
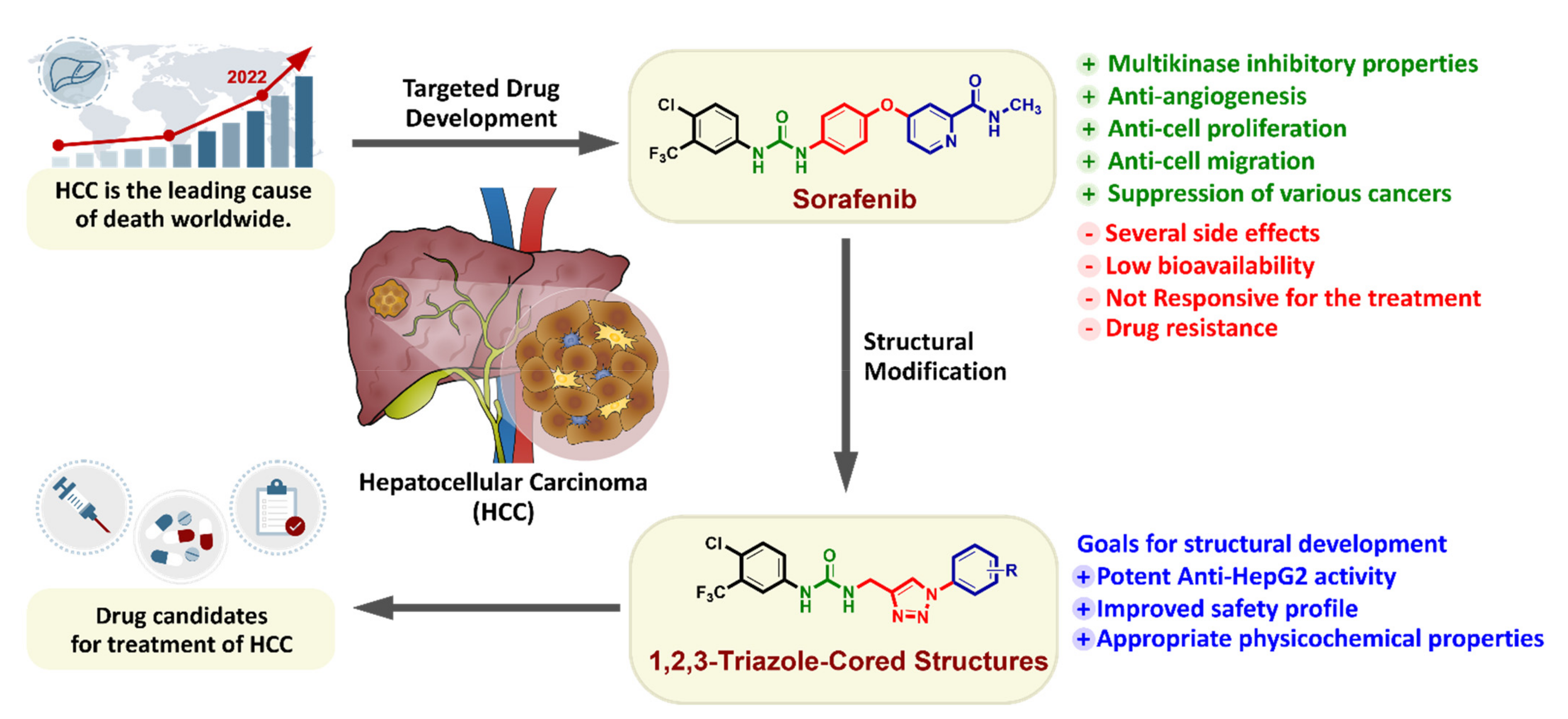
The Design and Synthesis of a New Series of 1,2,3-Triazole-Cored Structures Tethering Aryl Urea and Their Highly Selective Cytotoxicity toward HepG2

 , , and
, , and
Abstract
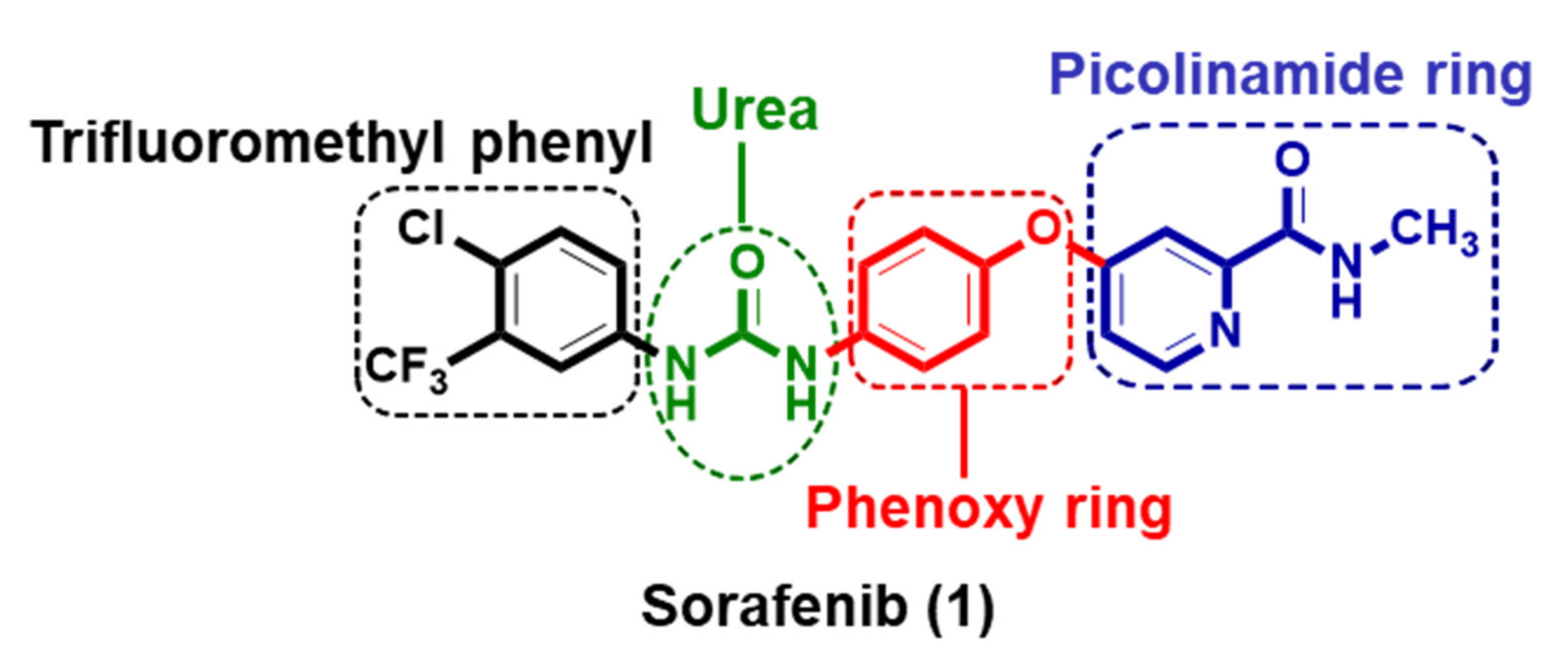
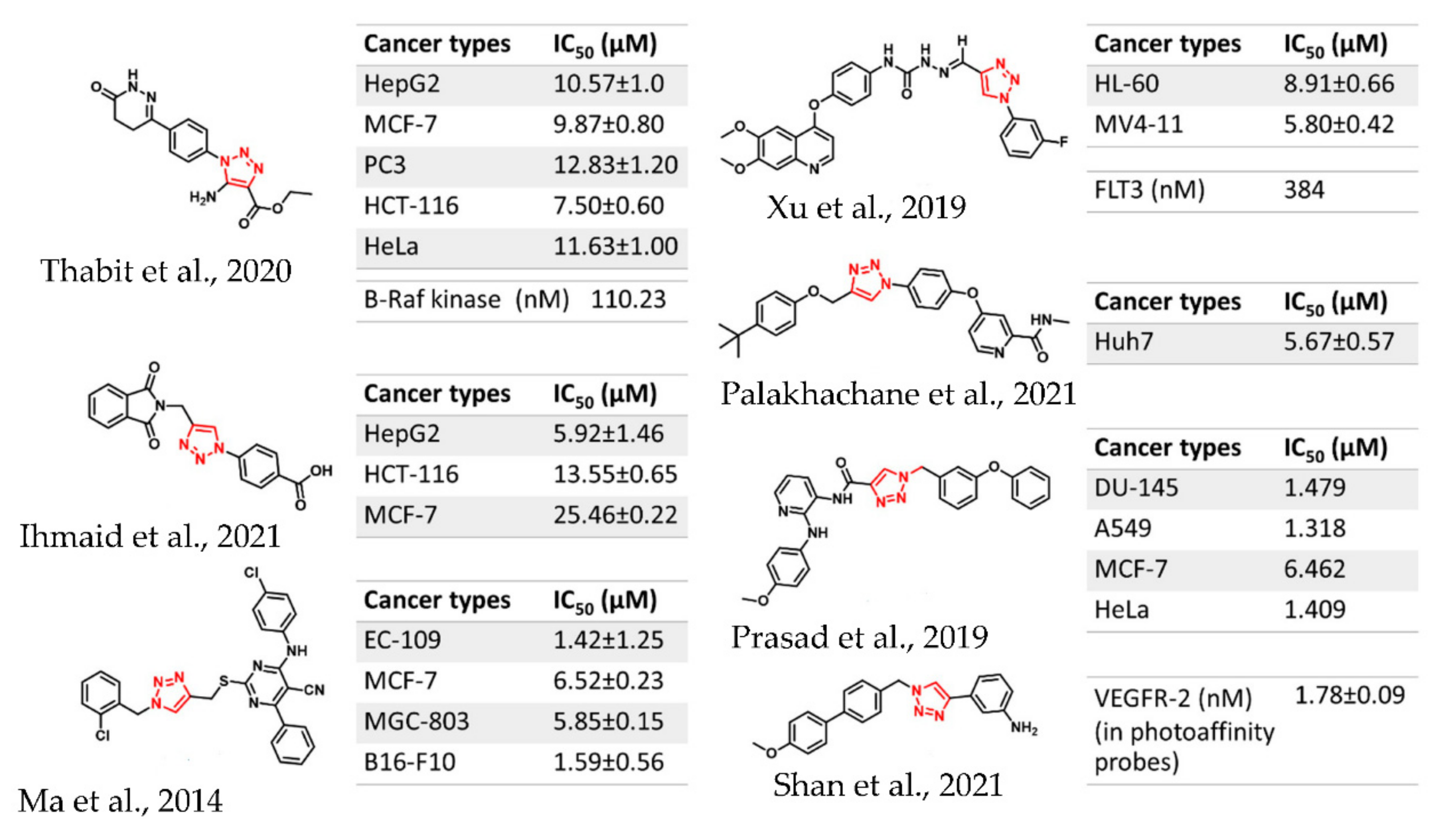
:1. Introduction
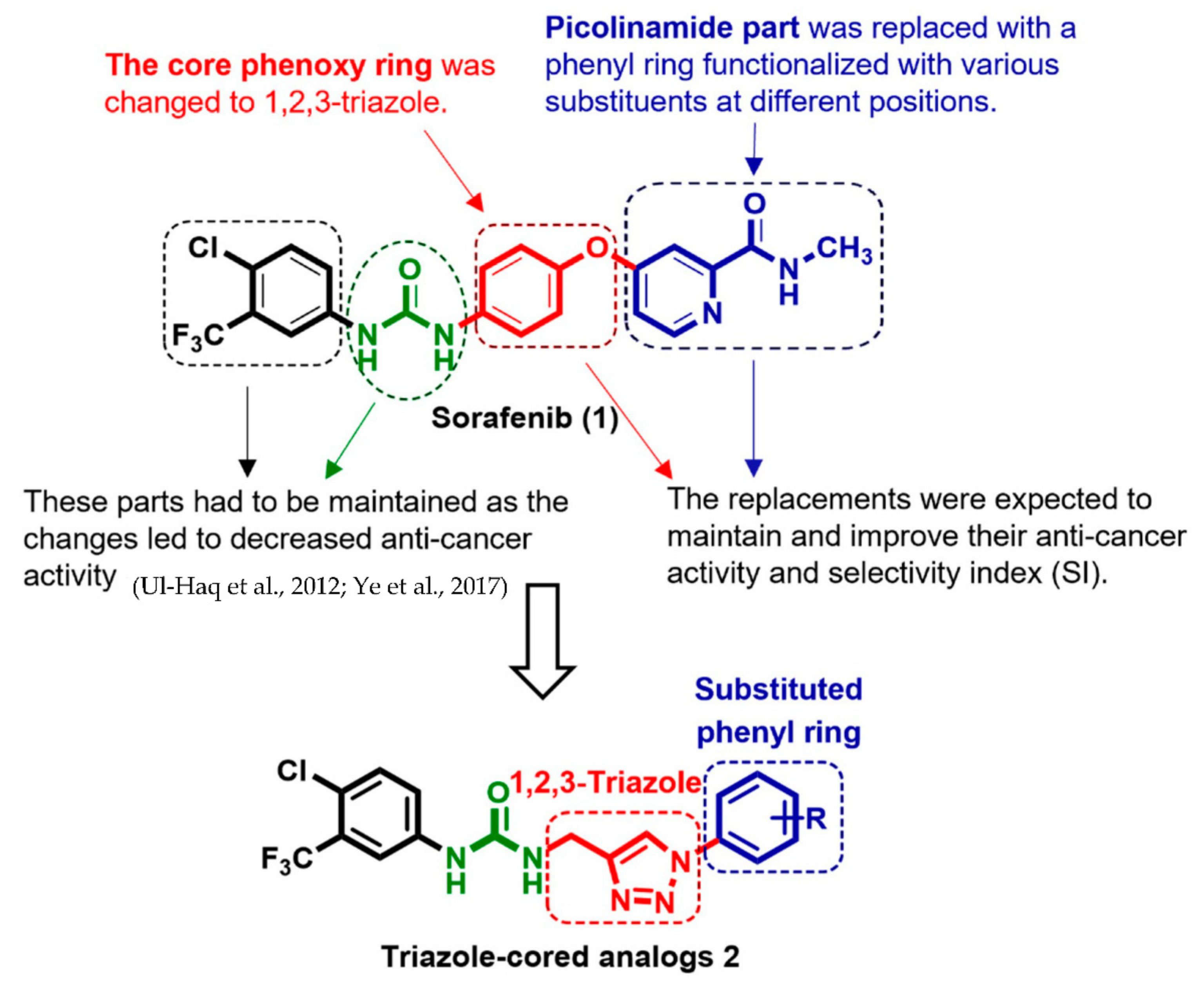
2. Results and Discussion
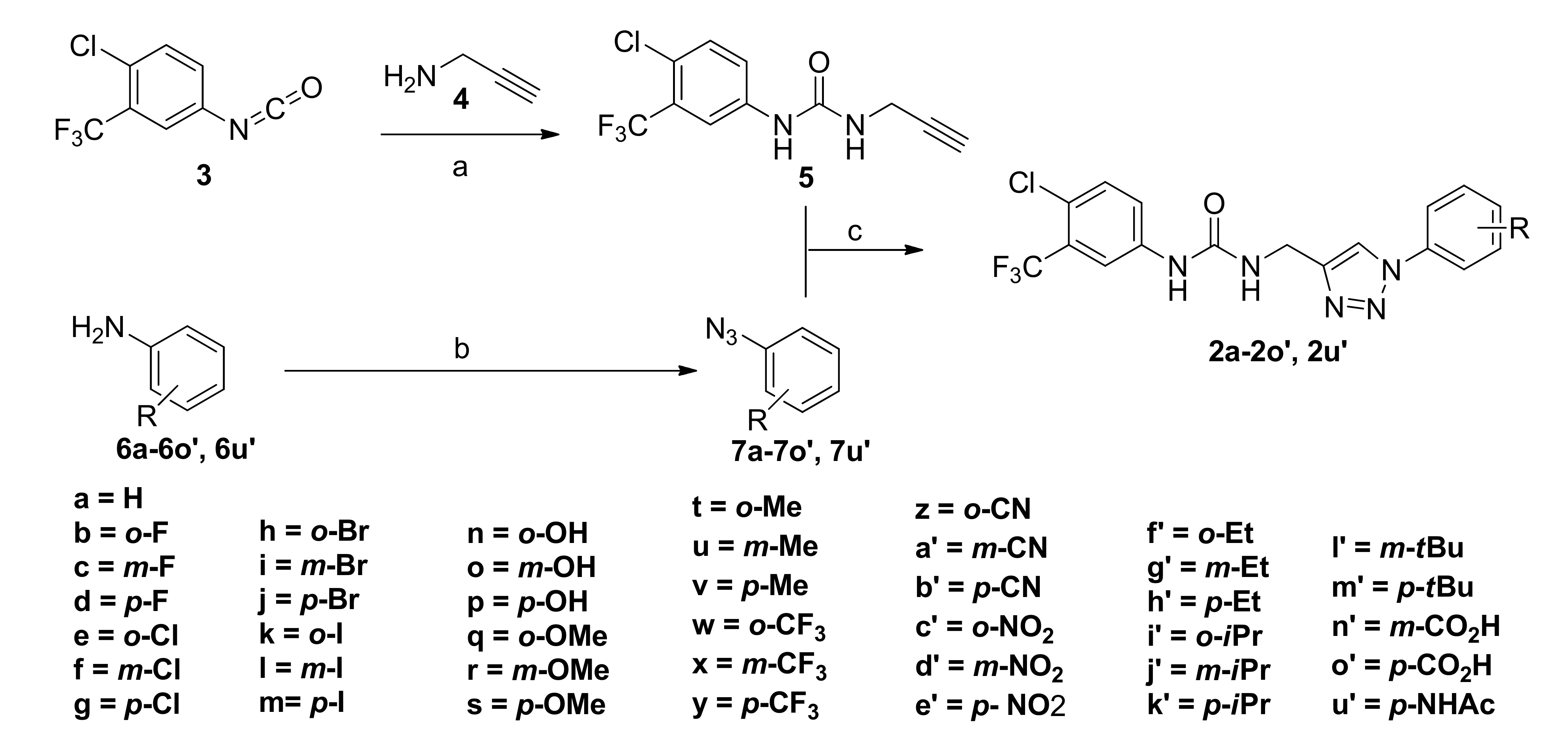
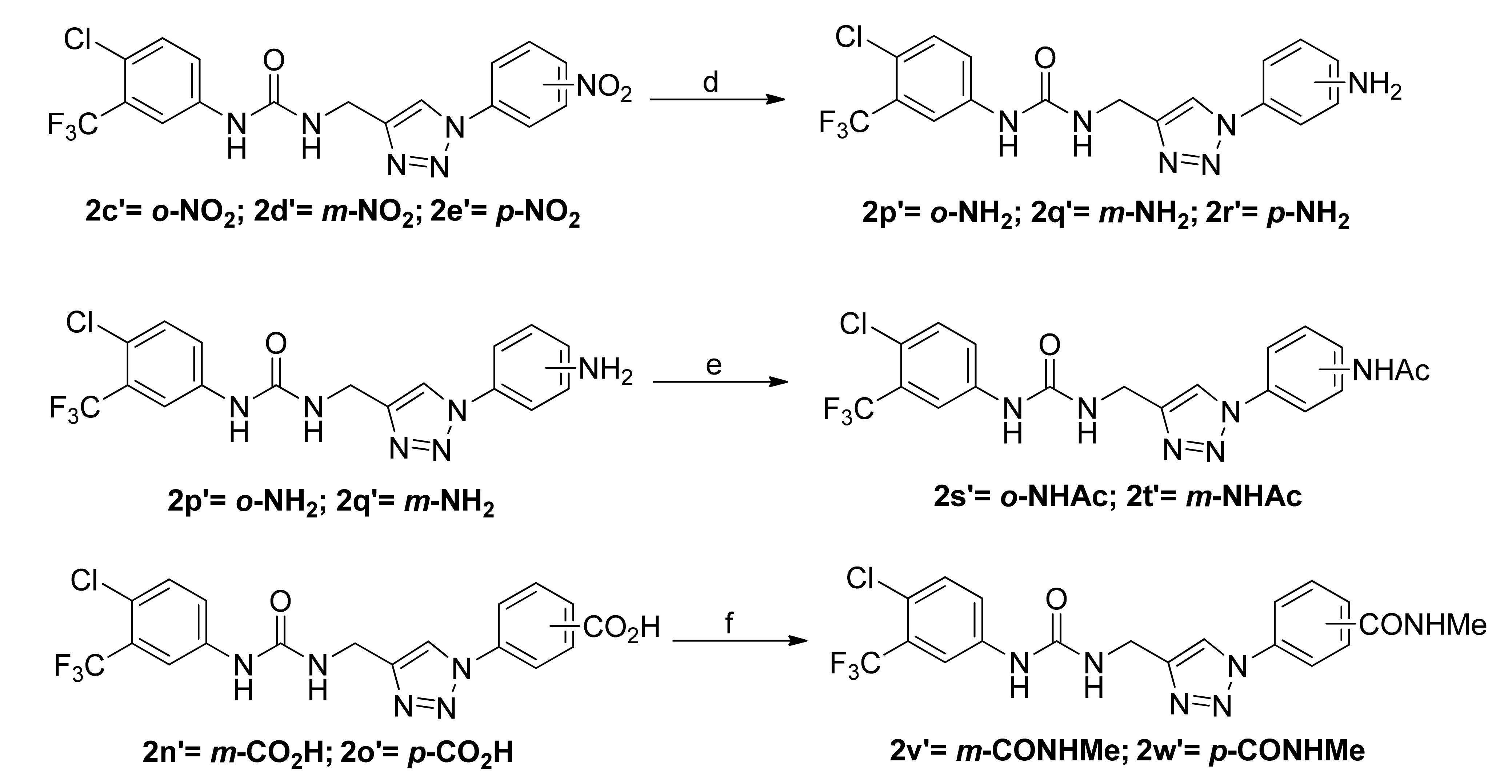
2.1. Chemistry
2.2. Cytotoxicity toward Cancer Cell Lines
2.3. Structure–Activity Relationships (SARs)
2.4. Cytotoxicity toward MRC-5 Cells and Selectivity Index (SI)
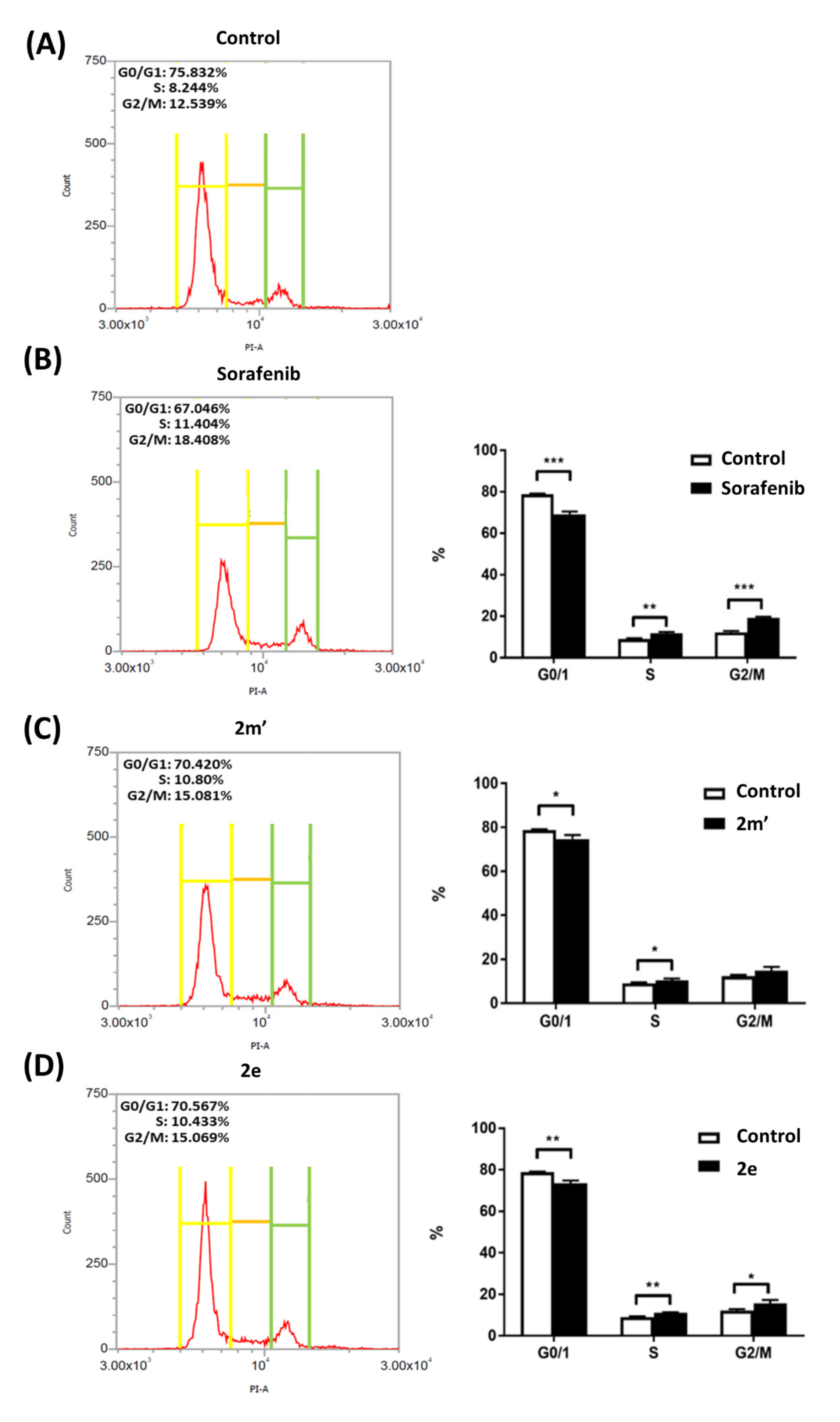
2.5. Cell-Cycle Analysis
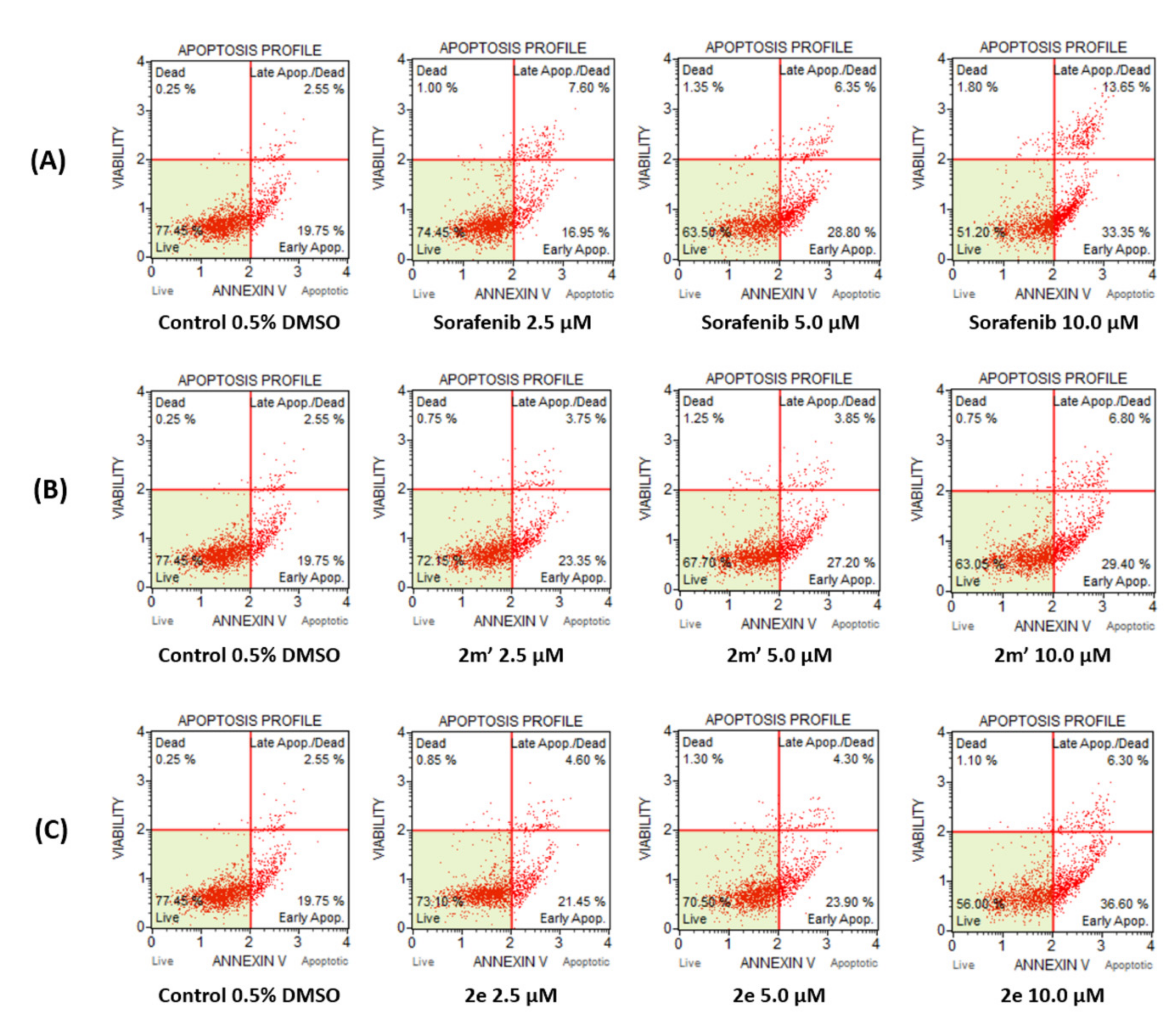
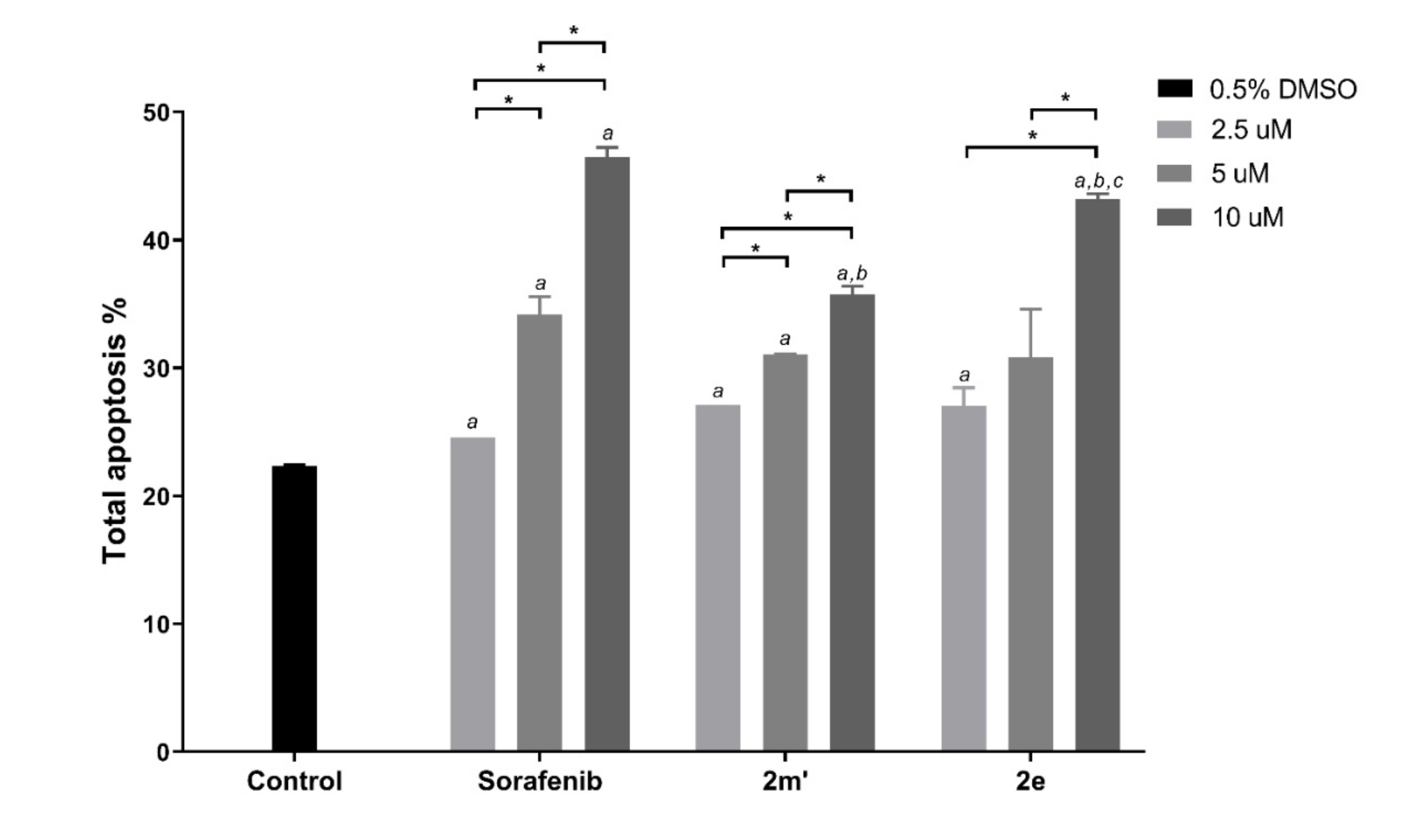
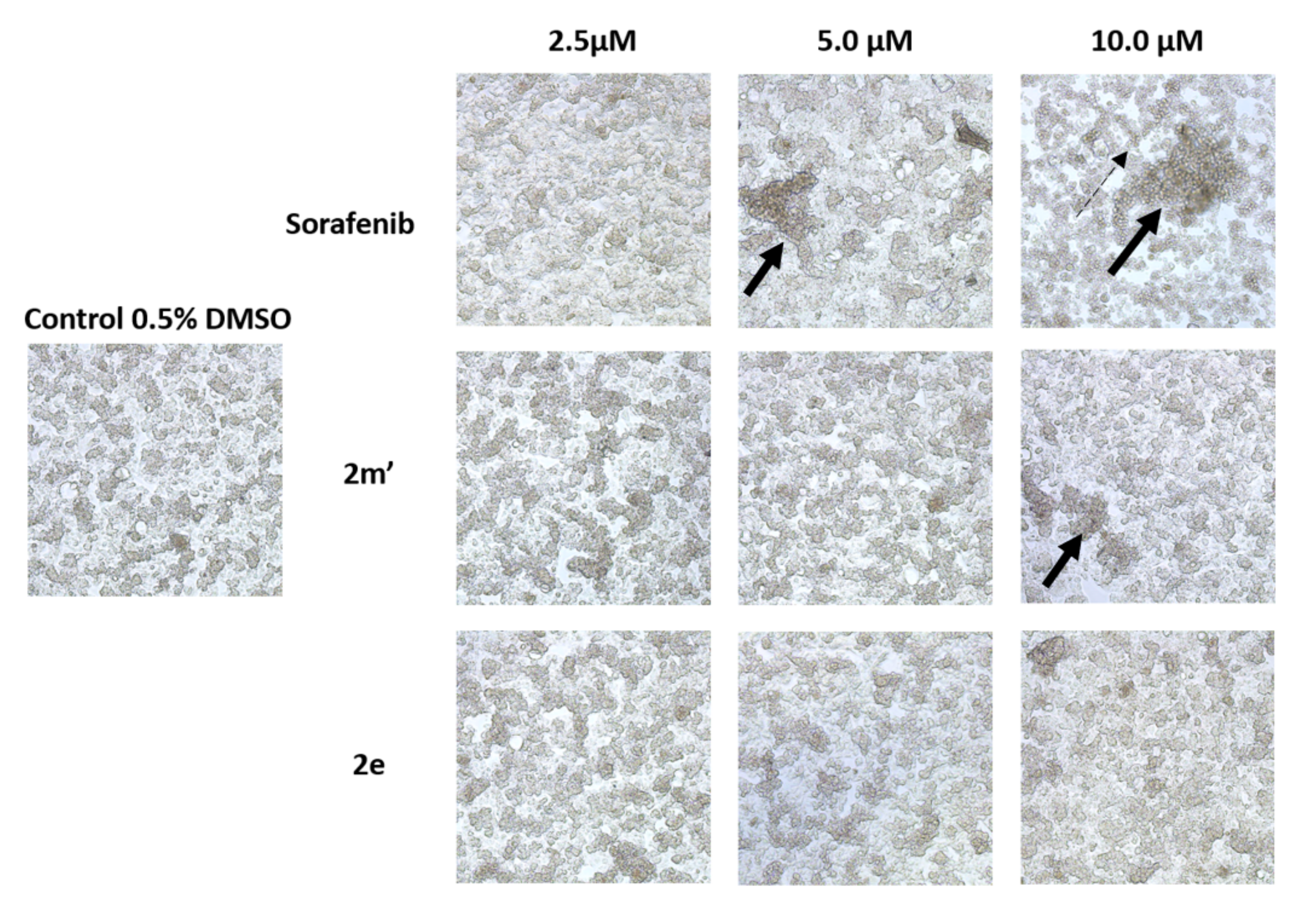
2.6. Detection of Apoptosis
2.7. Physicochemical Properties and Lipinski’s Rule of Five
3. Materials and Methods
3.1. Chemistry
3.1.1. General
3.1.2. The Procedure for the Preparation of Ureido Alkyne 5
3.1.3. General Procedure for the Preparation of Phenyl Azide 7a-7o’ and 7u’
3.1.4. General Procedure for the Preparation of Sorafenib Derivatives 2a-2o’ and 2u’
3.1.5. General Procedure for the Preparation of Sorafenib Derivatives 2p’–2r’
3.1.6. General Procedure for the Preparation of Sorafenib Derivatives 2s’ and 2t’
3.1.7. General Procedure for the Preparation of Sorafenib Derivatives 2v’ and 2w’
3.2. Cytotoxicity
3.3. Selectivity Index (SI)
3.4. Cell-Cycle Analysis
3.5. Detection of Apoptosis
3.6. Physicochemical Property Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Number of New Cases of Cancer in 2020, Both Sexes, all Ages, Search Date: 18 March 2021. Available online: https://gco.iarc.fr/today/data/factsheets/populations/900-world-fact-sheets.pdf (accessed on 27 July 2021).
- Siegel, R.; DeSantis, C.; Virgo, K.; Stein, K.; Mariotto, A.; Smith, T.; Cooper, D.; Gansler, T.; Lerro, C.; Fedewa, S.; et al. Cancer treatment and survivorship statistics, 2012. CA A Cancer J. Clin. 2012, 62, 220–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.-W.; Song, Y.; Kim, K.M.; Kim, J.-S.; Choi, E.K.; Kim, J.; Seo, H. Hepatocellular carcinoma-targeted drug discovery through image-based phenotypic screening in co-cultures of HCC cells with hepatocytes. BMC Cancer 2016, 16, 810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palakhachane, S.; Ketkaew, Y.; Chuaypen, N.; Sirirak, J.; Boonsombat, J.; Ruchirawat, S.; Tangkijvanich, P.; Suksamrarn, A.; Limpachayaporn, P. Synthesis of sorafenib analogues incorporating a 1,2,3-triazole ring and cytotoxicity towards hepatocellular carcinoma cell lines. Bioorganic Chem. 2021, 112, 104831. [Google Scholar] [CrossRef] [PubMed]
- Ul-Haq, Z.; Mahmood, U.; Reza, S. A combined 3D-QSAR and molecular docking strategy to understand the binding mechanism of V600EB-RAF inhibitors. Mol. Divers. 2012, 16, 771–785. [Google Scholar] [CrossRef]
- Wu, C.; Wang, M.; Tang, Q.; Luo, R.; Chen, L.; Zheng, P.; Zhu, W. Design, Synthesis, Activity and Docking Study of Sorafenib Analogs Bearing Sulfonylurea Unit. Molecules 2015, 20, 19361–19371. [Google Scholar] [CrossRef] [Green Version]
- Zhan, W.; Li, Y.; Huang, W.; Zhao, Y.; Yao, Z.; Yu, S.; Yuan, S.; Jiang, F.; Yao, S.; Li, S. Design, synthesis and antitumor activities of novel bis-aryl ureas derivatives as Raf kinase inhibitors. Bioorganic Med. Chem. 2012, 20, 4323–4329. [Google Scholar] [CrossRef]
- Mauri, D.; Polyzos, N.P.; Salanti, G.; Pavlidis, N.; Ioannidis, J.P.A. Multiple-Treatments Meta-analysis of Chemotherapy and Targeted Therapies in Advanced Breast Cancer. JNCI J. Natl. Cancer Inst. 2008, 100, 1780–1791. [Google Scholar] [CrossRef] [Green Version]
- Kumar, L.; Harish, P.; Malik, P.S.; Khurana, S. Chemotherapy and targeted therapy in the management of cervical cancer. Curr. Probl. Cancer 2018, 42, 120–128. [Google Scholar] [CrossRef]
- de Gramont, A.; de Gramont, A.; Chibaudel, B.; Bachet, J.-B.; Larsen, A.K.; Tournigand, C.; Louvet, C.; André, T. From Chemotherapy to Targeted Therapy in Adjuvant Treatment for Stage III Colon Cancer. Semin. Oncol. 2011, 38, 521–532. [Google Scholar] [CrossRef]
- Sawyers, C. Targeted cancer therapy. Nature 2004, 432, 294–297. [Google Scholar] [CrossRef]
- Sternberg, C.N.; Donat, S.M.; Bellmunt, J.; Millikan, R.E.; Stadler, W.; De Mulder, P.; Sherif, A.; von der Maase, H.; Tsukamoto, T.; Soloway, M.S. Chemotherapy for Bladder Cancer: Treatment Guidelines for Neoadjuvant Chemotherapy, Bladder Preservation, Adjuvant Chemotherapy, and Metastatic Cancer. Urology 2007, 69, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Bibby, M. Orthotopic models of cancer for preclinical drug evaluation: Advantages and disadvantages. Eur. J. Cancer 2004, 40, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Widmer, N.; Bardin, C.; Chatelut, E.; Paci, A.; Beijnen, J.; Levêque, D.; Veal, G.; Astier, A. Review of therapeutic drug monitoring of anticancer drugs part two targeted therapies. Eur. J. Cancer 2014, 50, 2020–2036. [Google Scholar] [CrossRef]
- Xu, R.; Wang, Q. Large-scale automatic extraction of side effects associated with targeted anticancer drugs from full-text oncological articles. J. Biomed. Inform. 2015, 55, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug Resistance in Cancer: An Overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonavida, B. Sensitizing activities of nitric oxide donors for cancer resistance to anticancer therapeutic drugs. Biochem. Pharmacol. 2020, 176, 113913. [Google Scholar] [CrossRef]
- Daher, S.; Massarwa, M.; Benson, A.A.; Khoury, T. Current and Future Treatment of Hepatocellular Carcinoma: An Updated Comprehensive Review. J. Clin. Transl. Hepatol. 2017, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Chen, Z.; Chen, Y.; Lu, J.; Li, Y.; Wang, S.; Wu, G.; Qian, F. Improving Oral Bioavailability of Sorafenib by Optimizing the “Spring” and “Parachute” Based on Molecular Interaction Mechanisms. Mol. Pharm. 2016, 13, 599–608. [Google Scholar] [CrossRef]
- Zhu, Y.-J.; Zheng, B.; Wang, H.-Y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Chavda, J.; Bhatt, H. Systemic review on B-RafV600E mutation as potential therapeutic target for the treatment of cancer. Eur. J. Med. Chem. 2020, 206, 112675. [Google Scholar] [CrossRef]
- El-Adl, K.; El-Helby, A.-G.A.; Sakr, H.; Eissa, I.H.; El-Hddad, S.S.; Shoman, F.M. Design, synthesis, molecular docking and anticancer evaluations of 5-benzylidenethiazolidine-2,4-dione derivatives targeting VEGFR-2 enzyme. Bioorganic Chem. 2020, 102, 104059. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masłowska, K.; Halik, P.; Tymecka, D.; Misicka, A.; Gniazdowska, E. The Role of VEGF Receptors as Molecular Target in Nuclear Medicine for Cancer Diagnosis and Combination Therapy. Cancers 2021, 13, 1072. [Google Scholar] [CrossRef]
- Aziz, M.A.; Serya, R.A.T.; Lasheen, D.; Abdel-Aziz, A.K.; Esmat, A.; Mansour, A.M.; Singab, A.N.B.; Abouzid, K.A.M. Discovery of Potent VEGFR-2 Inhibitors based on Furopyrimidine and Thienopyrimidne Scaffolds as Cancer Targeting Agents. Sci. Rep. 2016, 6, 24460. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A crucial target for anti-and pro-angiogenic therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Gao, Z.-H.; Qu, X.-J. The Adverse Effects of Sorafenib in Patients with Advanced Cancers. Basic Clin. Pharmacol. Toxicol. 2015, 116, 216–221. [Google Scholar] [CrossRef]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: Theoretical basis and therapeutic aspects. Signal Transduct. Target. Ther. 2020, 5, 1–15. [Google Scholar] [CrossRef]
- Zeidan, M.A.; Mostafa, A.S.; Gomaa, R.M.; Abou-Zeid, L.A.; El-Mesery, M.; El-Sayed, M.A.-A.; Selim, K.B. Design, synthesis and docking study of novel picolinamide derivatives as anticancer agents and VEGFR-2 inhibitors. Eur. J. Med. Chem. 2019, 168, 315–329. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Thabit, M.G.; Mostafa, A.S.; Selim, K.B.; Elsayed, M.A.; Nasr, M.N. Design, synthesis and molecular modeling of phenyl dihydropyridazinone derivatives as B-Raf inhibitors with anticancer activity. Bioorganic Chem. 2020, 103, 104148. [Google Scholar] [CrossRef]
- Gollob, J.A.; Wilhelm, S.; Carter, C.; Kelley, S.L. Role of Raf Kinase in Cancer: Therapeutic Potential of Targeting the Raf/MEK/ERK Signal Transduction Pathway. Semin. Oncol. 2006, 33, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Maurer, G.; Tarkowski, B.; Baccarini, M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene 2011, 30, 3477–3488. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Batt, D.; Warmuth, M. B-Raf kinase inhibitors for cancer treatment. Curr. Opin. Investig. Drugs 2007, 8, 452–456. [Google Scholar] [PubMed]
- Pan, X.; Liang, L.; Si, R.; Wang, J.; Zhang, Q.; Zhou, H.; Zhang, L.; Zhang, J. Discovery of novel anti-angiogenesis agents. Part 10: Multi-target inhibitors of VEGFR-2, Tie-2 and EphB4 incorporated with 1,2,3-triazol. Eur. J. Med. Chem. 2019, 163, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sanphanya, K.; Wattanapitayakul, S.K.; Phowichit, S.; Fokin, V.V.; Vajragupta, O. Novel VEGFR-2 kinase inhibitors identified by the back-to-front approach. Bioorganic Med. Chem. Lett. 2013, 23, 2962–2967. [Google Scholar] [CrossRef] [Green Version]
- Qin, M.; Yan, S.; Wang, L.; Zhang, H.; Zhao, Y.; Wu, S.; Wu, D.; Gong, P. Discovery of novel diaryl urea derivatives bearing a triazole moiety as potential antitumor agents. Eur. J. Med. Chem. 2016, 115, 1–13. [Google Scholar] [CrossRef]
- El-Din, M.M.G.; Gamal, M.; Abdel-Maksoud, M.; Yoo, K.H.; Oh, C.-H. Synthesis and broad-spectrum antiproliferative activity of diarylamides and diarylureas possessing 1,3,4-oxadiazole derivatives. Bioorganic Med. Chem. Lett. 2015, 25, 1692–1699. [Google Scholar] [CrossRef]
- El-Damasy, A.K.; Lee, J.-H.; Seo, S.H.; Cho, N.-C.; Pae, A.N.; Keum, G. Design and synthesis of new potent anticancer benzothiazole amides and ureas featuring pyridylamide moiety and possessing dual B-RafV600E and C-Raf kinase inhibitory activities. Eur. J. Med. Chem. 2016, 115, 201–216. [Google Scholar] [CrossRef]
- Tang, K.; Luo, C.; Li, Y.; Lu, C.; Zhou, W.; Huang, H.; Chen, X. The Study of a Novel Sorafenib Derivative HLC-080 as an Antitumor Agent. PLoS ONE 2014, 9, e101889. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-N.; Wang, X.-F.; Li, T.; Wu, D.-W.; Fu, X.-B.; Zhang, G.-J.; Shen, X.-C.; Wang, H.-S. Design, synthesis, and biological evaluation of novel quinazolinyl-diaryl urea derivatives as potential anticancer agents. Eur. J. Med. Chem. 2016, 107, 12–25. [Google Scholar] [CrossRef]
- Sun, S.; He, Z.; Huang, M.; Wang, N.; He, Z.; Kong, X.; Yao, J. Design and discovery of thioether and nicotinamide containing sorafenib analogues as multikinase inhibitors targeting B-Raf, B-RafV600E and VEGFR-2. Bioorganic Med. Chem. 2018, 26, 2381–2391. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, D.S.; Abell, A. ChemInform Abstract: 1,2,3-Triazoles in Peptidomimetic Chemistry. Eur. J. Organic Chem. 2011, 42, 2399–2411. [Google Scholar] [CrossRef]
- Perczel, A.; Atanasov, A.G.; Sklenář, V.; Nováček, J.; Papoušková, V.; Kadeřávek, P.; Žídek, L.; Kozłowski, H.; Wątły, J.; Hecel, A.; et al. The Eighth Central European Conference “Chemistry towards Biology”: Snapshot. Molecules 2016, 21, 1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashooriha, M.; Khoshneviszadeh, M.; Khoshneviszadeh, M.; Moradi, S.E.; Rafiei, A.; Kardan, M.; Emami, S. 1,2,3-Triazole-based kojic acid analogs as potent tyrosinase inhibitors: Design, synthesis and biological evaluation. Bioorganic Chem. 2019, 82, 414–422. [Google Scholar] [CrossRef]
- Ye, W.; Yao, Q.; Yu, S.; Gong, P.; Qin, M. Synthesis and Antitumor Activity of Triazole-Containing Sorafenib Analogs. Molecules 2017, 22, 1759. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.S. 1,2,3-Triazoles: Scaffold With Medicinal Significance. Inflamm. Cell Signal. 2014, 1, 10. [Google Scholar] [CrossRef]
- Yang, J.; Shibu, M.A.; Kong, L.; Luo, J.; BadrealamKhan, F.; Huang, Y.; Tu, Z.-C.; Yun, C.-H.; Huang, C.-Y.; Ding, K.; et al. Design, Synthesis, and Structure-Activity Relationships of 1,2,3-Triazole Benzenesulfonamides as New Selective Leucine-Zipper and Sterile-α Motif Kinase (ZAK) Inhibitors. J. Med. Chem. 2020, 63, 2114–2130. [Google Scholar] [CrossRef]
- Ma, L.-Y.; Pang, L.-P.; Wang, B.; Zhang, M.; Hu, B.; Xue, D.-Q.; Shao, K.-P.; Zhang, B.-L.; Liu, Y.; Zhang, E.; et al. Design and synthesis of novel 1,2,3-triazole-pyrimidine hybrids as potential anticancer agents. Eur. J. Med. Chem. 2014, 86, 368–380. [Google Scholar] [CrossRef]
- Ihmaid, S.K.; Alraqa, S.Y.; Aouad, M.R.; Aljuhani, A.; Elbadawy, H.M.; Salama, S.A.; Rezki, N.; Ahmed, H.E. Design of molecular hybrids of phthalimide-triazole agents with potent selective MCF-7/HepG2 cytotoxicity: Synthesis, EGFR inhibitory effect, and metabolic stability. Bioorganic Chem. 2021, 111, 104835. [Google Scholar] [CrossRef]
- Xu, Q.; Dai, B.; Li, Z.; Xu, L.; Yang, D.; Gong, P.; Hou, Y.; Liu, Y. Design, synthesis, and biological evaluation of 4-((6,7-dimethoxyquinoline-4-yl)oxy)aniline derivatives as FLT3 inhibitors for the treatment of acute myeloid leukemia. Bioorganic Med. Chem. Lett. 2019, 29, 126630. [Google Scholar] [CrossRef]
- Shan, Y.; Wang, J.; Si, R.; Ma, Y.; Li, J.; Zhang, Q.; Lu, W.; Zhang, J. Exploring the potential intracellular targets of vascular normalization based on active candidates. Bioorganic Chem. 2021, 108, 104551. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.; Nayak, V.L.; Srikanth, P.; Baig, M.F.; Reddy, N.S.; Babu, K.S.; Kamal, A. Synthesis and biological evaluation of 1-benzyl-N-(2-(phenylamino)pyridin-3-yl)-1H-1,2,3-triazole-4-carboxamides as antimitotic agents. Bioorganic Chem. 2019, 83, 535–548. [Google Scholar] [CrossRef] [PubMed]
- Sirirak, J.; Suttayasorranakhom, S.; Limpachayaporn, P.; Oekchuae, S. In Silico, Design, and Development: Molecular Modeling towards B-RAF and VEGFR-2 of Novel Sorafenib Derivatives for Targeted Hepatocellular Carcinoma Cancer Inhibitors. Key Eng. Mater. 2021, 901, 3–8. [Google Scholar] [CrossRef]
- Chen, F.; Fang, Y.; Zhao, R.; Le, J.; Zhang, B.; Huang, R.; Chen, Z.; Shao, J. Evolution in medicinal chemistry of sorafenib derivatives for hepatocellular carcinoma. Eur. J. Med. Chem. 2019, 179, 916–935. [Google Scholar] [CrossRef] [PubMed]
- Anwer, K.E.; El-Sattar, N.E.A.A.; Shamaa, M.M.; Zakaria, M.Y.; Beshay, B.Y. Design, Green Synthesis and Tailoring of Vitamin E TPGS Augmented Niosomal Nano-Carrier of Pyrazolopyrimidines as Potential Anti-Liver and Breast Cancer Agents with Accentuated Oral Bioavailability. Pharmaceuticals 2022, 15, 330. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mohsen, H.T.; Abdullaziz, M.A.; El Kerdawy, A.M.; Ragab, F.A.F.; Flanagan, K.J.; Mahmoud, A.E.E.; Ali, M.M.; El Diwani, H.I.; Senge, M.O. Targeting Receptor Tyrosine Kinase VEGFR-2 in Hepatocellular Cancer: Rational Design, Synthesis and Biological Evaluation of 1,2-Disubstituted Benzimidazoles. Molecules 2020, 25, 770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdel-Mohsen, H.T.; Omar, M.A.; El Kerdawy, A.M.; Mahmoud, A.E.; Ali, M.M.; El Diwani, H.I. Novel potent substituted 4-amino-2-thiopyrimidines as dual VEGFR-2 and BRAF kinase inhibitors. Eur. J. Med. Chem. 2019, 179, 707–722. [Google Scholar] [CrossRef] [PubMed]
- Abdullaziz, M.A.; Abdel-Mohsen, H.T.; El Kerdawy, A.M.; Ragab, F.A.; Ali, M.M.; Abu-Bakr, S.; Girgis, A.S.; El Diwani, H.I. Design, synthesis, molecular docking and cytotoxic evaluation of novel 2-furybenzimidazoles as VEGFR-2 inhibitors. Eur. J. Med. Chem. 2017, 136, 315–329. [Google Scholar] [CrossRef]
- Ryu, B.-Y.; Emrick, T. Bisphenol-1,2,3-triazole (BPT) Epoxies and Cyanate Esters: Synthesis and Self-Catalyzed Curing. Macromolecules 2011, 44, 5693–5700. [Google Scholar] [CrossRef]
- Budhathoki-Uprety, J.; Jena, P.V.; Roxbury, D.; Heller, D.A. Helical Polycarbodiimide Cloaking of Carbon Nanotubes Enables Inter-Nanotube Exciton Energy Transfer Modulation. J. Am. Chem. Soc. 2014, 136, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Nagai, A.; Guo, Z.; Feng, X.; Jin, S.; Chen, X.; Ding, X.; Jiang, D. Pore surface engineering in covalent organic frameworks. Nat. Commun. 2011, 2, 536. [Google Scholar] [CrossRef] [PubMed]
- Bolje, A.; Urankar, D.; Košmrlj, J. Synthesis and NMR Analysis of 1, 4-Disubstituted 1, 2, 3-Triazoles Tethered to Pyridine, Pyrimidine, and Pyrazine Rings. Eur. J. Org. Chem. 2014, 36, 8167–8181. [Google Scholar] [CrossRef]
- Thongnest, S.; Chawengrum, P.; Keeratichamroen, S.; Lirdprapamongkol, K.; Eurtivong, C.; Boonsombat, J.; Kittakoop, P.; Svasti, J.; Ruchirawat, S. Vernodalidimer L, a sesquiterpene lactone dimer from Vernonia extensa and anti-tumor effects of vernodalin, vernolepin, and vernolide on HepG2 liver cancer cells. Bioorganic Chem. 2019, 92, 103197. [Google Scholar] [CrossRef] [PubMed]
- Bozorov, K.; Zhao, J.; Aisa, H.A. 1,2,3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview. Bioorganic Med. Chem. 2019, 27, 3511–3531. [Google Scholar] [CrossRef]
- Prayong, P.; Barusrux, S.; Weerapreeyakul, N. Cytotoxic activity screening of some indigenous Thai plants. Fitoterapia 2008, 79, 598–601. [Google Scholar] [CrossRef]
- Mansour, G.H.; El-Magd, M.A.; Mahfouz, D.H.; Abdelhamid, I.A.; Mohamed, M.F.; Ibrahim, N.S.; Wahab, A.H.A.A.; Elzayat, E.M. Bee venom and its active component Melittin synergistically potentiate the anticancer effect of Sorafenib against HepG2 cells. Bioorganic Chem. 2021, 116, 105329. [Google Scholar] [CrossRef]
- Sudan, S.; Rupasinghe, H.P.V. Flavonoid-Enriched Apple Fraction AF4 Induces Cell Cycle Arrest, DNA Topoisomerase II Inhibition, and Apoptosis in Human Liver Cancer HepG2 Cells. Nutr. Cancer 2014, 66, 1237–1246. [Google Scholar] [CrossRef]
- Lee, H.-A.; Chu, K.-B.; Moon, E.-K.; Kim, S.S.; Quan, F.-S. Sensitization to oxidative stress and G2/M cell cycle arrest by histone deacetylase inhibition in hepatocellular carcinoma cells. Free Radic. Biol. Med. 2020, 147, 129–138. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Pajouhesh, H.; Lenz, G.R. Medicinal chemical properties of successful central nervous system drugs. NeuroRX 2005, 2, 541–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, J.; Gattass, C.R. Topological Polar Surface Area Defines Substrate Transport by Multidrug Resistance Associated Protein 1 (MRP1/ABCC1). J. Med. Chem. 2009, 52, 1214–1218. [Google Scholar] [CrossRef]
- Iniyavan, P.; Balaji, G.; Sarveswari, S.; Vijayakumar, V. CuO nanoparticles: Synthesis and application as an efficient reusable catalyst for the preparation of xanthene substituted 1,2,3-triazoles via click chemistry. Tetrahedron Lett. 2015, 56, 5002–5009. [Google Scholar] [CrossRef]
- Birkenfelder, I.; Gurke, J.; Grubert, L.; Hecht, S.; Schmidt, B.M. Click Chemistry Derived Pyridazines: Electron-Deficient Building Blocks with Defined Conformation and Packing Structure. Chem. Asian J. 2017, 12, 3156–3161. [Google Scholar] [CrossRef]
- Liu, M.; Hou, Y.; Yin, W.; Zhou, S.; Qian, P.; Guo, Z.; Xu, L.; Zhao, Y. Discovery of a novel 6,7-disubstituted-4-(2-fluorophenoxy)quinolines bearing 1,2,3-triazole-4-carboxamide moiety as potent c-Met kinase inhibitors. Eur. J. Med. Chem. 2016, 119, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Hu, L.; Shi, W.; Cui, G.; Zhang, X.; Zhang, Q.-W. Design, Synthesis and Anti-Platelet Aggregation Activity Study of Ginkgolide-1,2,3-triazole Derivatives. Molecules 2019, 24, 2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwok, S.W.; Fotsing, J.R.; Fraser, R.J.; Rodionov, V.O.; Fokin, V.V. Transition-Metal-Free Catalytic Synthesis of 1,5-Diaryl-1,2,3-triazoles. Org. Lett. 2010, 12, 4217–4219. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, H.C.; Schaap, M.; Baird, L.; Georgakopoulos, N.D.; Fowkes, A.; Thiollier, C.; Kachi, H.; Dinkova-Kostova, A.T.; Wells, G. Design, Synthesis, and Evaluation of Triazole Derivatives That Induce Nrf2 Dependent Gene Products and Inhibit the Keap1–Nrf2 Protein-Protein Interaction. J. Med. Chem. 2015, 58, 7186–7194. [Google Scholar] [CrossRef]
- Zhang, Z.; Xiao, F.; Huang, B.; Hu, J.; Fu, B.; Zhang, Z. Cyclization of Alkyne-Azide with Isonitrile/CO via Self-Relay Rhodium Catalysis. Org. Lett. 2016, 18, 908–911. [Google Scholar] [CrossRef]
- Ren, L.; Jiao, N. PdCl2 catalyzed efficient assembly of organic azides, CO, and alcohols under mild conditions: A direct approach to synthesize carbamates. Chem. Commun. 2014, 50, 3706–3709. [Google Scholar] [CrossRef]
- Sasaki, K.; Kurumi, M.; Takata, H.; Nakayama, T. Synthesis and Chemiluminescent Activity of Pyridazino[4,5-b]indole- 1,4(2H,3H)-diones. Heterocycles 2000, 53, 2809–2819. [Google Scholar] [CrossRef]
- Zhou, S.; Liao, H.; Liu, M.; Feng, G.; Fu, B.; Li, R.; Cheng, M.; Zhao, Y.; Gong, P. Discovery and biological evaluation of novel 6,7-disubstituted-4-(2-fluorophenoxy)quinoline derivatives possessing 1,2,3-triazole-4-carboxamide moiety as c-Met kinase inhibitors. Bioorganic Med. Chem. 2014, 22, 6438–6452. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, V.D.; Parello, J.; Kutonova, K.V.; Trusova, M.E.; Postnikov, P.S. A Simple and Effective Synthesis of Aryl Azides via Arenediazonium Tosylates. Synthesis 2013, 45, 2706–2710. [Google Scholar] [CrossRef] [Green Version]
- Kanabar, D.; Farrales, P.; Kabir, A.; Juang, D.; Gnanmony, M.; Almasri, J.; Torrents, N.; Shukla, S.; Gupta, V.; Dukhande, V.V.; et al. Optimizing the aryl-triazole of cjoc42 for enhanced gankyrin binding and anti-cancer activity. Bioorganic Med. Chem. Lett. 2020, 30, 127372. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Li, J.; Yao, S.Q. In Situ “Click” Assembly of Small Molecule Matrix Metalloprotease Inhibitors Containing Zinc-Chelating Groups. Org. Lett. 2008, 10, 5529–5531. [Google Scholar] [CrossRef]
- Sebest, F.; Casarrubios, L.; Rzepa, H.S.; White, A.J.P.; Díez-González, S. Thermal azide–alkene cycloaddition reactions: Straightforward multi-gram access to Δ2-1,2,3-triazolines in deep eutectic solvents. Green Chem. 2018, 20, 4023–4035. [Google Scholar] [CrossRef] [Green Version]
- Pokhodylo, N.T.; Matiichuk, V.S.; Obushak, M. Synthesis and transformations of 1-(azidophenyl)-1H-tetrazoles. Russ. J. Org. Chem. 2010, 46, 556–560. [Google Scholar] [CrossRef]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, K.B.; Fokin, V.V. Copper(I)-Catalyzed Synthesis of Azoles. DFT Study Predicts Unprecedented Reactivity and Intermediates. J. Am. Chem. Soc. 2004, 127, 210–216. [Google Scholar] [CrossRef]
- Vershinina, I.A.; Gornukhina, O.V.; Lubimova, T.; Golubchikov, O.A.; Semeikin, A.S. 4-(8-quinolylazo)resorcinol and 1-(8-quinolylazo)-2-naphthol: Synthesis and sorption properties. Russ. J. Gen. Chem. 2016, 86, 2232–2235. [Google Scholar] [CrossRef]
- Beutner, G.L.; Young, I.S.; Davies, M.L.; Hickey, M.R.; Park, H.; Stevens, J.M.; Ye, Q. TCFH-NMI: Direct Access to N-Acyl Imidazoliums for Challenging Amide Bond Formations. Org. Lett. 2018, 20, 4218–4222. [Google Scholar] [CrossRef] [Green Version]
- Carmichael, J.; DeGraff, W.G.; Gazdar, A.F.; Minna, J.D.; Mitchell, J.B. Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res. 1987, 47, 936–942. [Google Scholar] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Doyle, A.; Griffiths, J.B. Mammalian Cell Culture-Essential Techniques; John Wiley and Sons Ltd.: New York, NY, USA, 1997. [Google Scholar]
- Tominaga, H.; Ishiyama, M.; Ohseto, F.; Sasamoto, K.; Hamamoto, T.; Suzuki, K.; Watanabe, M. A water-soluble tetrazolium salt useful for colorimetric cell viability assay. Anal. Commun. 1999, 36, 47–50. [Google Scholar] [CrossRef]
- Fernando, J.; Sancho, P.; Fernández-Rodriguez, C.M.; Lledó, J.L.; Caja, L.; Campbell, J.S.; Fausto, N.; Fabregat, I. Sorafenib sensitizes hepatocellular carcinoma cells to physiological apoptotic stimuli. J. Cell. Physiol. 2012, 227, 1319–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | Cytotoxicity | SI | ||
|---|---|---|---|---|---|
| HepG2 | MRC-5 | ||||
| Cell Viability at 25 µM | IC50 (µM) | IC50 (µM) | |||
| 2a | H | 83% | - | >100 | - |
| 2b | o-F | 79% | - | 73.5 ± 9.14 | - |
| 2c | m-F | 60% | - | 61.6 ± 3.63 | - |
| 2d | p-F | 58% | - | 59.3 ± 1.96 | - |
| 2e | o-Cl | 53% | 5.02 ± 2.07 | 61.4 ± 3.51 | 12.2 |
| 2f | m-Cl | 52% | 9.81 ± 5.02 | 63.8 ± 3.53 | 6.50 |
| 2g | p-Cl | 72% | - | 14.6 ± 4.17 | - |
| 2h | o-Br | 61% | - | 52.9 ± 3.10 | - |
| 2i | m-Br | 81% | - | 53.0 ± 3.31 | - |
| 2j | p-Br | 75% | - | 60.7 ± 1.88 | - |
| 2k | o-I | 60% | - | 49.1 ± 2.95 | - |
| 2l | m-I | 88% | - | 49.2 ± 2.13 | - |
| 2m | p-I | 63% | - | 49.9 ± 6.61 | - |
| 2n | o-OH | 75% | - | >100 | - |
| 2o | m-OH | 89% | - | >100 | - |
| 2p | p-OH | 66% | - | 69.6 ± 5.66 | - |
| 2q | o-OMe | 74% | - | 68.0 ± 5.14 | - |
| 2r | m-OMe | 76% | - | 76.1 ± 9.18 | - |
| 2s | p-OMe | 64% | - | 21.2 ± 2.51 | - |
| 2t | o-Me | 72% | - | 61.7 ± 3.22 | - |
| 2u | m-Me | 69% | - | >100 | - |
| 2v | p-Me | 91% | - | 99.9 ± 0.90 | - |
| 2w | o-CF3 | 58% | - | 58.8 ± 0.39 | - |
| 2x | m-CF3 | 81% | - | 63.2 ± 1.57 | - |
| 2y | p-CF3 | 39% | 5.97 ± 2.14 | 58.6 ± 3.51 | 9.81 |
| 2z | o-CN | 66% | - | 59.7 ± 6.63 | - |
| 2a’ | m-CN | 59% | - | 99.3 ± 2.99 | - |
| 2b’ | p-CN | 113% | - | 64.1 ± 0.36 | - |
| 2c’ | o-NO2 | 78% | - | 60.6 ± 5.26 | - |
| 2d’ | m-NO2 | 62% | - | 58.4 ± 5.69 | - |
| 2e’ | p-NO2 | 70% | - | 66.1 ± 4.50 | - |
| 2f’ | o-Et | 60% | - | 57.7 ± 1.82 | - |
| 2g’ | m-Et | 79% | - | 94.7 ± 5.24 | - |
| 2h’ | p-Et | 63% | - | 75.4 ± 10.43 | - |
| 2i’ | o-iPr | 29% | 5.40 ± 0.35 | 54.5 ± 7.03 | 10.1 |
| 2j’ | m-iPr | 62% | - | 73.3 ± 15.60 | - |
| 2k’ | p-iPr | 50% | 20.95 ± 4.17 | 65.9 ± 10.21 | 3.15 |
| 2l’ | m-tBu | 46% | 9.88 ± 3.03 | 68.2 ± 11.91 | 6.91 |
| 2m’ | p-tBu | 44% | 5.57 ± 0.91 | 81.6 ± 5.07 | 14.7 |
| 2n’ | m-CO2H | 96% | - | >100 | - |
| 2o’ | p-CO2H | 81% | - | >100 | - |
| 2p’ | o-NH2 | 65% | - | >100 | - |
| 2q’ | m-NH2 | 57% | - | 62.3 ± 3.38 | - |
| 2r’ | p-NH2 | 79% | - | >100 | - |
| 2s’ | o-NHAc | 60% | - | 53.6 ± 2.54 | - |
| 2t’ | m-NHAc | 93% | - | >100 | - |
| 2u’ | p-NHAc | 76% | - | >100 | - |
| 2v’ | m-CONHMe | 74% | - | >100 | - |
| 2w’ | p-CONHMe | 61% | - | 62.2 ± 7.88 | - |
| Doxorubicin | - | 0.59 ± 0.10 | 2.3 ± 0.22 | 3.83 | |
| Sorafenib (1) | - | 5.97 ± 0.71 | 19.7 ± 1.68 | 3.30 | |
| Compounds | aMW | bTPSA | cCLog P | dLog S | enHBA | fnHBD | gnVio | hnRB |
|---|---|---|---|---|---|---|---|---|
| 2m’ | 451.87 | 71.84 | 4.74 | −6.32 | 6 | 2 | 1 | 8 |
| 2e | 430.21 | 71.84 | 4.01 | −5.23 | 6 | 2 | 0 | 7 |
| Sorafenib | 464.82 | 92.35 | 4.11 | −5.71 | 7 | 3 | 0 | 9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oekchuae, S.; Sirirak, J.; Charoensuksai, P.; Wongprayoon, P.; Chuaypen, N.; Boonsombat, J.; Ruchirawat, S.; Tangkijvanich, P.; Suksamrarn, A.; Limpachayaporn, P. The Design and Synthesis of a New Series of 1,2,3-Triazole-Cored Structures Tethering Aryl Urea and Their Highly Selective Cytotoxicity toward HepG2. Pharmaceuticals 2022, 15, 504. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050504
Oekchuae S, Sirirak J, Charoensuksai P, Wongprayoon P, Chuaypen N, Boonsombat J, Ruchirawat S, Tangkijvanich P, Suksamrarn A, Limpachayaporn P. The Design and Synthesis of a New Series of 1,2,3-Triazole-Cored Structures Tethering Aryl Urea and Their Highly Selective Cytotoxicity toward HepG2. Pharmaceuticals. 2022; 15(5):504. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050504
Chicago/Turabian StyleOekchuae, Sittisak, Jitnapa Sirirak, Purin Charoensuksai, Pawaris Wongprayoon, Natthaya Chuaypen, Jutatip Boonsombat, Somsak Ruchirawat, Pisit Tangkijvanich, Apichart Suksamrarn, and Panupun Limpachayaporn. 2022. "The Design and Synthesis of a New Series of 1,2,3-Triazole-Cored Structures Tethering Aryl Urea and Their Highly Selective Cytotoxicity toward HepG2" Pharmaceuticals 15, no. 5: 504. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15050504