
Discovery of Therapeutics Targeting Oxidative Stress in Autosomal Recessive Cerebellar Ataxia: A Systematic Review

 ,
,  , , and
, , and
Abstract
:
1. Introduction
2. Involvement of Oxidative Stress and Mitochondrial Dysfunction in the Pathogenesis of ARCAs
2.1. Ataxia-Telangiectasia (A-T)
2.2. Ataxia with Oculomotor Apraxia (AOA)
2.3. Ataxia with Vitamin E Deficiency (AVED)
2.4. Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS)
2.5. Friedreich’s Ataxia (FRDA)
3. Standard Therapeutic Options for ARCAs and Their Adverse Effects
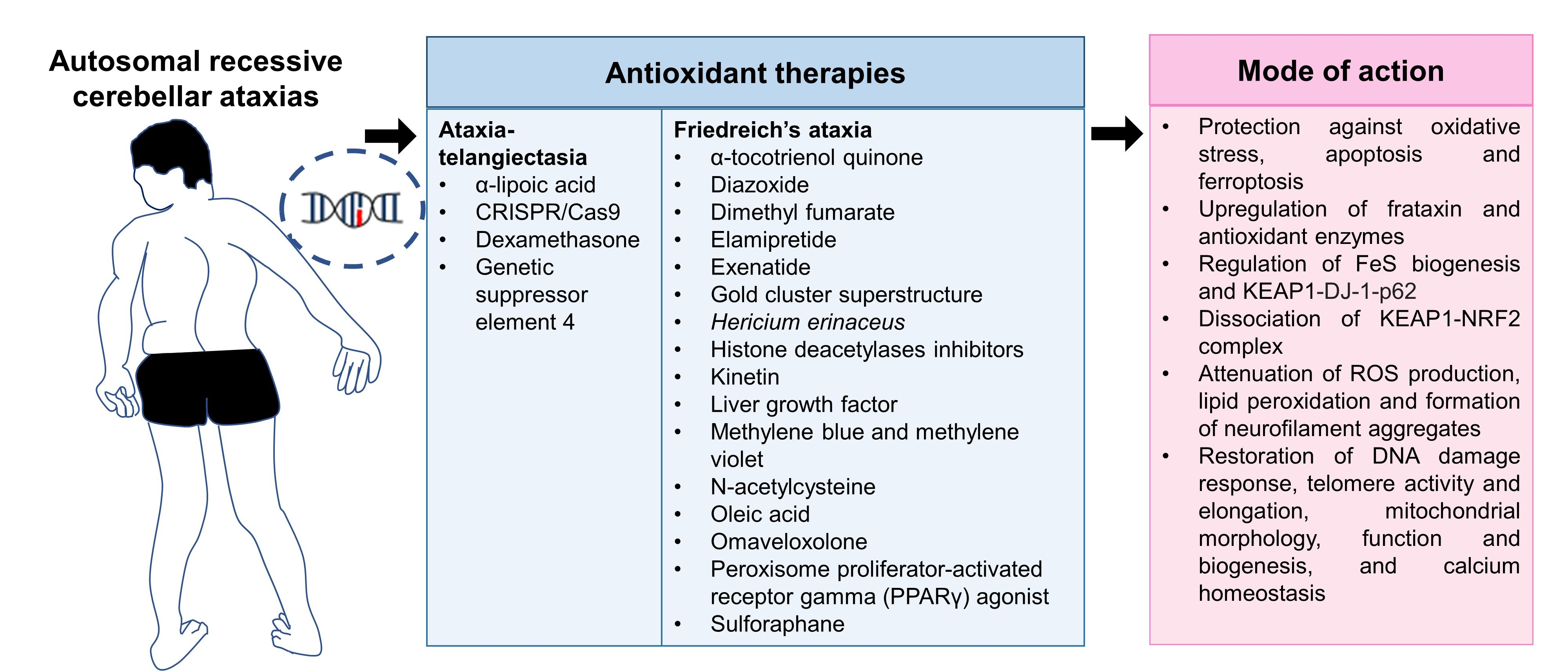
4. Involvement of Antioxidant Defense Mechanisms for the Management of Autosomal Recessive Cerebellar Ataxia
5. Materials and Methods
5.1. Search Strategy
5.2. Eligibility Criteria
5.3. Data Extraction and Analysis
6. Results
Study Selection
7. Discussion
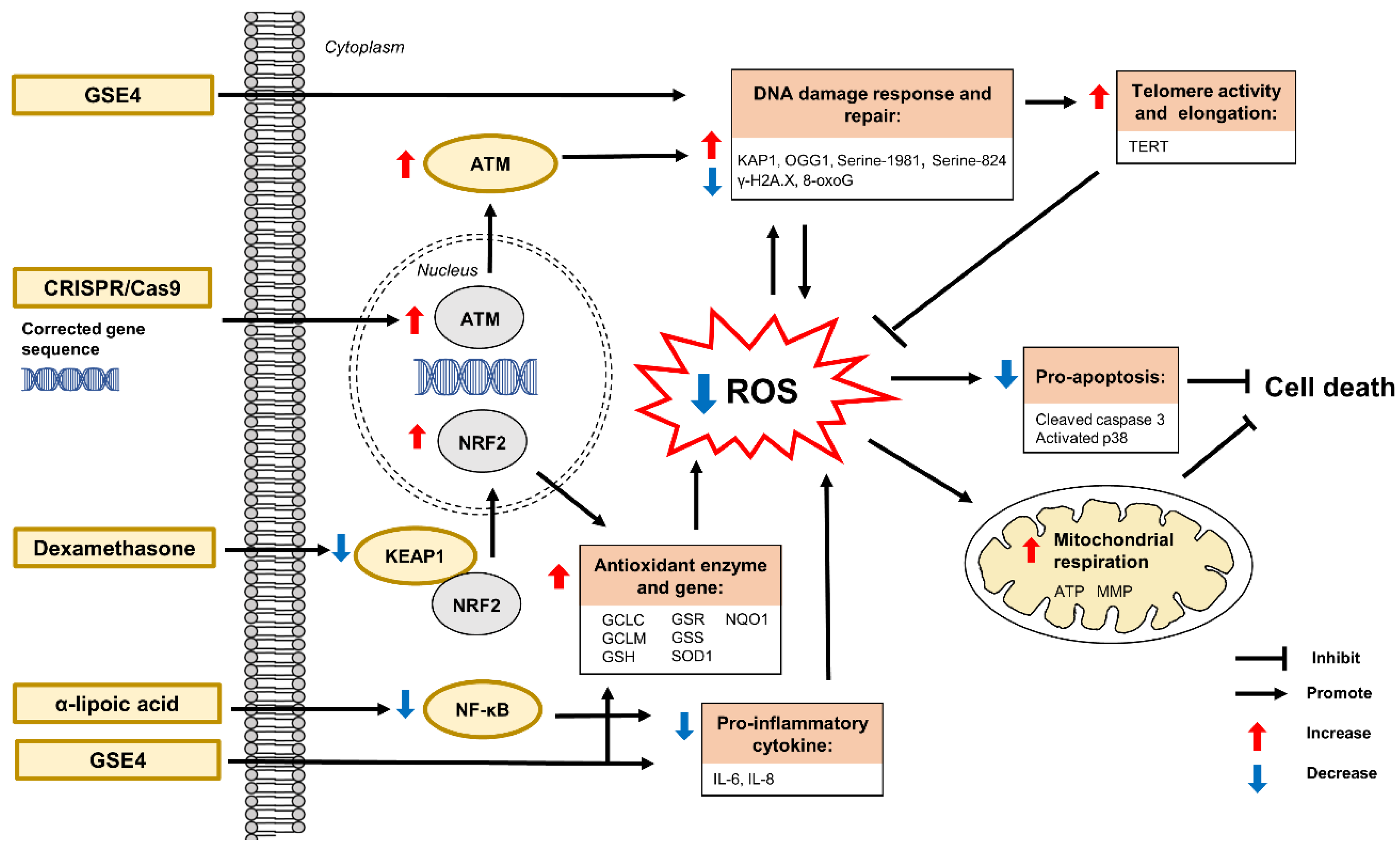
7.1. Antioxidant Defense Mechanisms and Antioxidant Therapies in Ataxia-Telangiectasia
7.1.1. α-Lipoic Acid
7.1.2. CRISPR/Cas9
7.1.3. Dexamethasone
7.1.4. Genetic Suppressor Element 4
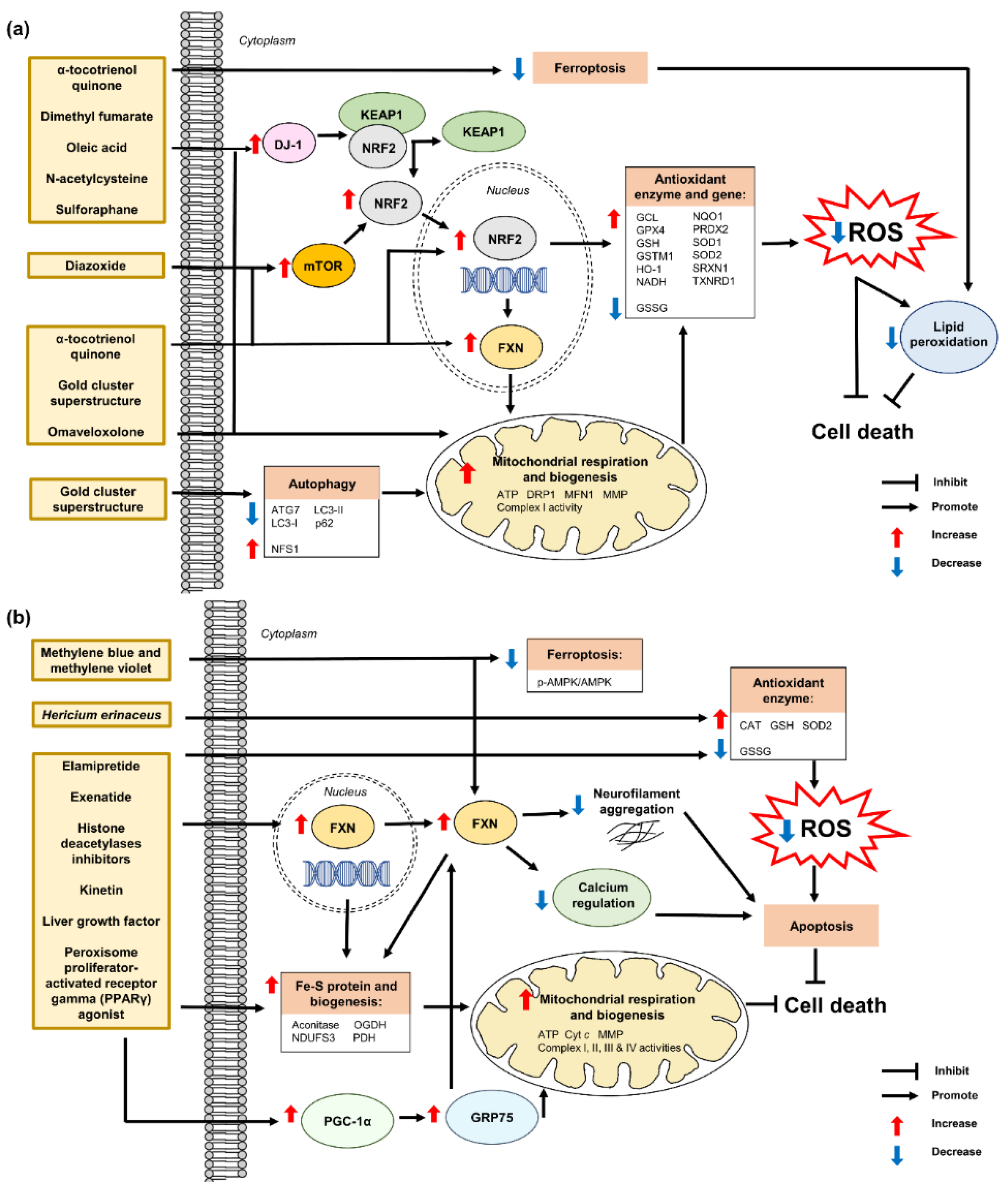
7.2. Antioxidant Defense Mechanisms and Antioxidant Therapies in Friedreich’s Ataxia
7.2.1. α-Tocotrienol Quinone
7.2.2. Diazoxide
7.2.3. Dimethyl Fumarate
7.2.4. Elamipretide
7.2.5. Exenatide
7.2.6. Gold Cluster Superstructure
7.2.7. Hericium erinaceus
7.2.8. Histone Deacetylases Inhibitors
7.2.9. Kinetin
7.2.10. Liver Growth Factor
7.2.11. Methylene Blue and Methylene Violet
7.2.12. N-Acetylcysteine
7.2.13. Oleic Acid
7.2.14. Omaveloxolone
7.2.15. Peroxisome Proliferator-Activated Receptor Gamma Agonist
7.2.16. Sulforaphane
8. Limitations and Perspectives for Future Research
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviation
| ATM | Ataxia-telangiectasia mutated; |
| DNA | deoxyribonucleic acid; |
| GCLC | glutamyl-cysteine ligase catalytic subunit; |
| GCLM | glutamyl-cysteine ligase modifier subunit; |
| GRE | genetic suppressor element; |
| GSH | glutathione; |
| GSR | glutathione reductase; |
| GSS | glutathione synthetase; |
| IL | interleukin; |
| iPSCs | induced pluripotent stem cells; |
| KAP1 | KRAB-associated protein 1; |
| KEAP1 | Kelch-like ECH-associated protein 1; |
| mRNA | messenger ribonucleic acid; |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells; |
| NRF2 | nuclear factor erythroid 2–related factor 2; |
| OGG1 | 8-oxoguanine DNA glycosylase-1; |
| ROS | reactive oxygen species; |
| SOD | superoxide dismutase; |
| TERT | telomerase reverse transcriptase; |
| γ-H2A.X | gamma-H2A histone family member X; |
| 2DG | deoxy-D-glucose; |
| 8-oxoG | 8-oxoguanineAkt, protein kinase B; |
| AMPK | AMP-activated protein kinase; |
| ARE | antioxidant response element; |
| ATP | adenosine triphosphate; |
| Bax | Bcl2 associated X; |
| Bcl-2 | B-cell lymphoma 2; |
| BSO | L-buthionine (S,R)-sulfoximine; |
| CAT | catalase; |
| COX1 | cytochrome c oxidase I; |
| DRG | dorsal root ganglion; |
| FAC | ferric ammonium citrate; |
| FXN | frataxin; |
| GCL | γ-glutamyl cysteine ligase; |
| GCLC | glutamyl-cysteine ligase catalytic subunit; |
| GCLM | glutamyl-cysteine ligase modifier subunit; |
| GPX4 | glutathione peroxidase 4; |
| GRP75 | glucose-regulated protein 75; |
| GSH | glutathione; |
| GSSG | oxidised glutathione; |
| HO-1 | heme oxygenase 1; |
| iPSCs | induced pluripotent stem cells; |
| ISC | iron-sulfur clusters; |
| ISCU | iron-sulfur cluster assembly enzyme; |
| KEAP1 | Kelch-like ECH-associated protein 1; |
| KIKO | knock-in/knock-out; |
| MMP | mitochondrial membrane potential; |
| mRNA | messenger ribonucleic acid; |
| mTFA | mitochondrial transcription factor A; |
| mTOR | mammalian target of rapamycin; |
| NDUFS3 | NADH: ubiquinone oxidoreductase core subunit s3; |
| NE | not evaluated; |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells; |
| NQO1 | NAD(P)H quinone oxidoreductase 1; |
| NRF1 | nuclear respiratory factor 1; |
| NRF2 | nuclear factor erythroid 2–related factor 2; |
| NSCs | neural stem cells; |
| OGDH | 8-oxoglutarate dehydrogenase E1 component; |
| PDH | pyruvate dehydrogenase; |
| PGC-1α | peroxisome proliferator-activated receptor-γ (PPARγ) coactivator 1 alpha; |
| ROS | reactive oxygen species; |
| RSL-3 | RAS-selective lethal 3; |
| SDH-A | succinate dehydrogenase complex, subunit A; |
| SOD | superoxide dismutase; |
| TERT | telomerase reverse transcriptase; |
| 4-HNE | 4-hydroxynonenal; |
| 8-oxodG | 8-oxo-2′-deoxyguanosine |
References
- Palau, F.; Espinós, C. Autosomal recessive cerebellar ataxias. Orphanet J. Rare Dis. 2006, 1, 47. [Google Scholar] [CrossRef]
- Embiruçu, E.K.; Martyn, M.L.; Schlesinger, D.; Kok, F. Autosomal recessive ataxias: 20 types, and counting. Arq. Neuropsiquiatr. 2009, 67, 1143–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruano, L.; Melo, C.; Silva, M.C.; Coutinho, P. The global epidemiology of hereditary ataxia and spastic paraplegia: A systematic review of prevalence studies. Neuroepidemiology 2014, 42, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Manto, M.; Gandini, J.; Feil, K.; Strupp, M. Cerebellar ataxias: An update. Curr. Opin. Neurol. 2020, 33, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Manto, M.U. (Ed.) Autosomal recessive cerebellar ataxias. In Cerebellar Disorders: A Practical Approach to Diagnosis and Management; Cambridge University Press: Cambridge, UK, 2010; pp. 189–228. [Google Scholar] [CrossRef] [Green Version]
- Fogel, B.L.; Geschwind, D.H. Clinical neurogenetics. In Bradley’s Neurology in Clinical Practice, 7th ed.; Daroff, R., Jankovic, J., Mazziotta, J., Pomeroy, S., Eds.; Elsevier: Philadelphia, PA, USA, 2015; pp. 648–675. [Google Scholar]
- Anheim, M.; Tranchant, C.; Koenig, M. The autosomal recessive cerebellar ataxias. N. Engl. J. Med. 2012, 366, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Fogel, B.L. Autosomal-recessive cerebellar ataxias. Handb. Clin. Neurol. 2018, 147, 187–209. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Anheim, M.; Durr, A.; Klein, C.; Koenig, M.; Synofzik, M.; Marras, C.; van de Warrenburg, B.P.; International Parkinson and Movement Disorder Society Task Force on Classification and Nomenclature of Genetic Movement Disorders. The genetic nomenclature of recessive cerebellar ataxias. Mov. Disord. 2018, 33, 1056–1076. [Google Scholar] [CrossRef] [PubMed]
- Beaudin, M.; Matilla-Dueñas, A.; Soong, B.W.; Pedroso, J.L.; Barsottini, O.G.; Mitoma, H.; Tsuji, S.; Schmahmann, J.D.; Manto, M.; Rouleau, G.A.; et al. The classification of autosomal recessive cerebellar ataxias: A consensus statement from the society for research on the cerebellum and ataxias task force. Cerebellum 2019, 18, 1098–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phang, M.W.L.; Lew, S.Y.; Chung, I.; Lim, W.K.; Lim, L.W.; Wong, K.H. Therapeutic roles of natural remedies in combating hereditary ataxia: A systematic review. Chin. Med. 2021, 16, 15. [Google Scholar] [CrossRef]
- Guevara-García, M.; Gil-del Valle, L.; Velásquez-Pérez, L.; García-Rodríguez, J.C. Oxidative stress as a cofactor in spinocerebellar ataxia type 2. Redox Rep. 2012, 17, 84–89. [Google Scholar] [CrossRef] [Green Version]
- Sarva, H.; Shanker, V.L. Treatment options in degenerative cerebellar ataxia: A systematic review. Mov. Disord. Clin. Pract. 2014, 1, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Braga Neto, P.; Pedroso, J.L.; Kuo, S.H.; Marcondes Junior, C.F.; Teive, H.A.; Barsottini, O.G. Current concepts in the treatment of hereditary ataxias. Arq. Neuropsiquiatr. 2016, 74, 244–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearney, M.; Orrell, R.W.; Fahey, M.; Brassington, R.; Pandolfo, M. Pharmacological treatments for Friedreich ataxia. Cochrane Database Syst. Rev. 2016, 8, CD007791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picher-Martel, V.; Dupré, N. Current and promising therapies in autosomal recessive ataxias. CNS Neurol. Disord. Drug Targets 2018, 17, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.A.; Hancock, J.; Ghanekar, S.D.; Kuo, S.H.; Dohse, C.A.; Vega, J. Emerging therapies in Friedreich’s ataxia. Expert Rev. Neurother. 2020, 20, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Subramony, S.H.; Moscovich, M.; Ashizawa, T. Genetics and clinical features of inherited ataxias. In Movement Disorders: Genetics and Models, 2nd ed.; LeDoux, M., Ed.; Academic Press: London, UK, 2015; pp. 939–978. [Google Scholar]
- Gatti, R.; Perlman, S. Ataxia-Telangiectasia. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 1999. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK26468/ (accessed on 27 October 2016).
- Riboldi, G.M.; Samanta, D.; Frucht, S. Ataxia Telangiectasia. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK519542/ (accessed on 9 May 2022).
- Amirifar, P.; Ranjouri, M.R.; Lavin, M.; Abolhassani, H.; Yazdani, R.; Aghamohammadi, A. Ataxia-telangiectasia: Epidemiology, pathogenesis, clinical phenotype, diagnosis, prognosis and management. Expert Rev. Clin. Immunol. 2020, 16, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Biagiotti, S.; Bianchi, M.; Rossi, L.; Chessa, L.; Magnani, M. Activation of NRF2 by dexamethasone in ataxia telangiectasia cells involves KEAP1 inhibition but not the inhibition of p38. PLoS ONE 2019, 14, e0216668. [Google Scholar] [CrossRef] [Green Version]
- Blignaut, M.; Harries, S.; Lochner, A.; Huisamen, B. Ataxia telangiectasia mutated protein kinase: A potential master puppeteer of oxidative stress-induced metabolic recycling. Oxidative Med. Cell. Longev. 2021, 2021, 8850708. [Google Scholar] [CrossRef] [PubMed]
- Semlitsch, M.; Shackelford, R.E.; Zirkl, S.; Sattler, W.; Malle, E. ATM protects against oxidative stress induced by oxidized low-density lipoprotein. DNA Repair 2011, 10, 848–860. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, M.; Gatti, R.A. Pathogenesis of ataxia-telangiectasia: The next generation of ATM functions. Blood 2013, 121, 4036–4045. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Lee, H.; Lim, J.W.; Kim, H. Inhibitory effect of alpha-lipoic acid on mitochondrial dysfunction and interleukin-8 expression in interleukin-1beta-stimulated ataxia telangiectasia fibroblasts. J. Physiol. Pharmacol. 2020, 71, 155–165. [Google Scholar] [CrossRef]
- Ambrose, M.; Goldstine, J.V.; Gatti, R.A. Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Hum. Mol. Genet. 2007, 16, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Le Ber, I.; Moreira, M.C.; Rivaud-Péchoux, S.; Chamayou, C.; Ochsner, F.; Kuntzer, T.; Tardieu, M.; Saïd, G.; Habert, M.O.; Demarquay, G.; et al. Cerebellar ataxia with oculomotor apraxia type 1: Clinical and genetic studies. Brain 2003, 126, 2761–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amouri, R.; Moreira, M.C.; Zouari, M.; El Euch, G.; Barhoumi, C.; Kefi, M.; Belal, S.; Koenig, M.; Hentati, F. Aprataxin gene mutations in Tunisian families. Neurology 2004, 63, 928–929. [Google Scholar] [CrossRef]
- Habeck, M.; Zühlke, C.; Bentele, K.H.; Unkelbach, S.; Kress, W.; Bürk, K.; Schwinger, E.; Hellenbroich, Y. Aprataxin mutations are a rare cause of early onset ataxia in Germany. J. Neurol. 2004, 251, 591–594. [Google Scholar] [CrossRef]
- Castellotti, B.; Mariotti, C.; Rimoldi, M.; Fancellu, R.; Plumari, M.; Caimi, S.; Uziel, G.; Nardocci, N.; Moroni, I.; Zorzi, G.; et al. Ataxia with oculomotor apraxia type1 (AOA1): Novel and recurrent aprataxin mutations, coenzyme Q10 analyses, and clinical findings in Italian patients. Neurogenetics 2011, 12, 193–201. [Google Scholar] [CrossRef]
- Tsao, C.Y.; Paulson, G. Type 1 ataxia with oculomotor apraxia with aprataxin gene mutations in two American children. J. Child Neurol. 2005, 20, 619–620. [Google Scholar] [CrossRef]
- Quinzii, C.M.; Kattah, A.G.; Naini, A.; Akman, H.O.; Mootha, V.K.; DiMauro, S.; Hirano, M. Coenzyme Q deficiency and cerebellar ataxia associated with an aprataxin mutation. Neurology 2005, 64, 539–541. [Google Scholar] [CrossRef]
- Haj Salem, I.; Noreau, A.; Bouchard, J.P.; Dion, P.A.; Rouleau, G.A.; Dupré, N. Autosomal recessive cerebellar ataxias. In Handbook of the Cerebellum and Cerebellar Disorders; Manto, M., Gruol, D., Schmahmann, J., Koibuchi, N., Sillitoe, R., Eds.; Springer: Cham, Denmark, 2020. [Google Scholar] [CrossRef]
- Anheim, M.; Fleury, M.; Monga, B.; Laugel, V.; Chaigne, D.; Rodier, G.; Ginglinger, E.; Boulay, C.; Courtois, S.; Drouot, N.; et al. Epidemiological, clinical, paraclinical and molecular study of a cohort of 102 patients affected with autosomal recessive progressive cerebellar ataxia from Alsace, Eastern France: Implications for clinical management. Neurogenetics 2010, 11, 1–12. [Google Scholar] [CrossRef]
- Coutinho, P.; Barbot, C.; Coutinho, P. Ataxia with oculomotor apraxia type 1. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 2002. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK1456/ (accessed on 19 March 2015).
- Ahel, I.; Rass, U.; El-Khamisy, S.F.; Katyal, S.; Clements, P.M.; McKinnon, P.J.; Caldecott, K.W.; West, S.C. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 2006, 443, 713–716. [Google Scholar] [CrossRef]
- Zheng, J.; Croteau, D.L.; Bohr, V.A.; Akbari, M. Diminished OPA1 expression and impaired mitochondrial morphology and homeostasis in aprataxin-deficient cells. Nucleic Acids Res. 2019, 47, 4086–4110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavin, M.F.; Yeo, A.J.; Becherel, O.J. Senataxin protects the genome: Implications for neurodegeneration and other abnormalities. Rare Dis. 2013, 1, e25230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Ber, I.; Bouslam, N.; Rivaud-Péchoux, S.; Guimarães, J.; Benomar, A.; Chamayou, C.; Goizet, C.; Moreira, M.C.; Klur, S.; Yahyaoui, M.; et al. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: A clinical and genetic study in 18 patients. Brain 2004, 127, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tazir, M.; Ali-Pacha, L.; M’Zahem, A.; Delaunoy, J.P.; Fritsch, M.; Nouioua, S.; Benhassine, T.; Assami, S.; Grid, D.; Vallat, J.M.; et al. Ataxia with oculomotor apraxia type 2: A clinical and genetic study of 19 patients. J. Neurol. Sci. 2009, 278, 77–81. [Google Scholar] [CrossRef]
- Hammer, M.B.; El Euch-Fayache, G.; Nehdi, H.; Saidi, D.; Nasri, A.; Nabli, F.; Bouhlal, Y.; Maamouri-Hicheri, W.; Hentati, F.; Amouri, R. Clinical and molecular findings of ataxia with oculomotor apraxia type 2 (AOA2) in 5 Tunisian families. Diagn. Mol. Pathol. 2012, 21, 241–245. [Google Scholar] [CrossRef]
- Nakamura, K.; Yoshida, K.; Makishita, H.; Kitamura, E.; Hashimoto, S.; Ikeda, S. A novel nonsense mutation in a Japanese family with ataxia with oculomotor apraxia type 2 (AOA2). J. Hum. Genet. 2009, 54, 746–748. [Google Scholar] [CrossRef]
- Bohlega, S.A.; Shinwari, J.M.; Al Sharif, L.J.; Khalil, D.S.; Alkhairallah, T.S.; Al Tassan, N.A. Clinical and molecular characterization of ataxia with oculomotor apraxia patients in Saudi Arabia. BMC Med. Genet. 2011, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Bernard, V.; Minnerop, M.; Bürk, K.; Kreuz, F.; Gillessen-Kaesbach, G.; Zühlke, C. Exon deletions and intragenic insertions are not rare in ataxia with oculomotor apraxia 2. BMC Med. Genet. 2009, 10, 87. [Google Scholar] [CrossRef] [Green Version]
- Datta, N.; Hohler, A. A new SETX mutation producing AOA2 in two siblings. Int. J. Neurosci. 2013, 123, 670–673. [Google Scholar] [CrossRef]
- Suraweera, A.; Becherel, O.J.; Chen, P.; Rundle, N.; Woods, R.; Nakamura, J.; Gatei, M.; Criscuolo, C.; Filla, A.; Chessa, L.; et al. Senataxin, defective in ataxia oculomotor apraxia type 2, is involved in the defense against oxidative DNA damage. J. Cell Biol. 2007, 177, 969–979. [Google Scholar] [CrossRef] [Green Version]
- Gueven, N.; Becherel, O.J.; Howe, O.; Chen, P.; Haince, J.F.; Ouellet, M.E.; Poirier, G.G.; Waterhouse, N.; Fusser, M.; Epe, B.; et al. A novel form of ataxia oculomotor apraxia characterized by oxidative stress and apoptosis resistance. Cell Death Differ. 2007, 14, 1149–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotoda, T.; Arita, M.; Arai, H.; Inoue, K.; Yokota, T.; Fukuo, Y.; Yazaki, Y.; Yamada, N. Adult-onset spinocerebellar dysfunction caused by a mutation in the gene for the alpha-tocopherol-transfer protein. N. Engl. J. Med. 1995, 333, 1313–1318. [Google Scholar] [CrossRef] [PubMed]
- Zortea, M.; Armani, M.; Pastorello, E.; Nunez, G.F.; Lombardi, S.; Tonello, S.; Rigoni, M.T.; Zuliani, L.; Mostacciuolo, M.L.; Gellera, C.; et al. Prevalence of inherited ataxias in the province of Padua, Italy. Neuroepidemiology 2004, 23, 275–280. [Google Scholar] [CrossRef] [PubMed]
- El Euch-Fayache, G.; Bouhlal, Y.; Amouri, R.; Feki, M.; Hentati, F. Molecular, clinical and peripheral neuropathy study of Tunisian patients with ataxia with vitamin E deficiency. Brain 2014, 137, 402–410. [Google Scholar] [CrossRef]
- Elkamil, A.; Johansen, K.K.; Aasly, J. Ataxia with vitamin E deficiency in Norway. J. Mov. Disord. 2015, 8, 33–36. [Google Scholar] [CrossRef]
- Arita, M.; Sato, Y.; Miyata, A.; Tanabe, T.; Takahashi, E.; Kayden, H.J.; Arai, H.; Inoue, K. Human α-tocopherol transfer protein: cDNA cloning, expression and chromosomal localization. Biochem. J. 1995, 306, 437–443. [Google Scholar] [CrossRef]
- Copp, R.P.; Wisniewski, T.; Hentati, F.; Larnaout, A.; Hamida, M.B.; Kayden, H.J. Localization of α-tocopherol transfer protein in the brains of patients with ataxia with vitamin E deficiency and other oxidative stress related neurodegenerative disorders. Brain Res. 1999, 822, 80–87. [Google Scholar] [CrossRef]
- Sato, Y.; Arai, H.; Miyata, A.; Tokita, S.; Yamamoto, K.; Tanabe, T.; Inoue, K. Primary structure of alpha-tocopherol transfer protein from rat liver. Homology with cellular retinaldehyde-binding protein. J. Biol. Chem. 1993, 268, 17705–17710. [Google Scholar] [CrossRef]
- Müller-Schmehl, K.; Beninde, J.; Finckh, B.; Florian, S.; Dudenhausen, J.W.; Brigelius-Flohé, R.; Schuelke, M. Localization of α-tocopherol transfer protein in trophoblast, fetal capillaries’ endothelium and amnion epithelium of human term placenta. Free Radic. Res. 2004, 38, 413–420. [Google Scholar] [CrossRef]
- Bouchard, J.P.; Richter, A.; Mathieu, J.; Brunet, D.; Hudson, T.J.; Morgan, K.; Melançon, S.B. Autosomal recessive spastic ataxia of Charlevoix–Saguenay. Neuromuscul. Disord. 1998, 8, 474–479. [Google Scholar] [CrossRef]
- El Euch-Fayache, G.; Lalani, I.; Amouri, R.; Turki, I.; Ouahchi, K.; Hung, W.Y.; Belal, S.; Siddique, T.; Hentati, F. Phenotypic features and genetic findings in sacsin-related autosomal recessive ataxia in Tunisia. Arch. Neurol. 2003, 60, 982–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouhlal, Y.; El Euch-Fayeche, G.; Hentati, F.; Amouri, R. A novel SACS gene mutation in a Tunisian family. J. Mol. Neurosci. 2009, 39, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.M.; Ozgul, R.K.; Poisson, V.C.; Topaloglu, H. Private SACS mutations in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) families from Turkey. Neurogenetics 2004, 5, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Takiyama, Y.; Sakoe, K.; Mori, K.; Namekawa, M.; Shimazaki, H.; Nakano, I.; Nishizawa, M. Identification of a SACS gene missense mutation in ARSACS. Neurology 2004, 62, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Grieco, G.S.; Malandrini, A.; Comanducci, G.; Leuzzi, V.; Valoppi, M.; Tessa, A.; Palmeri, S.; Benedetti, L.; Pierallini, A.; Gambelli, S.; et al. Novel SACS mutations in autosomal recessive spastic ataxia of Charlevoix-Saguenay type. Neurology 2004, 62, 103–106. [Google Scholar] [CrossRef]
- Criscuolo, C.; Saccà, F.; De Michele, G.; Mancini, P.; Combarros, O.; Infante, J.; Garcia, A.; Banfi, S.; Filla, A.; Berciano, J. Novel mutation of SACS gene in a Spanish family with autosomal recessive spastic ataxia. Mov. Disord. 2005, 20, 1358–1361. [Google Scholar] [CrossRef]
- Vermeer, S.; Meijer, R.P.; Pijl, B.J.; Timmermans, J.; Cruysberg, J.R.; Bos, M.M.; Schelhaas, H.J.; van de Warrenburg, B.P.; Knoers, N.V.; Scheffer, H.; et al. ARSACS in the Dutch population: A frequent cause of early-onset cerebellar ataxia. Neurogenetics 2008, 9, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Breckpot, J.; Takiyama, Y.; Thienpont, B.; Van Vooren, S.; Vermeesch, J.R.; Ortibus, E.; Devriendt, K. A novel genomic disorder: A deletion of the SACS gene leading to spastic ataxia of Charlevoix-Saguenay. Eur. J. Human. Genet. 2008, 16, 1050–1054. [Google Scholar] [CrossRef]
- Ouyang, Y.; Segers, K.; Bouquiaux, O.; Wang, F.C.; Janin, N.; Andris, C.; Shimazaki, H.; Sakoe, K.; Nakano, I.; Takiyama, Y. Novel SACS mutation in a Belgian family with sacsin-related ataxia. J. Neurol. Sci. 2008, 264, 73–76. [Google Scholar] [CrossRef]
- Baets, J.; Deconinck, T.; Smets, K.; Goossens, D.; Van den Bergh, P.; Dahan, K.; Schmedding, E.; Santens, P.; Rasic, V.M.; Van Damme, P.; et al. Mutations in SACS cause atypical and late-onset forms of ARSACS. Neurology 2010, 75, 1181–1188. [Google Scholar] [CrossRef]
- Kuchay, R.A.H.; Mir, Y.R.; Zeng, X.; Hassan, A.; Musarrat, J.; Parwez, I.; Kernstock, C.; Traschütz, A.; Synofzik, M. ARSACS as a worldwide disease: Novel SACS mutations identified in a consanguineous family from the remote tribal Jammu and Kashmir Region in India. Cerebellum 2019, 18, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.P.; Rommel, N.; Oettinger, A.; Stoll, L.H.; Kraus, E.M.; Gagnon, C.; Horger, M.; Krumm, P.; Timmann, D.; Storey, E.; et al. Coordination and timing deficits in speech and swallowing in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). J. Neurol. 2018, 265, 2060–2070. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, S.; van de Warrenburg, B.P.; Kamsteeg, E.J.; Brais, B.; Synofzik, M. ARSACS. In GeneReviews® [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 2003. Available online: https://pubmed.ncbi.nlm.nih.gov/20301432/ (accessed on 2 January 2020).
- Parfitt, D.A.; Michael, G.J.; Vermeulen, E.G.; Prodromou, N.V.; Webb, T.R.; Gallo, J.M.; Cheetham, M.E.; Nicoll, W.S.; Blatch, G.L.; Chapple, J.P. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum. Mol. Genet. 2009, 18, 1556–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girard, M.; Larivière, R.; Parfitt, D.A.; Deane, E.C.; Gaudet, R.; Nossova, N.; Blondeau, F.; Prenosil, G.; Vermeulen, E.G.; Duchen, M.R.; et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl. Acad. Sci. USA 2012, 109, 1661–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duncan, E.J.; Larivière, R.; Bradshaw, T.Y.; Longo, F.; Sgarioto, N.; Hayes, M.J.; Romano, L.; Nethisinghe, S.; Giunti, P.; Bruntraeger, M.B.; et al. Altered organization of the intermediate filament cytoskeleton and relocalization of proteostasis modulators in cells lacking the ataxia protein sacsin. Hum. Mol. Genet. 2017, 26, 3130–3143. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, C.; Procaccini, C.; Meschini, M.C.; Cianflone, A.; Carbone, R.; Doccini, S.; Devos, D.; Nesti, C.; Vuillaume, I.; Pellegrino, M.; et al. Powerhouse failure and oxidative damage in autosomal recessive spastic ataxia of Charlevoix-Saguenay. J. Neurol. 2015, 262, 2755–2763. [Google Scholar] [CrossRef] [Green Version]
- Schulz, J.B.; Boesch, S.; Bürk, K.; Dürr, A.; Giunti, P.; Mariotti, C.; Pousset, F.; Schöls, L.; Vankan, P.; Pandolfo, M. Diagnosis and treatment of Friedreich ataxia: A European perspective. Nat. Rev. Neurol. 2009, 5, 222–234. [Google Scholar] [CrossRef]
- Vankan, P. Prevalence gradients of Friedreich’s ataxia and R1b haplotype in Europe co-localize, suggesting a common palaeolithic origin in the Franco-Cantabrian ice age refuge. J. Neurochem. 2013, 126, 11–20. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Gille, G.; Reichmann, H. Iron-dependent functions of mitochondria--relation to neurodegeneration. J. Neural Transm. (Vienna) 2011, 118, 349–359. [Google Scholar] [CrossRef]
- Cotticelli, M.G.; Crabbe, A.M.; Wilson, R.B.; Shchepinov, M.S. Insights into the role of oxidative stress in the pathology of Friedreich ataxia using peroxidation resistant polyunsaturated fatty acids. Redox Biol. 2013, 1, 398–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, T.E.; Yu, A.E.; Wen, Y.; Yang, S.H.; Simpkins, J.W. Estrogen prevents oxidative damage to the mitochondria in Friedreich’s ataxia skin fibroblasts. PLoS ONE 2012, 7, e34600. [Google Scholar] [CrossRef] [PubMed]
- Bolotta, A.; Pini, A.; Abruzzo, P.M.; Ghezzo, A.; Modesti, A.; Gamberi, T.; Ferreri, C.; Bugamelli, F.; Fortuna, F.; Vertuani, S.; et al. Effects of tocotrienol supplementation in Friedreich’s ataxia: A model of oxidative stress pathology. Exp. Biol. Med. (Maywood) 2020, 245, 201–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emond, M.; Lepage, G.; Vanasse, M.; Pandolfo, M. Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology 2000, 55, 1752–1753. [Google Scholar] [CrossRef]
- Schulz, J.B.; Dehmer, T.; Schöls, L.; Mende, H.; Hardt, C.; Vorgerd, M.; Bürk, K.; Matson, W.; Dichgans, J.; Beal, M.F.; et al. Oxidative stress in patients with Friedreich ataxia. Neurology 2000, 55, 1719–1721. [Google Scholar] [CrossRef]
- Piemonte, F.; Pastore, A.; Tozzi, G.; Tagliacozzi, D.; Santorelli, F.M.; Carrozzo, R.; Casali, C.; Damiano, M.; Federici, G.; Bertini, E. Glutathione in blood of patients with Friedreich’s ataxia. Eur. J. Clin. Invest. 2001, 31, 1007–1011. [Google Scholar] [CrossRef]
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The role of oxidative stress in Friedreich’s ataxia. FEBS Lett. 2018, 592, 718–727. [Google Scholar] [CrossRef]
- Van de Warrenburg, B.P.; van Gaalen, J.; Boesch, S.; Burgunder, J.M.; Dürr, A.; Giunti, P.; Klockgether, T.; Mariotti, C.; Pandolfo, M.; Riess, O. EFNS/ENS Consensus on the diagnosis and management of chronic ataxias in adulthood. Eur. J. Neurol. 2014, 21, 552–562. [Google Scholar] [CrossRef]
- Gohil, K.; Azzi, A. Reply to drug insight: Antioxidant therapy in inherited ataxias. Nat. Clin. Pract. Neurol. 2008, 4, E1–E2. [Google Scholar] [CrossRef]
- Pedroso, J.L.; Braga-Neto, P.; Abrahão, A.; Rivero, R.L.M.; Abdalla, C.; Abdala, N.; Barsottini, O.G.P. Autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS): Typical clinical and neuroimaging features in a Brazilian family. Arq. Neuropsiquiatr. 2011, 69, 288–291. [Google Scholar] [CrossRef] [Green Version]
- Lavin, M.F.; Gueven, N.; Bottle, S.; Gatti, R.A. Current and potential therapeutic strategies for the treatment of ataxia-telangiectasia. Br. Med. Bull. 2007, 81, 129–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, A.M.; Cochemé, H.M.; Smith, R.A.; Murphy, M.P. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J. Biol. Chem. 2005, 280, 21295–21312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paredes-Fuentes, A.J.; Cesar, S.; Montero, R.; Latre, C.; Genovès, J.; Martorell, L.; Cuadras, D.; Colom, H.; Pineda, M.; Del Mar O’Callaghan, M.; et al. Plasma idebenone monitoring in Friedreich’s ataxia patients during a long-term follow-up. Biomed. Pharmacother. 2021, 143, 112143. [Google Scholar] [CrossRef] [PubMed]
- Lynch, D.R.; Perlman, S.L.; Meier, T. A phase 3, double-blind, placebo-controlled trial of idebenone in Friedreich ataxia. Arch. Neurol. 2010, 67, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Lagedrost, S.J.; Sutton, M.S.; Cohen, M.S.; Satou, G.M.; Kaufman, B.D.; Perlman, S.L.; Rummey, C.; Meier, T.; Lynch, D.R. Idebenone in Friedreich ataxia cardiomyopathy-results from a 6-month phase III study (IONIA). Am. Heart. J. 2011, 161, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Meier, T.; Perlman, S.L.; Rummey, C.; Coppard, N.J.; Lynch, D.R. Assessment of neurological efficacy of idebenone in pediatric patients with Friedreich’s ataxia: Data from a 6-month controlled study followed by a 12-month open-label extension study. J. Neurol. 2012, 259, 284–291. [Google Scholar] [CrossRef]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975. [Google Scholar] [CrossRef]
- Pandolfo, M. Drug insight: Antioxidant therapy in inherited ataxias. Nat. Clin. Pract. Neurol. 2008, 4, 86–96. [Google Scholar] [CrossRef]
- Reliene, R.; Schiestl, R.H. Experimental antioxidant therapy in ataxia telangiectasia. Clin. Med. Oncol. 2008, 2, 431–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavian, N.; Mehlal, S.; Jeljeli, M.; Saidu, N.E.B.; Nicco, C.; Cerles, O.; Chouzenoux, S.; Cauvet, A.; Ait-Djoudi, M.; Chéreau, C. The Nrf2-antioxidant response element signaling pathway controls fibrosis and autoimmunity in scleroderma. Front. Immunol. 2018, 9, 1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Ward, K.; Xavier, C.; Jann, J.; Clark, A.F.; Pang, I.H.; Wu, H. The novel triterpenoid RTA 408 protects human retinal pigment epithelial cells against H2O2-induced cell injury via NF-E2-related factor 2 (Nrf2) activation. Redox Biol. 2016, 8, 98–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovchinnikov, D.A.; Withey, S.L.; Leeson, H.C.; Lei, U.W.; Sundarrajan, A.; Junday, K.; Pewarchuk, M.; Yeo, A.J.; Kijas, A.W.; Lavin, M.F.; et al. Correction of ATM mutations in iPS cells from two ataxia-telangiectasia patients restores DNA damage and oxidative stress responses. Hum. Mol. Genet. 2020, 29, 990–1001. [Google Scholar] [CrossRef]
- Biagiotti, S.; Menotta, M.; Orazi, S.; Spapperi, C.; Brundu, S.; Fraternale, A.; Bianci, M.; Rossi, L.; Chessa, L.; Magnani, M. Dexamethasone improves redox state in ataxia telangiectasia cells by promoting an NRF2-mediated antioxidant response. FEBS J. 2016, 283, 3962–3978. [Google Scholar] [CrossRef]
- Pintado-Berninches, L.; Fernandez-Varas, B.; Benitez-Buelga, C.; Manguan-Garcia, C.; Serrano-Benitez, A.; Iarriccio, L.; Carrillo, J.; Guenechea, G.; Egusquiaguirre, S.P.; Pedraz, J.L.; et al. GSE4 peptide suppresses oxidative and telomere deficiencies in ataxia telangiectasia patient cells. Cell Death Differ. 2019, 26, 1998–2014. [Google Scholar] [CrossRef]
- Inman, D.M.; Lambert, W.S.; Calkins, D.J.; Horner, P.J. α-lipoic acid antioxidant treatment limits glaucoma-related retinal ganglion cell death and dysfunction. PLoS ONE 2013, 8, e65389. [Google Scholar] [CrossRef] [Green Version]
- Kogan, M.; Jeong, H.S. Alzheimer disease. In Integrative Medicine, 4th ed.; Rakel, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 95–107. [Google Scholar] [CrossRef]
- Linker, E.; Humphreys, C. Insulin resistance and the metabolic syndrome. In Integrative Medicine, 4th ed.; Rakel, D., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 320–333. [Google Scholar] [CrossRef]
- Mendoza-Núñez, V.M.; García-Martínez, B.I.; Rosado-Pérez, J.; Santiago-Osorio, E.; Pedraza-Chaverri, J.; Hernández-Abad, V.J. The effect of 600 mg alpha-lipoic acid supplementation on oxidative stress, inflammation, and RAGE in older adults with type 2 diabetes mellitus. Oxidative Med. Cell. Longev. 2019, 2019, 3276958. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Rodríguez, D.R.; Ramírez-Solís, R.; Garza-Elizondo, M.A.; Garza-Rodríguez, M.L.; Barrera-Saldaña, H.A. Genome editing: A perspective on the application of CRISPR/Cas9 to study human diseases. Int. J. Mol. Med. 2019, 43, 1559–1574. [Google Scholar] [CrossRef] [Green Version]
- Zabirowicz, E.S.; Gan, T.J. Pharmacology of postoperative nausea and vomiting. In Pharmacology and Physiology for Anesthesia, 2nd ed.; Hemmings, H.C., Egan, T.D., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 671–692. [Google Scholar] [CrossRef]
- Leuzzi, V.; Micheli, R.; D’Agnano, D.; Molinaro, A.; Venturi, T.; Plebani, A.; Soresina, A.; Marini, M.; Leali, P.F.; Quinti, I.; et al. Positive effect of erythrocyte-delivered dexamethasone in ataxia-telangiectasia. Neurol. Neuroimmunol. NeuroInflamm. 2015, 2, e98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado-Pinilla, R.; Sánchez-Pérez, I.; Ramón Murguía, J.; Sastre, L.; Perona, R. A dyskerin motif reactivates telomerase activity in X-linked dyskeratosis congenita and in telomerase-deficient human cells. Blood 2008, 111, 2606–2614. [Google Scholar] [CrossRef] [PubMed]
- Iarriccio, L.; Manguán-García, C.; Pintado-Berninches, L.; Mancheño, J.M.; Molina, A.; Perona, R.; Sastre, L. GSE4, a small dyskerin-and GSE24.2-related peptide, induces telomerase activity, cell proliferation and reduces DNA damage, oxidative stress and cell senescence in dyskerin mutant cells. PLoS ONE 2015, 10, e0142980. [Google Scholar] [CrossRef] [PubMed]
- Pandita, T.K. ATM function and telomere stability. Oncogene 2002, 21, 611–618. [Google Scholar] [CrossRef] [Green Version]
- Opresko, P.L.; Shay, J.W. Telomere-associated aging disorders. Ageing Res. Rev. 2017, 33, 52–66. [Google Scholar] [CrossRef]
- Petrillo, S.; D’Amico, J.; La Rosa, P.; Bertini, E.S.; Piemonte, F. Targeting NRF2 for the treatment of Friedreich’s ataxia: A comparison among drugs. Int. J. Mol. Sci. 2019, 20, 5211. [Google Scholar] [CrossRef] [Green Version]
- La Rosa, P.; Petrillo, S.; Turchi, R.; Berardinelli, F.; Schirinzi, T.; Vasco, G.; Lettieri-Barbato, D.; Fiorenza, M.T.; Bertini, E.S.; Aquilano, K.; et al. The Nrf2 induction prevents ferroptosis in Friedreich’s Ataxia. Redox Biol. 2021, 38, 101791. [Google Scholar] [CrossRef]
- La Rosa, P.; Russo, M.; D’Amico, J.; Petrillo, S.; Aquilano, K.; Lettieri-Barbato, D.; Turchi, R.; Bertini, E.S.; Piemonte, F. Nrf2 induction re-establishes a proper neuronal differentiation program in Friedreich’s ataxia neural stem cells. Front. Cell. Neurosci. 2019, 13, 356. [Google Scholar] [CrossRef] [Green Version]
- Santoro, A.; Anjomani Virmouni, S.; Paradies, E.; Villalobos Coa, V.L.; Al-Mahdawi, S.; Khoo, M.; Porcelli, V.; Vozza, A.; Perrone, M.; Denora, N.; et al. Effect of diazoxide on Friedreich ataxia models. Hum. Mol. Gene 2018, 27, 992–1001. [Google Scholar] [CrossRef] [Green Version]
- Jasoliya, M.J.; McMackin, M.Z.; Henderson, C.K.; Perlman, S.L.; Cortopassi, G.A. Frataxin deficiency impairs mitochondrial biogenesis in cells, mice and humans. Hum. Mol. Genet. 2017, 26, 2627–2633. [Google Scholar] [CrossRef] [Green Version]
- Jasoliya, M.; Sacca, F.; Sahdeo, S.; Chedin, F.; Pane, C.; Brescia Morra, V.; Filla, A.; Pook, M.; Cortopassi, G. Dimethyl fumarate dosing in humans increases frataxin expression: A potential therapy for Friedreich’s ataxia. PLoS ONE 2019, 14, e0217776. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, H.; Hao, S.; Chen, J.; Wu, J.; Song, C.; Zhang, M.; Qiao, T.; Li, K. Peptide SS-31 upregulates frataxin expression and improves the quality of mitochondria: Implications in the treatment of Friedreich ataxia. Sci. Rep. 2017, 7, 9840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Cai, J.; Shen, J.; Dong, W.; Xu, L.; Fang, M.; Lin, Y.; Liu, J.; Ding, Y.; Qiao, T.; et al. SS-31 efficacy in a mouse model of Friedreich ataxia by upregulation of frataxin expression. Hum. Mol. Genet. 2022, 31, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Igoillo-Esteve, M.; Oliveira, A.F.; Cosentino, C.; Fantuzzi, F.; Demarez, C.; Toivonen, S.; Hu, A.; Chintawar, S.; Lopes, M.; Pachera, N.; et al. Exenatide induces frataxin expression and improves mitochondrial function in Friedreich ataxia. JCI Insight 2020, 5, e134221. [Google Scholar] [CrossRef] [Green Version]
- Villa, C.; Legato, M.; Umbach, A.; Riganti, C.; Jones, R.; Martini, B.; Boido, M.; Medana, C.; Facchinetti, I.; Barni, D.; et al. Treatment with ROS detoxifying gold quantum clusters alleviates the functional decline in a mouse model of Friedreich ataxia. Sci. Transl. Med. 2021, 13, eabe1633. [Google Scholar] [CrossRef] [PubMed]
- Lew, S.Y.; Yow, Y.Y.; Lim, L.W.; Wong, K.H. Antioxidant-mediated protective role of Hericium erinaceus (Bull.: Fr.) Pers. against oxidative damage in fibroblasts from Friedreich’s ataxia patient. Food Sci. Technol. 2020, 40, 264–272. [Google Scholar] [CrossRef] [Green Version]
- Codazzi, F.; Hu, A.; Rai, M.; Donatello, S.; Salerno Scarzella, F.; Mangiameli, E.; Pelizzoni, I.; Grohovaz, F.; Pandolfo, M. Friedreich ataxia-induced pluripotent stem cell-derived neurons show a cellular phenotype that is corrected by a benzamide HDAC inhibitor. Hum. Mol. Genet. 2016, 25, 4847–4855. [Google Scholar] [CrossRef] [Green Version]
- Maková, B.; Mik, V.; Lišková, B.; Gonzalez, G.; Vítek, D.; Medvedíková, M.; Monfort, B.; Ručilová, V.; Kadlecová, A.; Khirsariya, P.; et al. Cytoprotective activities of kinetin purine isosteres. Bioorg. Med. Chem. 2021, 33, 115993. [Google Scholar] [CrossRef]
- Calatrava-Ferreras, L.; Gonzalo-Gobernado, R.; Reimers, D.; Herranz, A.S.; Casarejos, M.J.; Jiménez-Escrig, A.; Regadera, J.; Velasco-Martín, J.; Vallejo-Muñoz, M.; Díaz-Gil, J.J.; et al. Liver growth factor (LGF) upregulates frataxin protein expression and reduces oxidative stress in Friedreich’s ataxia transgenic mice. Int. J. Mol. Sci. 2016, 17, 2066. [Google Scholar] [CrossRef] [Green Version]
- Khdour, O.M.; Bandyopadhyay, I.; Roy Chowdhury, S.; Visavadiya, N.P.; Hecht, S.M. Lipophilic methylene blue analogues enhance mitochondrial function and increase frataxin levels in a cellular model of Friedreich’s ataxia. Bioorg. Med. Chem. 2018, 26, 3359–3369. [Google Scholar] [CrossRef]
- Khdour, O.M.; Bandyopadhyay, I.; Visavadiya, N.P.; Roy Chowdhury, S.; Hecht, S.M. Phenothiazine antioxidants increase mitochondrial biogenesis and frataxin levels in Friedreich’s ataxia cells. MedChemComm 2018, 9, 1491–1501. [Google Scholar] [CrossRef] [PubMed]
- Roy Chowdhury, S.; Khdour, O.M.; Bandyopadhyay, I.; Hecht, S.M. Lipophilic methylene violet analogues as modulators of mitochondrial function and dysfunction. Bioorg. Med. Chem. 2017, 25, 5537–5547. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bandyopadhyay, I.; Zheng, L.; Khdour, O.M.; Hecht, S.M. Antiferroptotic activity of phenothiazine analogues: A novel therapeutic strategy for oxidative stress related disease. ACS Med. Chem. Lett. 2020, 11, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Cotticelli, M.G.; Forestieri, R.; Xia, S.; Joyasawal, S.; Lee, T.; Xu, K.; Smith III, A.B.; Huryn, D.M.; Wilson, R.B. Identification of a novel oleic acid analog with protective effects in multiple cellular models of Friedreich ataxia. ACS Chem. Neurosci. 2020, 11, 2535–2542. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Baccaro, A.; Esteras, N.; Giunti, P. Novel Nrf2-inducer prevents mitochondrial defects and oxidative stress in Friedreich’s ataxia models. Front. Cell. Neurosci. 2018, 12, 188. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pascau, L.; Britti, E.; Calap-Quintana, P.; Dong, Y.N.; Vergara, C.; Delaspre, F.; Medina-Carbonero, M.; Tamarit, J.; Pallardó, F.V.; Gonzalez-Cabo, P.; et al. PPAR gamma agonist leriglitazone improves frataxin-loss impairments in cellular and animal models of Friedreich ataxia. Neurobiol. Dis. 2021, 148, 105162. [Google Scholar] [CrossRef]
- Dong, Y.N.; McMillan, E.; Clark, E.M.; Lin, H.; Lynch, D.R. GRP75 overexpression rescues frataxin deficiency and mitochondrial phenotypes in Friedreich ataxia cellular models. Hum. Mol. Genet. 2019, 28, 1594–1607. [Google Scholar] [CrossRef]
- Petrillo, S.; Piermarini, E.; Pastore, A.; Vasco, G.; Schirinzi, T.; Carrozzo, R.; Bertini, E.; Piemonte, F. Nrf2-inducers counteract neurodegeneration in frataxin-silenced motor neurons: Disclosing new therapeutic targets for Friedreich’s ataxia. Int. J. Mol. Sci. 2017, 18, 2173. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, D.; Catteruccia, M.; Piemonte, F.; Pastore, A.; Tozzi, G.; Dionisi-Vici, C.; Pontrelli, G.; Corsetti, T.; Livadiotti, S.; Kheifets, V.; et al. EPI-743 reverses the progression of the pediatric mitochondrial disease–genetically defined Leigh syndrome. Mol. Genet. Metab. 2012, 107, 383–388. [Google Scholar] [CrossRef]
- Lynch, D.R.; Willi, S.M.; Wilson, R.B.; Cotticelli, M.G.; Brigatti, K.W.; Deutsch, E.C.; Kucheruk, O.; Shrader, W.; Rioux, P.; Miller, G.; et al. A0001 in Friedreich ataxia: Biochemical characterization and effects in a clinical trial. Mov. Disord. 2012, 27, 1026–1033. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campese, V.M.; Lakdawala, R. The challenges of blood pressure control in dialysis patients. Recent Adv. Cardiovasc. Drug Discov. 2015, 10, 34–59. [Google Scholar] [CrossRef] [PubMed]
- Virgili, N.; Mancera, P.; Wappenhans, B.; Sorrosal, G.; Biber, K.; Pugliese, M.; Espinosa-Parrilla, J.F. KATP channel opener diazoxide prevents neurodegeneration: A new mechanism of action via antioxidative pathway activation. PLoS ONE 2013, 8, e75189. [Google Scholar] [CrossRef] [PubMed]
- Arnon, S.; Lagerev, E.; Herzlich, J.; Eliakim, A.; Litmanovitz, I. Diazoxide treatment for persistent hypoglycemia in a small for gestational age preterm infant with adequate low insulin levels. Case Rep. Perinat. Med. 2014, 3, 83–85. [Google Scholar] [CrossRef]
- Gómez-Barroso, M.; Moreno-Calderón, K.M.; Sánchez-Duarte, E.; Cortés-Rojo, C.; Saavedra-Molina, A.; Rodríguez-Orozco, A.R.; Montoya-Pérez, R. Diazoxide and exercise enhance muscle contraction during obesity by decreasing ROS levels, lipid peroxidation, and improving glutathione redox status. Antioxidants 2020, 9, 1232. [Google Scholar] [CrossRef]
- Kempe, P.R.; Chiarotto, G.B.; Barraviera, B.; Ferreira, R.S.; de Oliveira, A.L. Neuroprotection and immunomodulation by dimethyl fumarate and a heterologous fibrin biopolymer after ventral root avulsion and reimplantation. J. Venom. Anim. Toxins Incl. Trop. Dis. 2020, 26, e20190093. [Google Scholar] [CrossRef]
- Garcia-Mesa, Y.; Xu, H.N.; Vance, P.; Gruenewald, A.L.; Garza, R.; Midkiff, C.; Alvarez-Hernandez, X.; Irwin, D.J.; Gill, A.J.; Kolson, D.L. Dimethyl fumarate, an approved multiple sclerosis treatment, reduces brain oxidative stress in SIV-infected rhesus macaques: Potential therapeutic repurposing for HIV neuroprotection. Antioxidants 2021, 10, 416. [Google Scholar] [CrossRef]
- Davinelli, S.; De Stefani, D.; De Vivo, I.; Scapagnini, G. Polyphenols as caloric restriction mimetics regulating mitochondrial biogenesis and mitophagy. Trends Endocrinol. Metab. 2020, 31, 536–550. [Google Scholar] [CrossRef]
- Szeto, H.H.; Liu, S.; Soong, Y.; Wu, D.; Darrah, S.F.; Cheng, F.Y.; Zhao, Z.; Ganger, M.; Tow, C.Y.; Seshan, S.V. Mitochondria-targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J. Am. Soc. Nephrol. 2011, 22, 1041–1052. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S. Advances in the treatment of mitochondrial epilepsies. Epilepsy Behav. 2019, 101, 106546. [Google Scholar] [CrossRef]
- Mitchell, W.; Ng, E.A.; Tamucci, J.D.; Boyd, K.J.; Sathappa, M.; Coscia, A.; Pan, M.; Han, X.; Eddy, N.A.; May, E.R.; et al. The mitochondria-targeted peptide SS-31 binds lipid bilayers and modulates surface electrostatics as a key component of its mechanism of action: SS-31 modulates lipid bilayer surface electrostatics. J. Biol. Chem. 2020, 295, 7452–7469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborti, C.K. Exenatide: A new promising antidiabetic agent. Indian J. Pharm. Sci. 2010, 72, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonia, T.A.; Sharma, C.P. Diabetes mellitus—An overview. In Oral Delivery of Insulin, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 1–57. [Google Scholar] [CrossRef]
- Santiago-Gonzalez, B.; Monguzzi, A.; Azpiroz, J.M.; Prato, M.; Erratico, S.; Campione, M.; Lorenzi, R.; Pedrini, J.; Santambrogio, C.; Torrente, Y.; et al. Permanent excimer superstructures by supramolecular networking of metal quantum clusters. Science 2016, 353, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Santiago-Gonzalez, B.; Monguzzi, A.; Caputo, M.; Villa, C.; Prato, M.; Santambrogio, C.; Torrente, Y.; Meinardi, F.; Brovelli, S. Metal nanoclusters with synergistically engineered optical and buffering activity of intracellular reactive oxygen species by compositional and supramolecular design. Sci. Rep. 2017, 7, 5976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, P.S.; Fung, M.-L.; Wong, K.H.; Lim, L.W. Therapeutic potential of Hericium erinaceus for depressive disorder. Int. J. Mol. Sci. 2020, 21, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, P.S.; Khairuddin, S.; Tse, A.C.K.; Hiew, L.F.; Lau, C.L.; Tipoe, G.L.; Fung, M.-L.; Wong, K.H.; Lim, L.W. Hericium erinaceus potentially rescues behavioural motor deficits through ERK-CREB-PSD95 neuroprotective mechanisms in rat model of 3-acetylpyridine-induced cerebellar ataxia. Sci. Rep. 2020, 10, 14945. [Google Scholar] [CrossRef]
- Chong, P.S.; Poon, C.H.; Roy, J.; Tsui, K.C.; Lew, S.Y.; Phang, M.W.L.; Tan, R.J.Y.; Cheng, P.G.; Fung, M.L.; Wong, K.H.; et al. Neurogenesis-dependent antidepressant-like activity of Hericium erinaceus in an animal model of depression. Chin. Med. 2021, 16, 132. [Google Scholar] [CrossRef]
- Lew, S.Y.; Lim, S.H.; Lim, L.W.; Wong, K.H. Neuroprotective effects of Hericium erinaceus (Bull.: Fr.) Pers. against high-dose corticosterone-induced oxidative stress in PC-12 cells. BMC Complement. Med. Ther. 2020, 20, 340. [Google Scholar] [CrossRef]
- Wong, K.H.; Naidu, M.; David, P.; Abdulla, M.A.; Abdullah, N.; Kuppusamy, U.R.; Sabaratnam, V. Peripheral nerve regeneration following crush injury to rat peroneal nerve by aqueous extract of medicinal mushroom Hericium erinaceus (Bull.: Fr.) Pers. (Aphyllophoromycetideae). Evid. Based Complement. Altern 2011, 2011, 580752. [Google Scholar] [CrossRef] [Green Version]
- Samberkar, S.; Gandhi, S.; Naidu, M.; Wong, K.H.; Raman, J.; Sabaratnam, V. Lion’s mane, Hericium erinaceus and Tiger Milk, Lignosus rhinocerotis (higher basidiomycetes) medicinal mushrooms stimulate neurite outgrowth in dissociated cells of brain, spinal cord, and retina: An in vitro study. Int. J. Med. Mushrooms 2015, 17, 1047–1054. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.H.; Ng, C.C.; Kanagasabapathy, G.; Yow, Y.Y.; Sabaratnam, V. An overview of culinary and medicinal mushrooms in neurodegeneration and neurotrauma research. Int. J. Med. Mushrooms 2017, 19, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.H.; Sabaratnam, V.; Abdullah, N.; Kuppusamy, U.R.; Naidu, M. Effects of cultivation techniques and processing on antimicrobial and antioxidant activities of Hericium erinaceus (Bull.: Fr.) Pers. extracts. Food Technol. Biotechnol. 2009, 47, 47–55. [Google Scholar]
- Sheikh, S.; Bekheet, M.; Olzscha, H.; La Thangue, N.B. Predicting and monitoring responses to epigenetic drugs. In Drug Discov. Cancer Epigenetics; Egger, G., Arimodo, P., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 373–406. [Google Scholar] [CrossRef]
- Qiu, X.; Rong, X.; Yang, J.; Lu, Y. Evaluation of the antioxidant effects of different histone deacetylase inhibitors (HDACis) on human lens epithelial cells (HLECs) after UVB exposure. BMC Ophthalmol. 2019, 19, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgin, S.C.; Grewal, S.I. Heterochromatin: Silence is golden. Curr. Biol. 2003, 13, R895–R898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, D.; Jenssen, K.; Burnett, R.; Soragni, E.; Perlman, S.L.; Gottesfeld, J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2006, 2, 551–558. [Google Scholar] [CrossRef]
- Chou, C.J.; Herman, D.; Gottesfeld, J.M. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J. Biol. Chem. 2008, 283, 35402–35409. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Soragni, E.; Chou, C.J.; Herman, D.; Plasterer, H.L.; Rusche, J.R.; Gottesfeld, J.M. Chemical probes identify a role for histone deacetylase 3 in Friedreich’s ataxia gene silencing. Chem. Biol. 2009, 16, 980–989. [Google Scholar] [CrossRef] [Green Version]
- Hertz, N.T.; Berthet, A.; Sos, M.L.; Thorn, K.S.; Burlingame, A.L.; Nakamura, K.; Shokat, K.M. A neo-substrate that amplifies catalytic activity of Parkinson’s-disease-related kinase PINK1. Cell 2013, 154, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Bowie, L.E.; Maiuri, T.; Alpaugh, M.; Gabriel, M.; Arbez, N.; Galleguillos, D.; Hung, C.L.; Patel, S.; Xia, J.; Hertz, N.T.; et al. N6-Furfuryladenine is protective in Huntington’s disease models by signaling huntingtin phosphorylation. Proc. Natl. Acad. Sci. USA 2018, 115, e7081–e7090. [Google Scholar] [CrossRef] [Green Version]
- Gonzalo-Gobernado, R.; Reimers, D.; Herranz, A.S.; Díaz-Gil, J.J.; Osuna, C.; Asensio, M.J.; Baena, S.; Rodríguez-Serrano, M.; Bazán, E. Mobilization of neural stem cells and generation of new neurons in 6-OHDA–lesioned rats by intracerebroventricular infusion of liver growth factor. J. Histochem. Cytochem. 2009, 57, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Gonzalo-Gobernado, R.; Calatrava-Ferreras, L.; Perucho, J.; Reimers, D.; Casarejos, M.J.; Herranz, A.S.; Jimenez-Escrig, A.; Diaz-Gil, J.J.; Bazan, E. Liver growth factor as a tissue regenerating factor in neurodegenerative diseases. Recent Pat. CNS Drug Discov. 2014, 9, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalo-Gobernado, R.; Calatrava-Ferreras, L.; Reimers, D.; Herranz, A.S.; Rodríguez-Serrano, M.; Miranda, C.; Jiménez-Escrig, A.; Díaz-Gil, J.J.; Bazán, E. Neuroprotective activity of peripherally administered liver growth factor in a rat model of Parkinson’s disease. PLoS ONE 2013, 8, e67771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalo-Gobernado, R.; Perucho, J.; Vallejo-Muñoz, M.; Casarejos, M.J.; Reimers, D.; Jiménez-Escrig, A.; Gómez, A.; Ulzurrun de Asanza, G.M.; Bazán, E. Liver growth factor “LGF” as a therapeutic agent for Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 9201. [Google Scholar] [CrossRef]
- Reimers, D.; Herranz, A.S.; Díaz-Gil, J.J.; Lobo, M.V.; Paíno, C.L.; Alonso, R.; Asensio, M.J.; Gonzalo-Gobernado, R.; Bazán, E. Intrastriatal infusion of liver growth factor stimulates dopamine terminal sprouting and partially restores motor function in 6-hydroxydopamine-lesioned rats. J. Histochem. Cytochem. 2006, 54, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Snyder, M.; Gangadhara, S.; Brohl, A.S.; Ludlow, S.; Nanjappa, S. Serotonin syndrome complicating treatment of ifosfamide neurotoxicity with methylene blue. Cancer Control 2017, 24, 1073274817729070. [Google Scholar] [CrossRef] [Green Version]
- Baddeley, T.C.; McCaffrey, J.; Storey, J.M.; Cheung, J.K.; Melis, V.; Horsley, D.; Harrington, C.R.; Wischik, C.M. Complex disposition of methylthioninium redox forms determines efficacy in tau aggregation inhibitor therapy for Alzheimer’s disease. J. Pharmacol. Exp. Ther. 2015, 352, 110–118. [Google Scholar] [CrossRef]
- Tucker, D.; Lu, Y.; Zhang, Q. From mitochondrial function to neuroprotection-an emerging role for methylene blue. Mol. Neurobiol. 2018, 55, 5137–5153. [Google Scholar] [CrossRef]
- Mokhtari, V.; Afsharian, P.; Shahhoseini, M.; Kalantar, S.M.; Moini, A.A. Review on various uses of N-acetyl cysteine. Cell J. 2017, 19, 11–17. [Google Scholar] [CrossRef]
- Shramko, V.S.; Polonskaya, Y.V.; Kashtanova, E.V.; Stakhneva, E.M.; Ragino, Y.I. The short overview on the relevance of fatty acids for human cardiovascular disorders. Biomolecules 2020, 10, 1127. [Google Scholar] [CrossRef]
- Probst, B.L.; Trevino, I.; McCauley, L.; Bumeister, R.; Dulubova, I.; Wigley, W.C.; Ferguson, D.A. RTA 408, A novel synthetic triterpenoid with broad anticancer and anti-inflammatory activity. PLoS ONE 2015, 10, e0122942. [Google Scholar] [CrossRef]
- Shekh-Ahmad, T.; Eckel, R.; Dayalan Naidu, S.; Higgins, M.; Yamamoto, M.; Dinkova-Kostova, A.T.; Kovac, S.; Abramov, A.Y.; Walker, M.C. KEAP1 inhibition is neuroprotective and suppresses the development of epilepsy. Brain 2018, 141, 1390–1403. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Xie, Z.; Hu, B.; Zhang, B.; Ma, Y.; Pan, X.; Huang, H.; Wang, J.; Zhao, X.; Jie, Z.; et al. The Nrf2 activator RTA-408 attenuates osteoclastogenesis by inhibiting STING dependent NF-κb signaling. Redox Biol. 2020, 28, 101309. [Google Scholar] [CrossRef] [PubMed]
- Baldelli, S.; Aquilano, K.; Ciriolo, M.R. PGC-1 α buffers ROS-mediated removal of mitochondria during myogenesis. Cell Death Dis. 2014, 5, e1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, inflammation, and oxidative stress: An integrative view in metabolism. Oxidative Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef] [Green Version]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane: Translational research from laboratory bench to clinic. Nutr. Rev. 2013, 71, 709–726. [Google Scholar] [CrossRef]
- Fofaria, N.M.; Ranjan, A.; Kim, S.H.; Srivastava, S.K. Mechanisms of the anticancer effects of isothiocyanates. In Enzyme; Bathaie, Z., Tamanoi, F., Eds.; Academic Press: Cambridge, MA, USA, 2015; pp. 111–137. [Google Scholar] [CrossRef]
- Lindroth, A.M.; Park, J.H.; Yoo, Y.; Park, Y.J. Nutriepigenomics: Personalized nutrition meets epigenetics. In Personalized Epigenetics, 1st ed.; Tollefsbol, T.O., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 313–347. [Google Scholar] [CrossRef]
- Pradhan, N.; Kar, S.; Parbin, S.; Sengupta, D.; Deb, M.; Das, L.; Patra, S.K. Epigenetic dietary interventions for prevention of cancer. In Epigenetics of Cancer Prevention; Bishayee, A., Bhatia, D., Eds.; Academic Press: Cambridge, MA, USA, 2019; pp. 23–48. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Clark, E.; Johnson, J.; Dong, Y.N.; Mercado-Ayon, E.; Warren, N.; Zhai, M.; McMillan, E.; Salovin, A.; Lin, H.; Lynch, D.R. Role of frataxin protein deficiency and metabolic dysfunction in Friedreich ataxia, an autosomal recessive mitochondrial disease. Neuronal Signal. 2018, 2, NS20180060. [Google Scholar] [CrossRef]
- Lynch, D.R.; Farmer, J.; Hauser, L.; Blair, I.A.; Wang, Q.Q.; Mesaros, C.; Snyder, N.; Boesch, S.; Chin, M.; Delatycki, M.B.; et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2018, 6, 15–26. [Google Scholar] [CrossRef]
- Galeano, D.; Li, S.; Gerstein, M.; Paccanaro, A. Predicting the frequencies of drug side effects. Nat. Commun. 2020, 11, 4575. [Google Scholar] [CrossRef]
- John, P.A.; Wong, K.H.; Naidu, M.; Sabaratnam, V.; David, P. Combination effects of curcumin and aqueous extract of Lignosus rhinocerotis mycelium on neurite outgrowth stimulation activity in PC-12 cells. Nat. Prod. Commun. 2013, 8, 711–714. [Google Scholar] [CrossRef] [Green Version]
- Beutler, J.A. Natural products as a foundation for drug discovery. Curr. Protoc. Pharmacol. 2019, 86, e67. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.-M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faouzi, A.; Roullin, V.G. Think big, start small: How nanomedicine could alleviate the burden of rare CNS diseases. Pharmaceuticals 2021, 14, 109. [Google Scholar] [CrossRef] [PubMed]
- Vannocci, T.; Dinarelli, S.; Girasole, M.; Pastore, A.; Longo, G.A. A new tool to determine the cellular metabolic landscape: Nanotechnology to the study of Friedreich’s ataxia. Sci. Rep. 2019, 9, 19282. [Google Scholar] [CrossRef]
- Uceda, A.B.; Donoso, J.; Frau, J.; Vilanova, B.; Adrover, M. Frataxins emerge as new players of the intracellular antioxidant machinery. Antioxidants 2021, 10, 315. [Google Scholar] [CrossRef]
- Chiang, S.; Huang, M.L.H.; Richardson, D.R. Treatment of dilated cardiomyopathy in a mouse model of Friedreich’s ataxia using N-acetylcysteine and identification of alterations in microRNA expression that could be involved in its pathogenesis. Pharmacol. Res. 2020, 159, 104994. [Google Scholar] [CrossRef]
- Ribai, P.; Pousset, F.; Tanguy, M.-L.; Rivaud-Pechoux, S.; Le Ber, I.; Gasparini, F.; Charles, P.; Béraud, A.S.; Schmitt, M.; Koenig, M.; et al. Neurological, cardiological, and oculomotor progression in 104 patients with Friedreich ataxia during long-term follow-up. Arch. Neurol. 2007, 64, 558–664. [Google Scholar] [CrossRef]
- Singh, S.V.; Srivastava, S.K.; Choi, S.; Lew, K.L.; Antosiewicz, J.; Xiao, D.; Zeng, Y.; Watkins, S.C.; Johnson, C.S.; Trump, D.L.; et al. Sulforaphane-induced cell death in human prostate cancer cells is initiated by reactive oxygen species. J. Biol. Chem. 2005, 280, 19911–19924. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Lee, S.H. Sulforaphane induces antioxidative and antiproliferative responses by generating reactive oxygen species in human bronchial epithelial BEAS-2B cells. J. Korean Med. Sci. 2011, 26, 1474–1482. [Google Scholar] [CrossRef] [Green Version]
- Spacey, S.; Szczygielski, B.; Young, S.; Hukin, J.; Selby, K.; Snutch, T. Malaysian siblings with Friedreich ataxia and chorea: A novel deletion in the frataxin gene. Can. J. Neurol. Sci. 2004, 31, 383–386. [Google Scholar] [CrossRef] [Green Version]
- Salehi, M.H.; Kamalidehghan, B.; Houshmand, M.; Yong Meng, G.; Sadeghizadeh, M.; Aryani, O.; Nafissi, S. Gene expression profiling of mitochondrial oxidative phosphorylation (OXPHOS) complex I in Friedreich ataxia (FRDA) patients. PLoS ONE 2014, 9, e94069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cendelin, J. From mice to men: Lessons from mutant ataxic mice. Cerebellum Ataxias 2014, 1, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.B.; Krieger, K.; Khan, F.M.; Huffman, W.; Chang, M.; Naik, A.; Yongle, R.; Hameed, I.; Krieger, K.; Girardi, L.N.; et al. The current state of animal models in research: A review. Int. J. Surg. 2019, 72, 9–13. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Therapy | Model | Finding | Mode of Action | Reference |
|---|---|---|---|---|
α-lipoic acid | IL-1β-induced oxidative stress in human fibroblasts | Attenuation of ROS production Restoration of mitochondrial function | ↓ mRNA expression and protein level of IL-8 ↓ NF-kB activation | [26] |
| CRISPR/Cas9 | H2O2-induced oxidative stress and 2DG-induced metabolic stress in human iPSCs | Restoration of DNA damage response, cell cycle control and ATM kinase | ↑ KAP1 ↓ γ-H2A.X and cleaved caspase 3 | [103] |
Dexamethasone | Human lymphoblasts | Attenuation of ROS production ↑ reduced GSH and total GSH | ↑ mRNA expression of GCLC, GCLM, GSS and GSR ↑ mRNA expression and protein level of NRF2 | [104] |
| Human lymphoblasts | Activation of NRF2 | ↓ mRNA expression and protein level of KEAP1 | [22] | |
| Genetic suppressor element 4 | Human fibroblasts and lymphoblasts | Protection against apoptosis Attenuation of oxidative stress-induced DNA damage Reconstitution of telomerase activity and elongation | ↑ OGG1 ↓ mRNA expression of IL-6, SOD1 and TERT ↓ 8-oxoG and p38 phosphorylation | [105] |
| Therapy | Model | Finding | Mode of Action | Reference |
|---|---|---|---|---|
α-tocotrienol quinone | Human fibroblasts | Modulation of NRF2 | ↑ mRNA expression and protein levels of frataxin, NRF2, NQO1, HO-1 and GCL Regulation of KEAP1-DJ-1-p62 | [117] |
| Human fibroblasts | Protection against ferroptosis Attenuation of lipid peroxidation Restoration of mitochondrial function | ↑ mRNA expression of FXN, SOD2 and GPX4, GCL ↑ mRNA expression and protein level of NRF2 | [118] | |
| NSCs derived from FXN KIKO mice | Attenuation of ROS production Restoration of morphology, differentiation and phenotypic defects | ↑ mRNA expression and protein levels of NRF2, NQO1 and HO-1 | [119] | |
Diazoxide | Human lymphoblasts | Protection against oxidative stress | ↑ mRNA expression and protein level of frataxin ↑ mTOR-S6K and nuclear translocation of NRF2 | [120] |
| YG8sR mice | ↑ fine motor coordination and balance, and stride length ↑ aconitase ↓protein oxidation in brain, liver and pancreas | ↑ mRNA expression and protein level of frataxin in cerebellum and heart ↑ mRNA expression of NRF2 in cerebellum and heart | ||
Dimethyl fumarate | Human fibroblasts | Modulation of NRF2 ↑ GSH | ↑ mRNA expression and protein levels of frataxin, NRF2, NQO1, HO-1 and GCL Regulation of KEAP1-DJ-1-p62 | [117] |
| Human fibroblasts and blood FXN KIKO mice | ↑ mitochondrial biogenesis | ↑ mRNA expression and protein of frataxin ↑ mRNA expression of NRF1 and mTFA | [121] | |
| Human fibroblasts | ↑ mitochondrial biogenesis | ↑ mRNA expression and protein level of frataxin | [122] | |
| Human lymphoblasts YG8 and FXN KIKO mice | ↑ protein level of frataxin | ↑ mRNA expression of frataxin ↓ R-loop formation and transcriptional silencing restricted FXN locus | ||
Elamipretide | Human fibroblasts and lymphoblasts | Attenuation of ROS production Restoration of MMP and mitochondrial morphology ↑ aconitase, complex II and III, SOD and CAT | ↑ frataxin, ATP and NAD+/NADH | [123] |
| Y47 and YG8R mice | ↑ motor function ↓ cytoplasmic vacuolization in DRG and lesions in the dentate nuclei Restoration of damaged myelin in the spinal cord | ↑ mRNA expression of frataxin | [124] | |
Exenatide | Human iPSC-derived β cells and sensory neurons | Restoration of mitochondrial function | ↑ frataxin, aconitase, NDUFS3, OGDH and PDH | [125] |
| FXN KIKO mice | ↑ glucose tolerance, β cell function and insulin secretion Neuroprotection against DRG | ↑ protein level of frataxin and ISC-containing protein ferrochelatase in cerebellum and cerebrum | ||
| Gold cluster superstructure | Human MSCs | Attenuation of ROS production Restoration of mitochondrial function and bioenergetic capacity, ATP, ETC function and MMP dissipation | ↑ frataxin Modulation of autophagic flux, frataxin-related proteins and dynamin-related proteins | [126] |
| YG8sR mice | Restoration of motor deficits, neuromuscular function, cardiac contractility, mitochondrial and ETC function Attenuation of ROS production ↑ ATP ↓ collagen deposition in the skeletal muscle and cardiac fibrosis | ↑ NSF and PGC-1α ↓ 4-HNE, 8-oxodG, LC3-II/LC3-1 and PPARγ Activation of NRF2-ARE | ||
| Hericium erinaceus | BSO-induced oxidative stress in human fibroblasts | Restoration of GSH/GSSG and plasma membrane integrity Prevention of apoptosis | NE | [127] |
Histone deacetylases inhibitors | Human iPSCs | Protection against oxidative stress | ↑ frataxin, ISCUs, aconitase 2, NDUFS3, OGDH and PDH ↓ ROS and SOD2 | [128] |
Kinetin | BSO-induced oxidative stress in human fibroblasts | Protection against secondary effects of frataxin deficiency | NE | [129] |
| Liver growth factor | YG8R mice | Restoration of motor coordination Attenuation of neuronal apoptosis Reversal of cardiac hypertrophy ↑ GSH ↓ GSSG | ↑ frataxin, complex IV and cytochrome c ↑ phospho-Akt/Akt and Bcl2/Bax | [130] |
Methylene blue analogs | BSO-induced oxidative stress in human fibroblasts Rotenone-induced oxidative stress in human lymphocytes | Attenuation of ROS production Restoration of mitochondrial function and biogenesis ↑ aconitase, ATP and MMP | ↑ frataxin and complex I | [131] |
| Methylene violet analogs Compound 1  Compound 2  Compound 4b  Compound 6b  | Diethyl maleate-induced oxidative stress in human lymphoblasts | Restoration of mitochondrial biogenesis ↑ aconitase, ATP and MMP | ↑ frataxin Regulation of SDH-A and COX-1 | [132] |
| Methylene violet analogs Compound 4  Compound 5  | BSO-induced oxidative stress in human fibroblasts Rotenone-induced oxidative stress in human lymphocytes | Attenuation of ROS production Restoration of mitochondrial function ↑ ATP and MMP | ↑ NADH:ubiquinone oxidoreductase (complex I) | [133] |
| Methylene violet analogs Compound 1b  Compound 2b  Compound 3b  Compound 4b  Compound 5b  | Erastin-induced oxidative stress in human fibroblasts RSL3-induced oxidative stress in human lymphocytes | Protection against ferroptosis Restoration of mitochondrial biogenesis | Regulation of AMPK ↑ pAMPK/AMPK | [134] |
N-acetylcysteine | Human fibroblasts | Modulation of NRF2 | ↑ mRNA expression and protein levels of frataxin, NRF2, NQO1, HO-1 and GCL Regulation of KEAP1-DJ-1-p62 | [117] |
Oleic acid | Erastin-induced ferroptosis in murine fibroblasts | Protection against ferroptosis | NE | [135] |
| Fatty acids and fatty-acid analogs Oleic acid derivatives  Cis-Vaccenic acid (7)  Petroselinic acid (8)  Gadoleic acid (10)  Erucic acid (11)  Heptadecenoic acid (12)  Palmitoleic acid (13)  | FAC- and BSO-induced oxidative stress in murine fibroblasts | Protection against cytotoxicity | NE | |
(R)-24 (S)-24  | FAC- and BSO-induced oxidative stress in human TERT-immortalized fibroblasts RSL-3-induced oxidative stress in siFXN-1 myoblast | Protection against cytotoxicity Protection against ferroptosis | NE | |
(R)-24 | Erastin-induced ferroptosis in human fibroblasts | Protection against ferroptosis | NE | |
Omaveloxolone | H2O2-induced oxidative stress in human fibroblasts FXN KIKO and Y8GR mice | Prevention of complex I inhibition Attenuation of ROS production and lipid peroxidation Restoration of GSH, mitochondrial function and MMP dissipation | ↑ mRNA expression and protein level of NRF2 ↑ NADH pool ↓ NADH redox state Regulation of KEAP1 | [136] |
| Human fibroblasts | Modulation of NRF2 ↑ GSH | ↑ mRNA expression and protein level of NRF2, NQO1, HO-1 and GCL Regulation of KEAP1-DJ-1-p62 | [117] | |
| Peroxisome proliferator-activated receptor gamma agonist Leriglitazone  | Human fibroblasts | Restoration of mitochondrial function and biogenesis | ↑ frataxin, PGC-1α and GRP75 | [137] |
| DRG sensory neurons | Attenuation of formation of neurofilament aggregates Restoration of mitochondrial function and calcium homeostasis | ↑ frataxin and NCLX ↓ cleavage of α-fodrin | ||
| YG8sR mice | Restoration of motor function | NE | ||
| Peroxisome proliferator-activated receptor gamma agonist GRP75 | Cortical homogenates, primary cortical neurons and HEK293 cells | ↑ accessibility of frataxin to MPP | ↑ ISCU2 | [138] |
| Human fibroblasts and HEK293 | Restoration of frataxin, mitochondrial network and ATP | ↑ frataxin and ISCU2 | ||
Sulforaphane | FXN-silenced NSC34 motor neurons | ↑ reduced GSH ↓ GSSG Reorganization of network formation and stimulation of neurite outgrowth | ↑ mRNA expression and protein levels of frataxin, NRF2, NQO1, NQO1, Cu/Zn SOD, SOD1, SOD2, GCL-C and GCL-M | [139] |
| Human fibroblasts | Modulation of NRF2 ↑ GSH | ↑ mRNA expression and protein levels of frataxin, NRF2, NQO1, HO-1 and GCL Regulation of KEAP1-DJ-1-p62 | [117] | |
| NSCs derived from FXN KIKO mice | Attenuation of ROS production Restoration of morphology, differentiation and phenotypic defects | ↑ mRNA expression and protein levels of NRF2, NQO1 and HO-1 | [119] | |
| Human fibroblasts and blood FXN KIKO mice | Protection against ferroptosis Attenuation of lipid peroxidation Regulation of mitochondrial morphology | ↑ mRNA expression of FXN, SOD2, GPX4 and GCL ↑ mRNA expression and protein level of NRF2 | [118] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lew, S.Y.; Phang, M.W.L.; Chong, P.S.; Roy, J.; Poon, C.H.; Yu, W.S.; Lim, L.W.; Wong, K.H. Discovery of Therapeutics Targeting Oxidative Stress in Autosomal Recessive Cerebellar Ataxia: A Systematic Review. Pharmaceuticals 2022, 15, 764. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15060764
Lew SY, Phang MWL, Chong PS, Roy J, Poon CH, Yu WS, Lim LW, Wong KH. Discovery of Therapeutics Targeting Oxidative Stress in Autosomal Recessive Cerebellar Ataxia: A Systematic Review. Pharmaceuticals. 2022; 15(6):764. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15060764
Chicago/Turabian StyleLew, Sze Yuen, Michael Weng Lok Phang, Pit Shan Chong, Jaydeep Roy, Chi Him Poon, Wing Shan Yu, Lee Wei Lim, and Kah Hui Wong. 2022. "Discovery of Therapeutics Targeting Oxidative Stress in Autosomal Recessive Cerebellar Ataxia: A Systematic Review" Pharmaceuticals 15, no. 6: 764. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15060764