C-1‴), 136.4 (C-13), 132.5 (C-8), 129.7 (C-2), 129.0 (C-3‴, C-5‴), 124.6 (C-11), 123.6 (C-4‴), 120.3 (C-2‴, C-6‴), 119.7 (C-10), 119.1 (C-9), 116.0 (C-7), 111.0 (C-12), 58.2 (C-5), 50.5 (–COOMe), 49.1 (C-16), 48.4 (C-21), 43.7 (C-20), 42.7 (N-Me), 31.4 (C-15), 24.1 (C-19), 23.9 (C-14), 20.0 (C-6), 12.1 (C-18) ppm. * Signals overlapped. ESIMS m/z 488 (M + H)+.
3-[(2″-(p-tolylcarbamoyl)hydrazineylidene]dregamine (26). Obtained from reaction of compound 2 (50 mg, 0.136 mmol, 1 equiv.) with 4-tolyl isocyanate (18 µL, 0.18 mmol, 1.3 equiv.). The mixture was stirred for 1 h at 70 °C and purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 24:1) followed by preparative TLC (CH2Cl2/MeOH, 19:1) to afford 14 mg (0.027 mmol, yield 21%) of an amorphous yellow powder. 1H NMR (300 MHz, CDCl3) δ 9.25 (1H, s, NH), 8.50 (1H, s, NH-2′), 8.11 (1H, s, NH-2″), 7.58 (1H, bd, J = 7.9 Hz, H-9), 7.44 (2H, d, J = 8.4 Hz, H-2‴, H-6‴), 7.28 (1H, bd, J = 8.0 Hz, H-12), 7.21 (1H, ddd, J = 8.0, 6.8, 0.9 Hz, H-11), 7.13–7.05* (3H, H-10, H-3‴, H-5‴), 3.95 (1H, m, H-5), 3.26 (1H, dd, J = 14.9, 8.1 Hz, H-6a), 3.12 (1H, dd, J = 14.9, 10.5 Hz, H-6b), 2.9–2.70* (3H, H-14a, H-15, H-16), 2.62 (3H, s, –COOMe), 2.57 (3H, s, N-Me), 2.56–2.45* (3H, H-14b, H-21), 2.30 (3H, s, 4‴-Me), 1.79 (1H, m, H-20), 1.34 (2H, m, H-19), 0.92 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, CDCl3) δ 171.2 (–COOMe), 154.4 (C-1″), 145.7 (C-3), 136.3 (C-13), 135.5 (C-1‴), 133.1 (C-4‴), 132.4 (C-8), 129.6 (C-2), 129.5 (C-3‴, C-5‴), 124.5 (C-11), 120.4 (C-2‴, C-6‴), 119.7 (C-10), 119.0 (C-9), 115.8 (C-7), 110.9 (C-12), 58.1 (C-5), 50.5 (–COOMe), 49.0 (C-16), 48.4 (C-21), 43.6 (C-20), 42.6 (N-Me), 31.3 (C-15), 23.9 (C-19), 23.9 (C-14), 20.9 (4‴-Me), 19.9 (C-6), 12.1 (C-18) ppm. * Signals overlapped. ESIMS m/z 502 (M + H)+.
3-[2″-((4‴-methoxyphenyl)carbamoyl)hydrazineylidene]dregamine (27). Obtained from reaction of compound 2 (50 mg, 0.136 mmol, 1 equiv.) with 4-methoxyphenyl isocyanate (21 µL, 0.16 mmol, 1.2 equiv.). The mixture was stirred for 1 h at 70 °C and purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 97:3) followed by preparative TLC (CH2Cl2/MeOH, 19:1) to afford 25 mg (0.048 mmol, yield 36%) of an amorphous light orange powder. 1H NMR (300 MHz, CDCl3) δ 9.35 (1H, s, N-H), 8.55 (1H, s, NH-2′), 8.15 (1H, s, NH-2″), 7.57 (1H, bd, J = 7.9 Hz, H-9), 7.43 (2H, d, J = 9.0 Hz, H-2‴, H-6‴), 7.25–7.16* (2H, H-11, H-12), 7.08 (1H, ddd, J = 8.0, 6.4, 1.7 Hz, H-10), 6.83 (2H, d, J = 9.0 Hz, H-3‴, H-5‴), 3.96 (1H, m, H-5), 3.75 (3H, s, 4‴-OMe), 3.23 (1H, dd, J = 14.8, 8.1 Hz, H-6a), 3.05 (1H, dd, J = 14.5, 10.2 Hz, H-6b), 2.87–2.68* (3H, H-14a, H-15, H-16), 2.61 (3H, s, –COOMe), 2.55 (3H, s, N-Me), 2.53–2.41* (3H, H-14b, H-21), 1.77 (1H, m, H-20), 1.30 (2H, m, H-19), 0.92 (3H, t, J = 7.3 Hz, H-18); 13C NMR (75 MHz, CDCl3) δ 171.1 (–COOMe), 156.2 (C-4‴), 154.8 (C-1″), 145.4 (C-3), 136.2 (C-13), 132.5 (C-8), 131.1 (C-1‴), 129.5 (C-2), 124.5 (C-11), 122.7 (C-2‴, C-6‴), 119.6 (C-10), 118.9 (C-9), 115.5 (C-7), 114.2 (C-3‴, C-5‴), 110.9 (C-12), 58.1 (C-5), 55.5 (C-5′-OMe), 50.4 (–COOMe), 48.9 (C-16), 48.3 (C-21), 43.5 (C-20), 42.5 (N-Me), 31.2 (C-15), 23.8 (C-19), 23.6 (C-14), 20.0 (C-6), 12.0 (C-18) ppm. * Signals overlapped. ESIMS m/z 518 (M + H)+.
3-[2″-((4‴-chlorophenyl)carbamoyl)hydrazineylidene]dregamine (28). Obtained from reaction of compound 2 (50 mg, 0.136 mmol, 1 equiv.) with 4-chlorophenyl isocyanate (27 mg, 0.18 mmol, 1.3 equiv.). The mixture was stirred for 1 h at 70 °C and purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 24:1) followed by preparative TLC (CH2Cl2/MeOH, 19:1) to afford 24 mg (0.045 mmol, yield 34%) of an amorphous yellow powder. 1H NMR (300 MHz, CDCl3) δ 9.48 (1H, s, NH), 8.66 (1H, s, NH-2′), 8.24 (1H, s, NH-2″), 7.58 (1H, bd, J = 8.0 Hz, H-9), 7.46 (2H, d, J = 8.9 Hz, H-2‴, H-6‴), 7.32 (1H, bd, J = 8.0 Hz, H-12), 7.26 (1H, m, H-11), 7.20 (2H, d, J = 8.8 Hz, H-3‴, H-5‴), 7.11 (1H, ddd, J = 8.0, 6.8, 1.0 Hz, H-10), 3.95 (1H, m, H-5), 3.25 (1H, dd, J = 14.8, 8.1 Hz, H-6a), 3.11 (1H, dd, J = 14.6, 10.3 Hz, H-6b), 2.88–2.67* (3H, H-14a, H-15, H-16), 2.59 (3H, s, –COOMe), 2.56 (3H, s, N-Me), 2.56–2.46* (3H, H-21, H-14b), 1.75 (1H, m, H-20), 1.25 (2H, m, H-19), 0.88 (3H, t, J = 7.4 Hz, H-18) ppm; 13C NMR (75 MHz, CDCl3) δ 171.4 (–COOMe), 154.6 (C-1″), 147.3 (C-3), 136.8 (C-1‴), 136.5 (C-13), 132.5 (C-8), 129.5 (C-2), 128.9 (C-3‴, C-5‴), 128.3 (C-4‴), 124.7 (C-11), 121.0 (C-2‴, C-6‴), 119.7 (C-10), 119.0 (C-9), 116.1 (C-7), 111.0 (C-12), 58.2 (C-5), 50.5 (–COOMe), 49.1 (C-16), 48.3 (C-21), 43.6 (C-20), 42.6 (N-Me), 31.4 (C-15), 24.2 (C-14), 23.8 (C-19), 19.9 (C-6), 12.0 (C-18) ppm. * Signals overlapped. ESIMS m/z 522 (M + H)+.
3-[2″-((4‴-bromophenyl)carbamoyl)hydrazineylidene]dregamine (29). Obtained from reaction of compound 2 (50 mg, 0.136 mmol, 1 equiv.) with 4-bromophenyl isocyanate (30 mg, 0.15 mmol, 1.1 equiv.). The mixture was stirred for 1 h at 70 °C and purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 24:1) followed by preparative TLC (CH2Cl2/MeOH, 19:1) to afford 36 mg (0.063 mmol, yield 47%) of an amorphous yellow powder. 1H NMR (300 MHz, CDCl3) δ 9.62 (1H, s, NH), 8.76 (1H, s, NH-2′), 8.34 (1H, s, NH-2″), 7.57 (1H, bd, J = 8.0 Hz, H-9), 7.40 (2H, d, J = 8.9 Hz, H-2‴, H-6‴), 7.35–7.28* (3H, H-12, H-3‴, H-5‴), 7.22 (1H, ddd, J = 8.0, 6.9, 0.9 Hz, H-11), 7.09 (1H, ddd, 8.0, 7.2, 1.0 Hz, H-10), 3.93 (1H, m, H-5), 3.22 (1H, dd, J = 14.8, 8.1 Hz, H-6a), 3.04 (1H, dd, J = 14.5, 10.3 Hz, H-6b), 2.85–2.64* (3H, H-14a, H-15, H-16), 2.57 (3H, s, –COOMe), 2.54 (3H, s, N-Me), 2.51–2.37* (3H, H-14b, H-21), 1.71 (1H, m, H-20), 1.23 (2H, m, H-19), 0.86 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, CDCl3) δ 171.4 (–COOMe), 154.7 (C-1″), 147.1 (C-3), 137.3 (C-1‴), 136.5 (C-13), 132.5 (C-8), 131.8* (C-3‴, C-5‴), 129.5 (C-2), 124.6 (C-11), 121.5 (C-2‴, C-6‴), 119.7 (C-10), 119.0 (C-9), 115.9* (C-7, C-4‴), 111.0 (C-12), 58.1 (C-5), 50.5 (–COOMe), 49.0 (C-16), 48.2 (C-21), 43.5 (C-20), 42.5 (N-Me), 31.3 (C-15), 24.0 (C-14), 23.7 (C-19), 19.9 (C-6), 12.0 (C-18) ppm. * Signals overlapped. ESIMS m/z 568 (M + H)+.
3-[2″-((4‴-nitrophenyl)carbamoyl)hydrazineylidene]dregamine (30). Obtained from reaction of compound 2 (50 mg, 0.136 mmol, 1 equiv.) with 4-nitrophenyl isocyanate (29 mg, 0.18 mmol, 1.3 equiv.). The mixture was stirred for 1 h at 70 °C and purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 24:1) followed by preparative TLC (CH2Cl2/MeOH, 19:1) to afford 17 mg (0.032 mmol, yield 24%) of an amorphous orange powder. 1H NMR (300 MHz, CDCl3) δ 9.49 (1H, s, NH), 8.69 (1H, s, NH-2′), 8.59 (1H, s, NH-2″), 8.07 (2H, d, J = 9.1 Hz, H-3‴, H-5‴), 7.68 (2H, d, J = 9.2 Hz, H-2‴, H-6‴), 7.60 (1H, bd, J = 8.0 Hz, H-9), 7.38 (1H, bd, J = 8.1 Hz, H-12), 7.27 (1H, ddd, J = 8.1, 6.9, 0.8 Hz, H-11), 7.13 (1H, ddd, J = 8.0, 6.8, 0.7 Hz, H-10), 3.95 (1H, m, H-5), 3.27 (1H, dd, J = 14.8, 8.1 Hz, H-6a), 3.13 (1H, dd, J = 14.7, 10.3 Hz, H-6b), 2.92–2.68* (3H, H-14a, H-15, H-16), 2.67–2.51* (9H, H-14b, H-21, –COOMe, N-Me), 1.77 (1H, m, H-20), 1.30 (2H, m, H-19), 0.91 (3H, t, J = 7.3 Hz, H-18) ppm; 13C NMR (75 MHz, CDCl3) δ 171.7 (–COOMe), 154.0 (C-1″), 149.1 (C-3), 144.3 (C-4‴), 142.8 (C-1‴), 136.7 (C-13), 132.1 (C-8), 129.5 (C-2), 125.1 (C-11), 125.0 (C-3‴, C-5‴), 119.9 (C-10), 119.2 (C-9), 118.5 (C-2‴, C-6‴), 116.9 (C-7), 111.1 (C-12), 58.2 (C-5), 50.6 (–COOMe), 49.1 (C-16), 48.3 (C-21), 43.6 (C-20), 42.6 (N-Me), 31.4 (C-15), 24.4 (C-14), 23.8 (C-19), 19.9 (C-6), 12.0 (C-18) ppm. * Signals overlapped. ESIMS m/z 533 (M + H)+.
3-[2″-((4‴-(trifluoromethyl)phenyl)carbamoyl)hydrazineylidene]dregamine (31). Obtained from reaction of compound 2 (50 mg, 0.136 mmol, 1 equiv.) with 4-(trifluoromethyl)phenyl isocyanate (25 µL, 0.18 mmol, 1.3 equiv.). The mixture was stirred for 1 h at 70 °C and purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 24:1) followed by preparative TLC (CH2Cl2/MeOH, 19:1) to afford 33 mg (0.059 mmol, yield 44%) of an amorphous yellow powder. 1H NMR (300 MHz, CDCl3) δ 9.61 (1H, s, NH), 8.67 (1H, s, NH-2′), 8.33 (1H, s, NH-2″), 7.67–7.57* (3H, H-9, H-2‴, H-6‴), 7.47–7.38* (3H, H-12, H-3‴, H-5‴), 7.28 (1H, ddd, J = 8.1, 7.0, 1.0 Hz, H-11), 7.12 (1H, ddd, J = 7.9, 7.0, 0.9 Hz, H-10), 3.96 (1H, m, H-5), 3.28 (1H, dd, J = 14.8, 8.1 Hz, H-6a), 3.17 (1H, dd, J = 14.6, 10.3 Hz, H-6b), 2.92–2.76* (2H, H-14a, H-15), 2.73–2.62* (4H, H-14b, H-16, H-21), 2.60 (3H, s, –COOMe), 2.58 (3H, s, N-Me), 1.74 (1H, m, H-20), 1.27 (2H, m, H-19), 0.89 (3H, t, J = 7.4 Hz, H-18) ppm; 13C NMR (75 MHz, CDCl3) δ 171.7 (–COOMe), 154.5 (C-1″), 148.4 (C-3), 141.4 (C-1‴), 136.9 (C-13), 132.5 (C-8), 129.6 (C-2), 126.2 (C-3‴), 126.1 (C-5‴), 125.1 (C-4‴), 124.8 (C-11), 124.7 (4‴-CF3), 119.8 (C-10), 119.1 (C-9), 118.6 (C-2‴, C-6‴), 116.4 (C-7), 111.2 (C-12), 58.3 (C-5), 50.7 (–COOMe), 49.2 (C-16), 48.3 (C-21), 43.8 (C-20), 42.6 (N-Me), 31.6 (C-15), 24.6 (C-14), 23.4 (C-19), 20.0 (C-6), 12.0 (C-18) ppm. * Signals overlapped. ESIMS m/z 556 (M + H)+.
3-[2″-((3-phenylpropyl)carbamoyl)hydrazineylidene]dregamine (32). Obtained from reaction of compound 2 (50 mg, 0.136 mmol, 1 equiv.) with 3-phenylpropyl isocyanate (25 µL, 0.16 mmol, 1.2 equiv.). The mixture was stirred for 1 h at 70 °C and purified by column chromatography (silica gel, CH2Cl2/MeOH, 1:0 to 19:1) followed by preparative TLC (CH2Cl2/MeOH, 93:7) to afford 6 mg (0.011 mmol, yield 8%) of an amorphous light yellow powder. 1H NMR (300 MHz, CDCl3) δ 8.85 (1H, s, NH), 7.95 (1H, s, NH-2′), 7.57 (1H, bd, J = 8.0 Hz, H-9), 7.32–7.18* (7H, H-11, H-12, H-2IV, H-3IV, H-4IV, H-5IV, H-6IV), 7.10 (1H, ddd, J = 8.0, 6.8, 1.2 Hz, H-10), 6.23 (1H, t, J = 6.0 Hz, NH-2″), 4.00 (1H, m, H-5), 3.42 (2H, m, H-1‴), 3.27 (1H, dd, J = 14.9, 8.3 Hz, H-6a), 3.13 (1H, dd, J = 14.8, 10.2 Hz, H-6b), 2.84–2.68* (5H, H-14a, H-15, H-16, H-3‴), 2.66–2.61* (2H, H-14b, H-21), 2.60 (3H, s, -COOMe), 2.60 (3H, s, N-Me), 2.00 (2H, m, H-2‴), 1.87 (1H, m, H-20), 1.41 (2H, m, H-19), 1.02 (3H, t, J = 7.4 Hz, H-18) ppm; 13C NMR (75 MHz, CDCl3) δ 171.0 (–COOMe), 156.5 (C-1″), 143.6 (C-3), 141.8 (C-1IV), 135.9 (C-13), 132.4 (C-8), 129.6 (C-2), 128.5* (C-2IV, C-3IV, C-5IV, C-6IV), 126.1 (C-4IV), 124.5 (C-11), 119.7 (C-10), 119.0 (C-9), 115.3 (C-7), 110.6 (C-12), 58.1 (C-5), 50.4 (–COOMe), 48.7 (C-16), 48.5 (C-21), 43.4 (C-20), 42.5 (N-Me), 39.8 (C-1‴), 33.4 (C-3‴), 31.9 (C-2‴), 31.2 (C-15), 23.7 (C-19), 23.3 (C-14), 19.5 (C-6), 11.8 (C-18) ppm. * Signals overlapped. ESIMS m/z 530 (M + H)+.


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}































