
Gemcitabine Direct Electrochemical Detection from Pharmaceutical Formulations Using a Boron-Doped Diamond Electrode

 , , , , ,
, , , , , 
Abstract
:
1. Introduction
2. Results and Discussions
2.1. Electrochemical Characterization of Gemcitabine
2.1.1. The Influence of the Electrode Material on the Detection of Gemcitabine
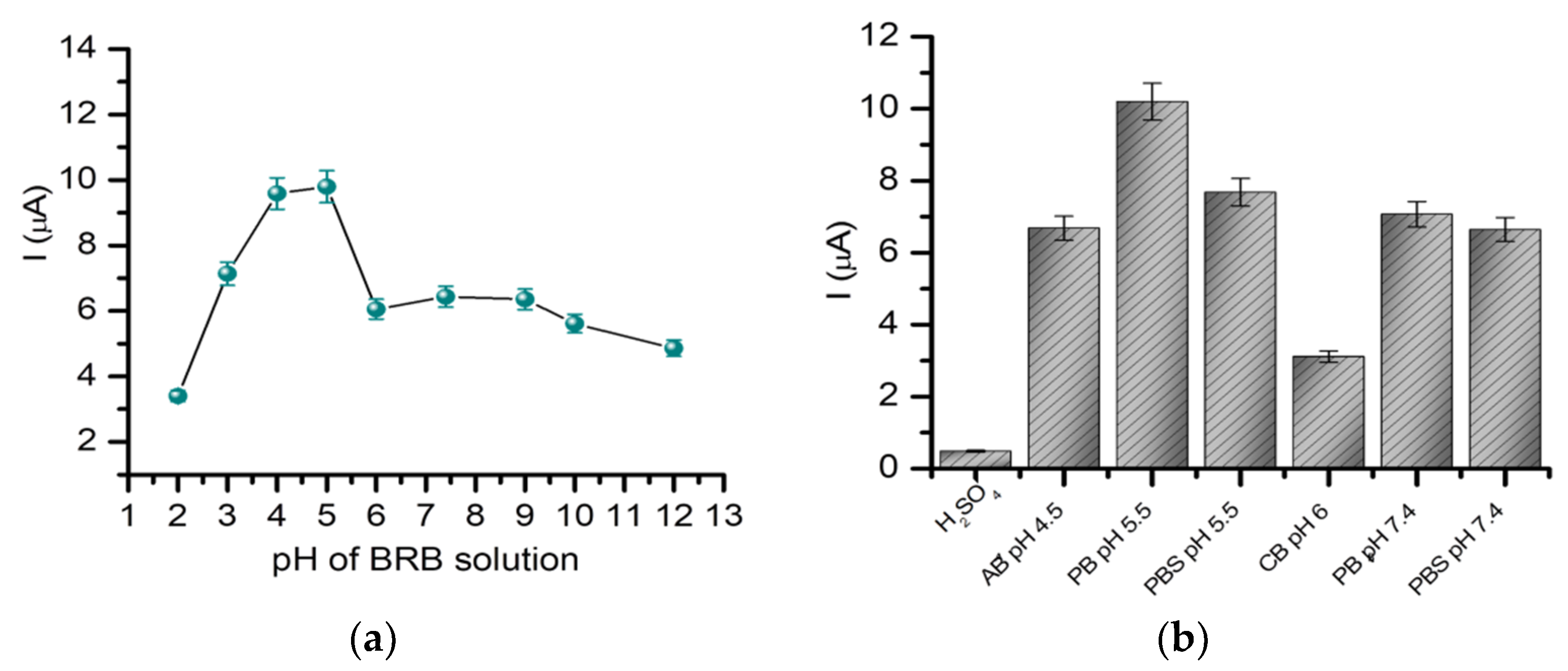
2.1.2. The Influence of the pH and Supporting Electrolyte on the Detection of Gemcitabine
2.1.3. The Influence of the Scan Rate on the Detection of Gemcitabine
2.2. Detection of Gemcitabine Using Voltammetry
2.3. Detection of Gemcitabine Using Amperometry
2.4. Real Sample Analysis
2.5. Robustness of the Applied Electrochemical Methods for Pharmaceutical Samples Analysis
2.6. Correlation and Comparison of Methods
3. Materials and Methods
3.1. Materials and Equipment
3.1.1. Chemicals and Reagents
3.1.2. Equipment
3.2. Methods
3.2.1. Buffer Preparation
3.2.2. Voltammetry Study
3.2.3. Amperometry Study
3.2.4. UV-Vis Spectrophotometry
3.2.5. HPLC-UV Analysis
3.2.6. Real Sample Analysis
3.2.7. Robustness of the Applied Electrochemical Methods for Pharmaceutical Sample Analysis
3.2.8. Correlation and Comparison of the Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alvarellos, M.L.; Lamba, J.; Sangkuhl, K.; Thorn, C.F.; Wang, L.; Klein, D.J.; Altman, R.B.; Klein, T.E.; Pharm, G.K.B. Summary: Gemcitabine Pathway. Pharmacogenet. Genom. 2014, 24, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Toschi, L.; Finocchiaro, G.; Bartolini, S.; Gioia, V.; Cappuzzo, F. Role of Gemcitabine in Cancer Therapy. Future Oncol. 2005, 1, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Noble, S.; Goa, K.L. Gemcitabine. A Review of Its Pharmacology and Clinical Potential in Non-Small Cell Lung Cancer and Pancreatic Cancer. Drugs 1997, 54, 447–472. [Google Scholar] [CrossRef] [PubMed]
- Ciccolini, J.; Serdjebi, C.; Peters, G.J.; Giovannetti, E. Pharmacokinetics and Pharmacogenetics of Gemcitabine as a Mainstay in Adult and Pediatric Oncology: An EORTC-PAMM Perspective. Cancer Chemother. Pharmacol. 2016, 78, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tamam, H.; Park, J.; Gadalla, H.H.; Masters, A.R.; Abdel-Aleem, J.A.; Abdelrahman, S.I.; Abdelrahman, A.A.; Lyle, L.T.; Yeo, Y. Development of Liposomal Gemcitabine with High Drug Loading Capacity. Mol. Pharm. 2019, 16, 2858–2871. [Google Scholar] [CrossRef] [PubMed]
- Tucci, S.T.; Kheirolomoom, A.; Ingham, E.S.; Mahakian, L.M.; Tam, S.M.; Foiret, J.; Hubbard, N.E.; Borowsky, A.D.; Baikoghli, M.; Cheng, R.H.; et al. Tumor-Specific Delivery of Gemcitabine with Activatable Liposomes. J. Control. Release 2019, 309, 277–288. [Google Scholar] [CrossRef]
- Öztürk, K.; Esendağlı, G.; Gürbüz, M.U.; Tülü, M.; Çalış, S. Effective Targeting of Gemcitabine to Pancreatic Cancer through PEG-Cored Flt-1 Antibody-Conjugated Dendrimers. Int. J. Pharm. 2017, 517, 157–167. [Google Scholar] [CrossRef]
- Soni, N.; Jain, K.; Gupta, U.; Jain, N.K. Controlled Delivery of Gemcitabine Hydrochloride Using Mannosylated Poly(Propyleneimine) Dendrimers. J. Nanopart. Res. 2015, 17, 1–17. [Google Scholar] [CrossRef]
- Affram, K.O.; Smith, T.; Ofori, E.; Krishnan, S.; Underwood, P.; Trevino, J.G.; Agyare, E. Cytotoxic Effects of Gemcitabine-Loaded Solid Lipid Nanoparticles in Pancreatic Cancer Cells. J. Drug Deliv. Sci. Technol. 2020, 55, 101374. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Shakya, A.K.; Naik, R.; Shalan, N. Stability-Indicating HPLC Determination of Gemcitabine in Pharmaceutical Formulations. Int. J. Anal. Chem. 2015, 2015, 862592. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Lu, L.; Xie, L.; Yang, J.; Zhang, X.; Ma, S. Concentration Changes in Gemcitabine and Its Metabolites after Hyperthermia in Pancreatic Cancer Cells Assessed Using RP-HPLC. Cell. Mol. Biol. Lett. 2019, 24, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Hiraki, R.; Nakamura, N.; Hayama, T.; Yasuda, T.; Miyata, Y.; Sakai, H.; Nakashima, M.N.; Nakashima, M.; Ohyama, K. Determination of Gemcitabine in Plasma of Bladder Cancer Patients by Hydrophilic Interaction Chromatography with Ultraviolet Detection. J. Chromatogr. Sci. 2020, 58, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Malatesta, L.; Cosco, D.; Paolino, D.; Cilurzo, F.; Costa, N.; Di Tullio, A.; Fresta, M.; Celia, C.; Di Marzio, L.; Locatelli, M. Simultaneous Quantification of Gemcitabine and Irinotecan Hydrochloride in Rat Plasma by Using High Performance Liquid Chromatography-Diode Array Detector. J. Pharm. Biomed. Anal. 2018, 159, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Gemcitabine European Pharmacopoeia 10th Edition Monograph. Available online: https://pheur.edqm.eu/subhome/10-5 (accessed on 1 June 2021).
- Menon, S.K.; Mistry, B.R.; Joshi, K.V.; Sutariya, P.G.; Patel, R.V. Analytical Detection and Method Development of Anticancer Drug Gemcitabine HCl Using Gold Nanoparticles. Spectrochim. Acta-Part A Mol. Biomol. Spectrosc. 2012, 94, 235–242. [Google Scholar] [CrossRef]
- Teradal, N.L.; Kalanur, S.S.; Prashanth, S.N.; Seetharamappa, J. Electrochemical Investigations of an Anticancer Drug in the Presence of Sodium Dodecyl Sulfate as an Enhancing Agent at Carbon Paste Electrode. J. Appl. Electrochem. 2012, 42, 917–923. [Google Scholar] [CrossRef]
- Naik, K.M.; Nandibewoor, S.T. Electro-Oxidation and Determination of Gemcitabine Hydrochloride, an Anticancer Drug at Gold Electrode. J. Ind. Eng. Chem. 2013, 19, 1933–1938. [Google Scholar] [CrossRef]
- Florea, A.; Guo, Z.; Cristea, C.; Bessueille, F.; Vocanson, F.; Goutaland, F.; Dzyadevych, S.; Săndulescu, R.; Jaffrezic-Renault, N. Anticancer Drug Detection Using a Highly Sensitive Molecularly Imprinted Electrochemical Sensor Based on an Electropolymerized Microporous Metal Organic Framework. Talanta 2015, 138, 71–76. [Google Scholar] [CrossRef]
- Tiğ, G.A.; Zeybek, B.; PekyardImcI, Ş. Electrochemical DNA Biosensor Based on Poly (2,6-Pyridinedicarboxylic Acid) Modified Glassy Carbon Electrode for the Determination of Anticancer Drug Gemcitabine. Talanta 2016, 154, 312–321. [Google Scholar] [CrossRef]
- Shoja, Y.; Kermanpur, A.; Karimzadeh, F.; Ghodsi, J.; Rafati, A.A.; Adhami, S. Electrochemical Molecularly Bioimprinted Siloxane Biosensor on the Basis of Core/Shell Silver Nanoparticles/EGFR Exon 21 L858R Point Mutant Gene/Siloxane Film for Ultra-Sensing of Gemcitabine as a Lung Cancer Chemotherapy Medication. Biosens. Bioelectron. 2019, 145, 111611. [Google Scholar] [CrossRef]
- Kalanur, S.S.; Katrahalli, U.; Seetharamappa, J. Electrochemical Studies and Spectroscopic Investigations on the Interaction of an Anticancer Drug with DNA and Their Analytical Applications. J. Electroanal. Chem. 2009, 636, 93–100. [Google Scholar] [CrossRef]
- Muzyka, K.; Sun, J.; Fereja, T.H.; Lan, Y.; Zhang, W.; Xu, G. Boron-Doped Diamond: Current Progress and Challenges in View of Electroanalytical Applications. Anal. Methods 2019, 11, 397–414. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | Detection Method | Linear Range (μg/mL) | LOD (μg/mL) | Ref |
|---|---|---|---|---|
| CPE, bare + surfactant | DPV | 0.140–89.900 | 0.0026 | 16 |
| AuE, bare | DPV | 0.026–3.947 | 0.0160 | 17 |
| AuE/MMOF | LSV | 1.1 × 10−9–0.011 | 9 × 10−10 | 18 |
| GCE/MIP/dsDNA | DPV | 1.000–30.000 | 0.2760 | 19 |
| CPE/MIP/MWCNT/AgNP/dsDNA | DPV | 0.394–24.470 | 0.0033 | 20 |
| BDDE | DPV | 2.500–50.000 | 0.8500 | This work |
| BDDE | AMP | 0.500–65.000 | 0.1500 | This work |
| (GMB) (μg/mL) | Method | Recovery (%) | RSD (%) |
|---|---|---|---|
| 2.5 | DPV | 93.2 | 0.40 |
| 10 | 100.6 | 9.98 | |
| 25 | 96.88 | 6.30 | |
| 2.5 | Amperometry | 83.33 | 1.26 |
| 10 | 82.35 | 7.41 | |
| 25 | 93.39 | 8.53 |
| (GMB) (μg/mL) | Method | Recovery (%) | RSD (%) |
|---|---|---|---|
| 2.5 | DPV | 92.22 | 12.35 |
| 10 | 105.12 | 9.48 | |
| 25 | 101.14 | 3.86 | |
| 2.5 | Amperometry | 89.47 | 4.54 |
| 10 | 95.40 | 2.68 | |
| 25 | 106.91 | 3.15 | |
| 2.5 | UV-Vis | 92.67 | 0.08 |
| 10 | 95.42 | 0.33 | |
| 25 | 90.65 | 0.83 | |
| 2.5 | HPLC-UV | 91.33 | 0.15 |
| 10 | 92.61 | 0.06 | |
| 25 | 91.90 | 0.23 |
| Source of Variation | SS | df | MS | F | p-Value | F Crit |
|---|---|---|---|---|---|---|
| Between Groups | 344.95 | 3 | 114.98 | 2.71 | 0.06114 | 2.90 |
| Within Groups | 1355.54 | 32 | 42.36 | |||
| p-value theoretical | ||||||
| Total | 1700.49 | 35 | 0.05 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rus, I.; Pusta, A.; Tertiș, M.; Barbălată, C.; Tomuță, I.; Săndulescu, R.; Cristea, C. Gemcitabine Direct Electrochemical Detection from Pharmaceutical Formulations Using a Boron-Doped Diamond Electrode. Pharmaceuticals 2021, 14, 912. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090912
Rus I, Pusta A, Tertiș M, Barbălată C, Tomuță I, Săndulescu R, Cristea C. Gemcitabine Direct Electrochemical Detection from Pharmaceutical Formulations Using a Boron-Doped Diamond Electrode. Pharmaceuticals. 2021; 14(9):912. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090912
Chicago/Turabian StyleRus, Iulia, Alexandra Pusta, Mihaela Tertiș, Cristina Barbălată, Ioan Tomuță, Robert Săndulescu, and Cecilia Cristea. 2021. "Gemcitabine Direct Electrochemical Detection from Pharmaceutical Formulations Using a Boron-Doped Diamond Electrode" Pharmaceuticals 14, no. 9: 912. https://0-doi-org.brum.beds.ac.uk/10.3390/ph14090912