
Design, Synthesis and Molecular Docking of Novel Acetophenone-1,2,3-Triazoles Containing Compounds as Potent Enoyl-Acyl Carrier Protein Reductase (InhA) Inhibitors

 ,
,  , , , and
, , , and
Abstract
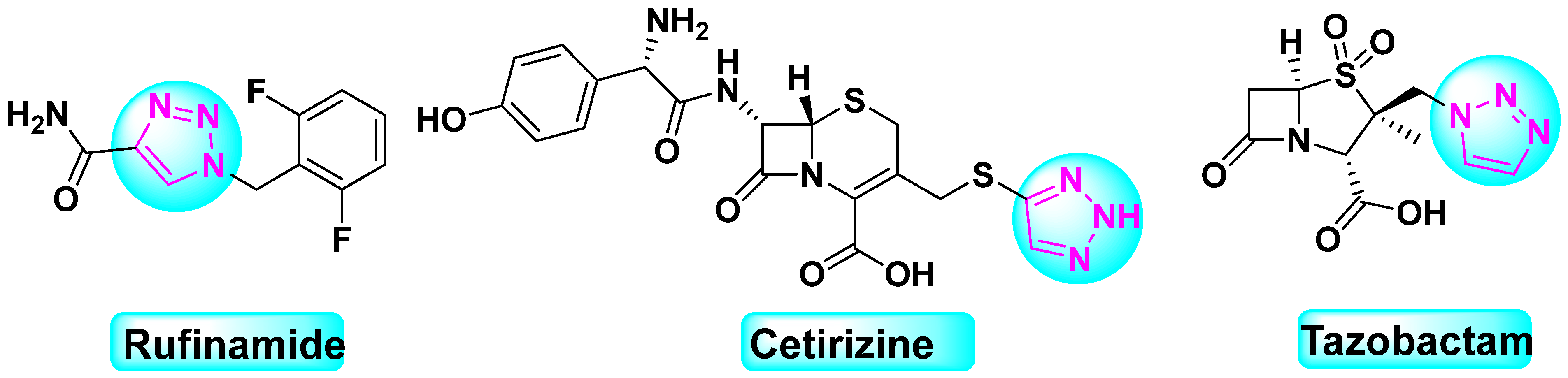
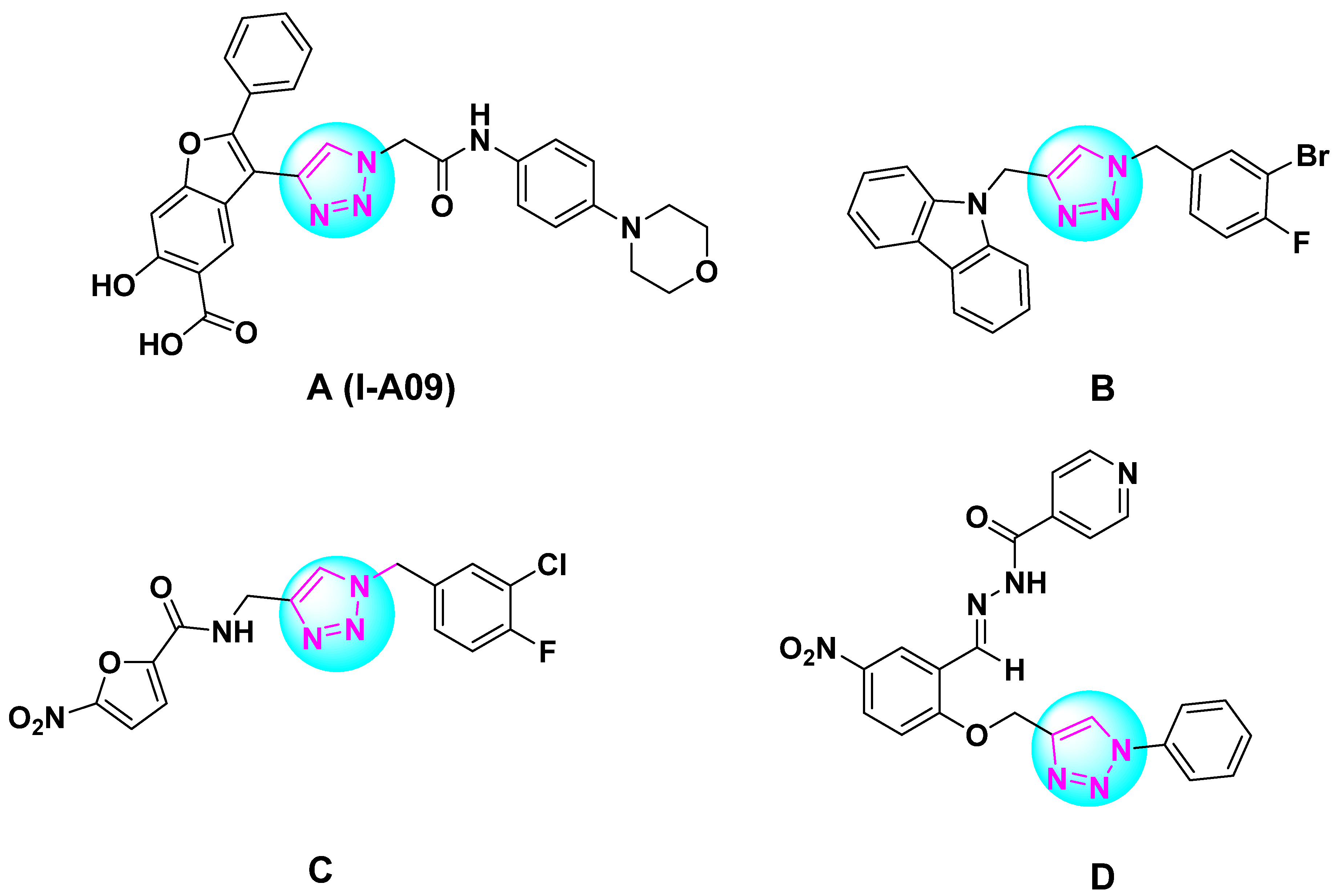
:1. Introduction
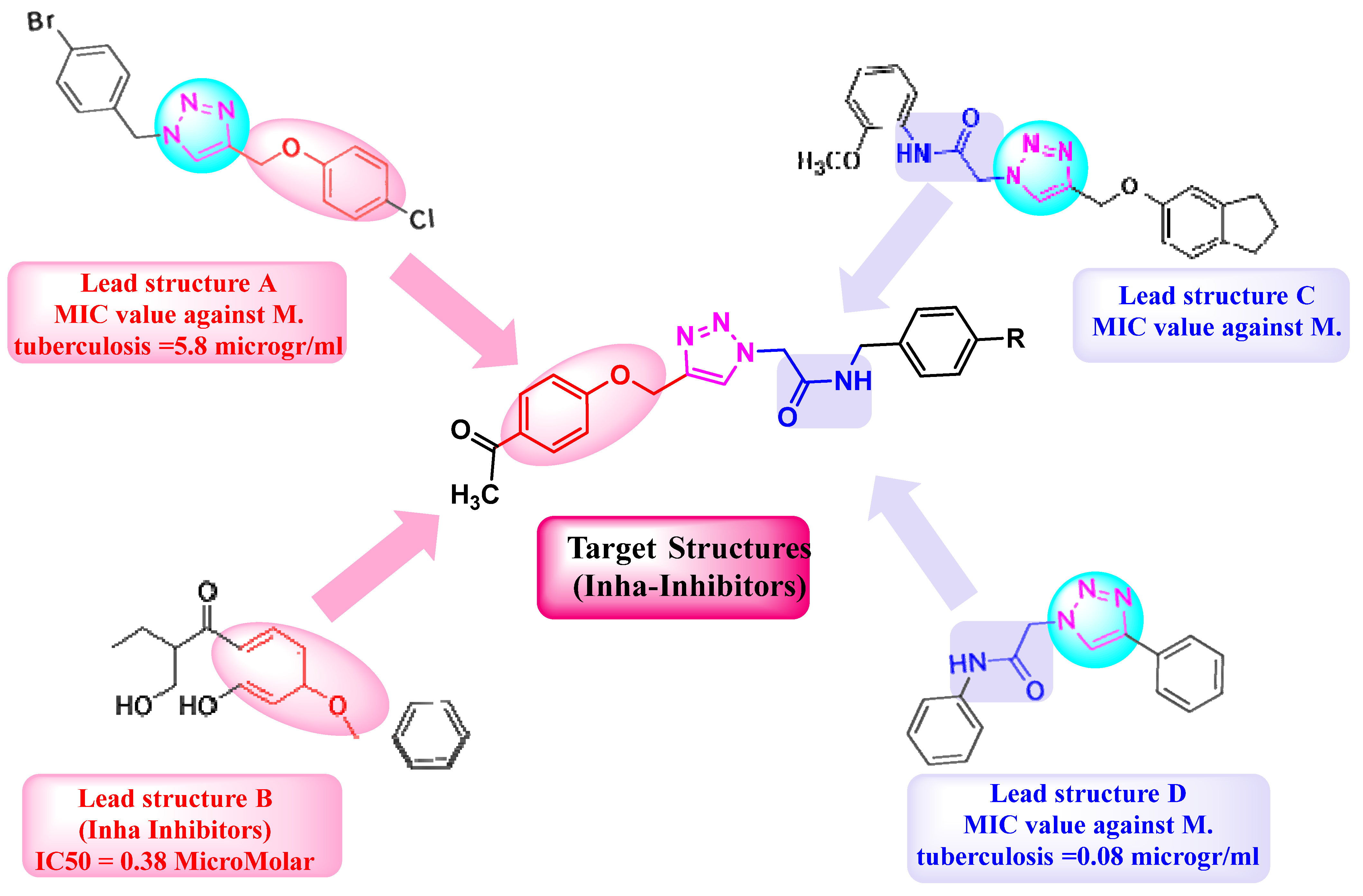
2. Results and Discussion
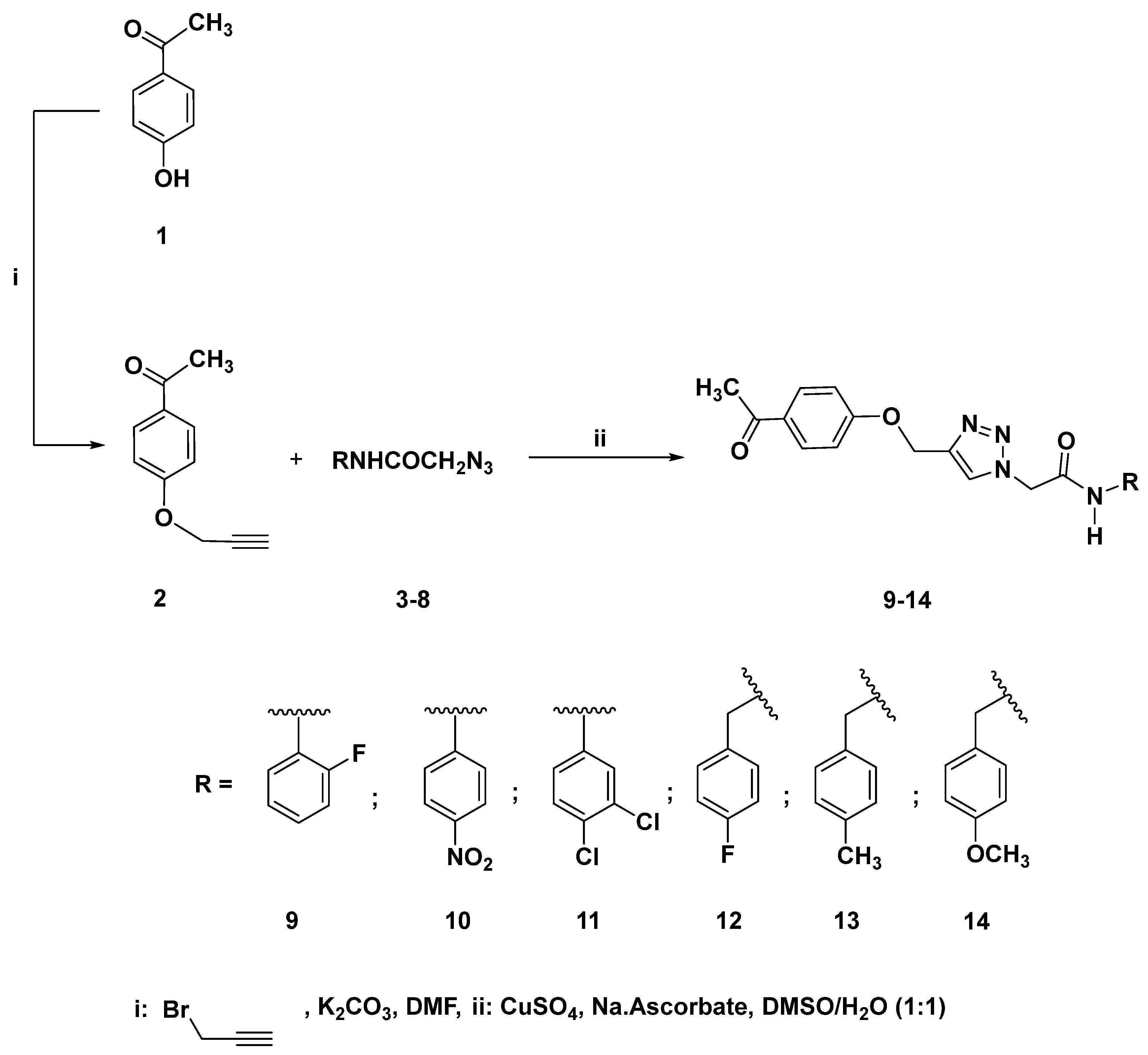
2.1. Chemistry
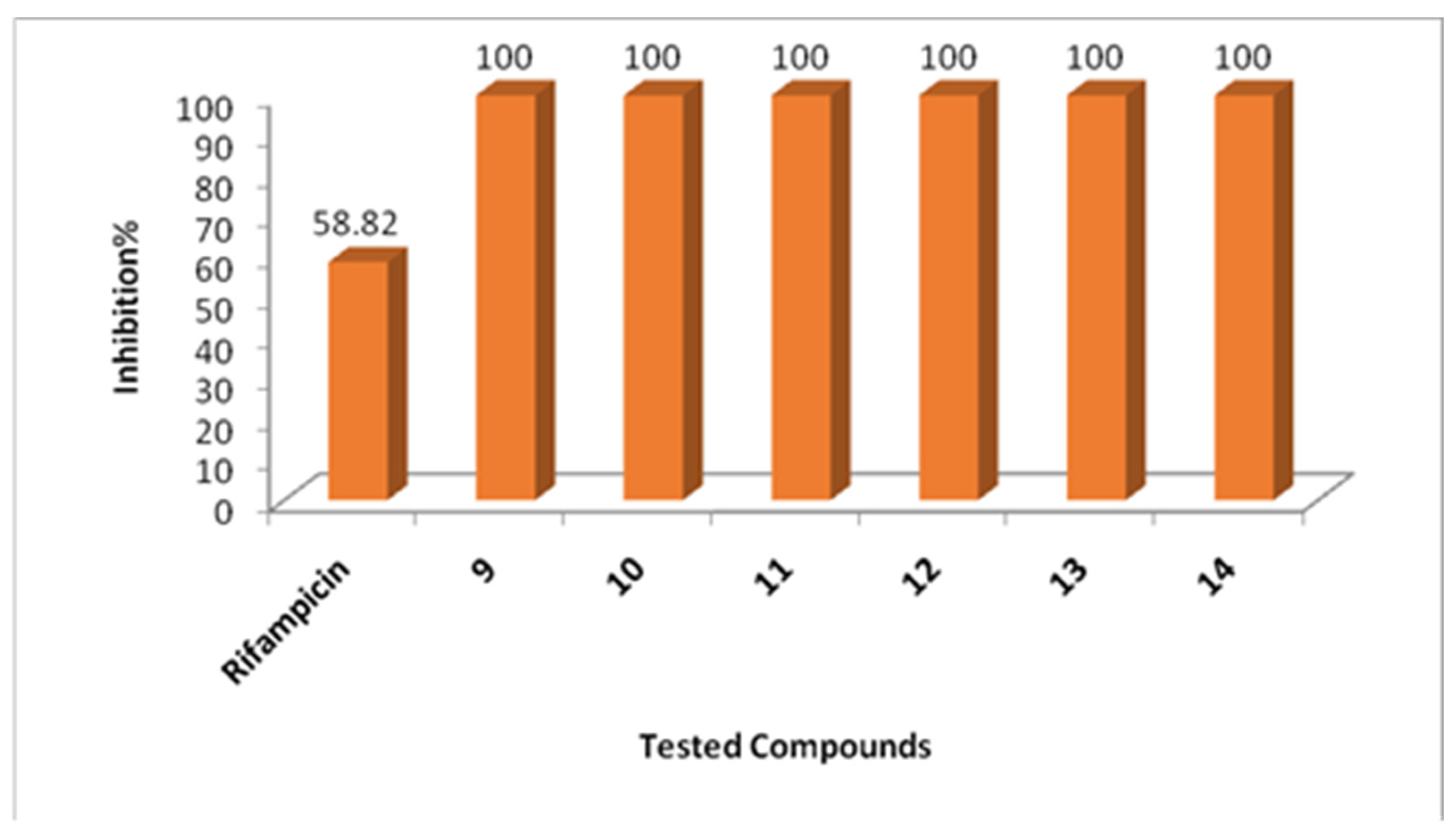
2.2. Biological Evaluation
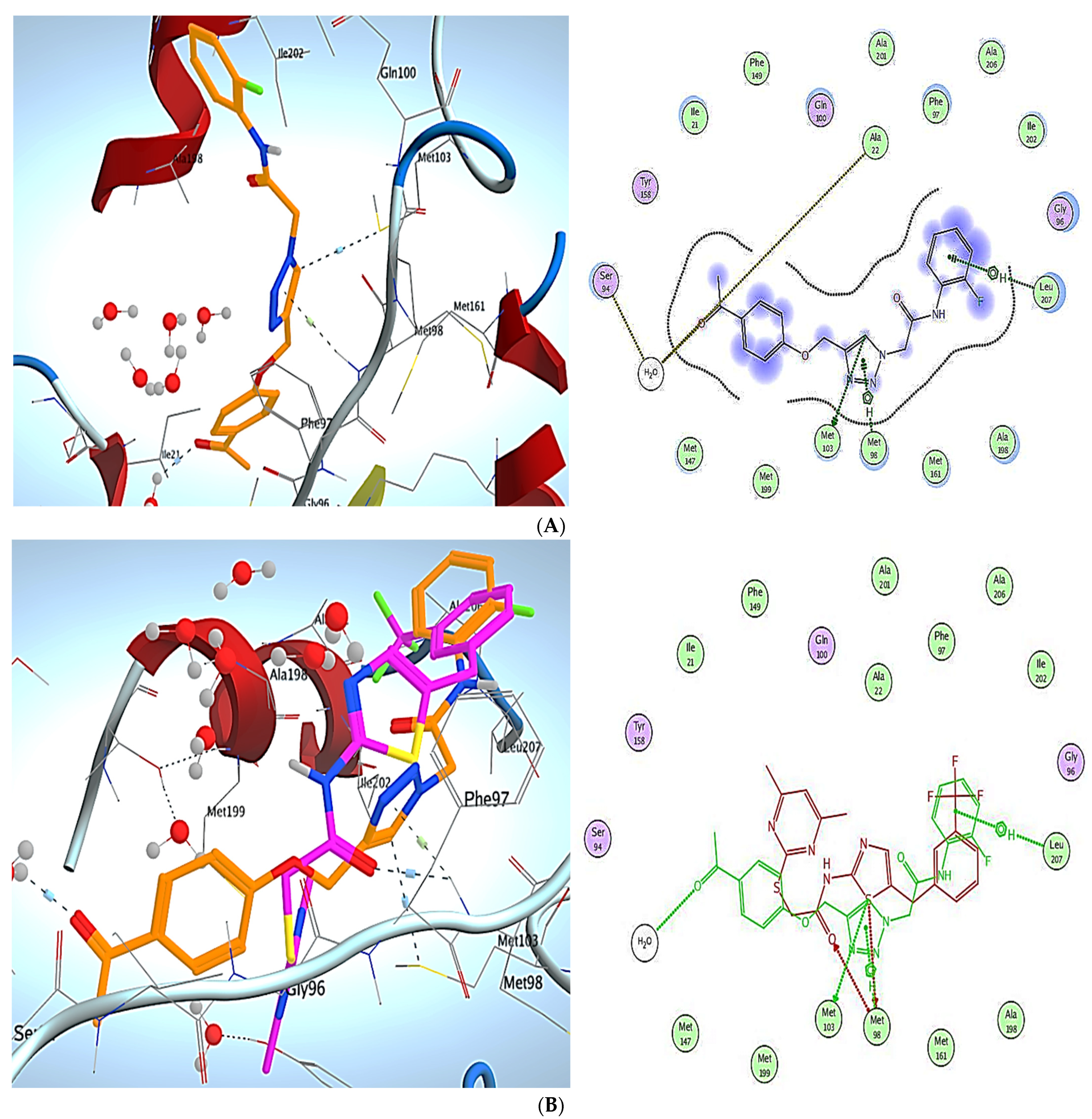
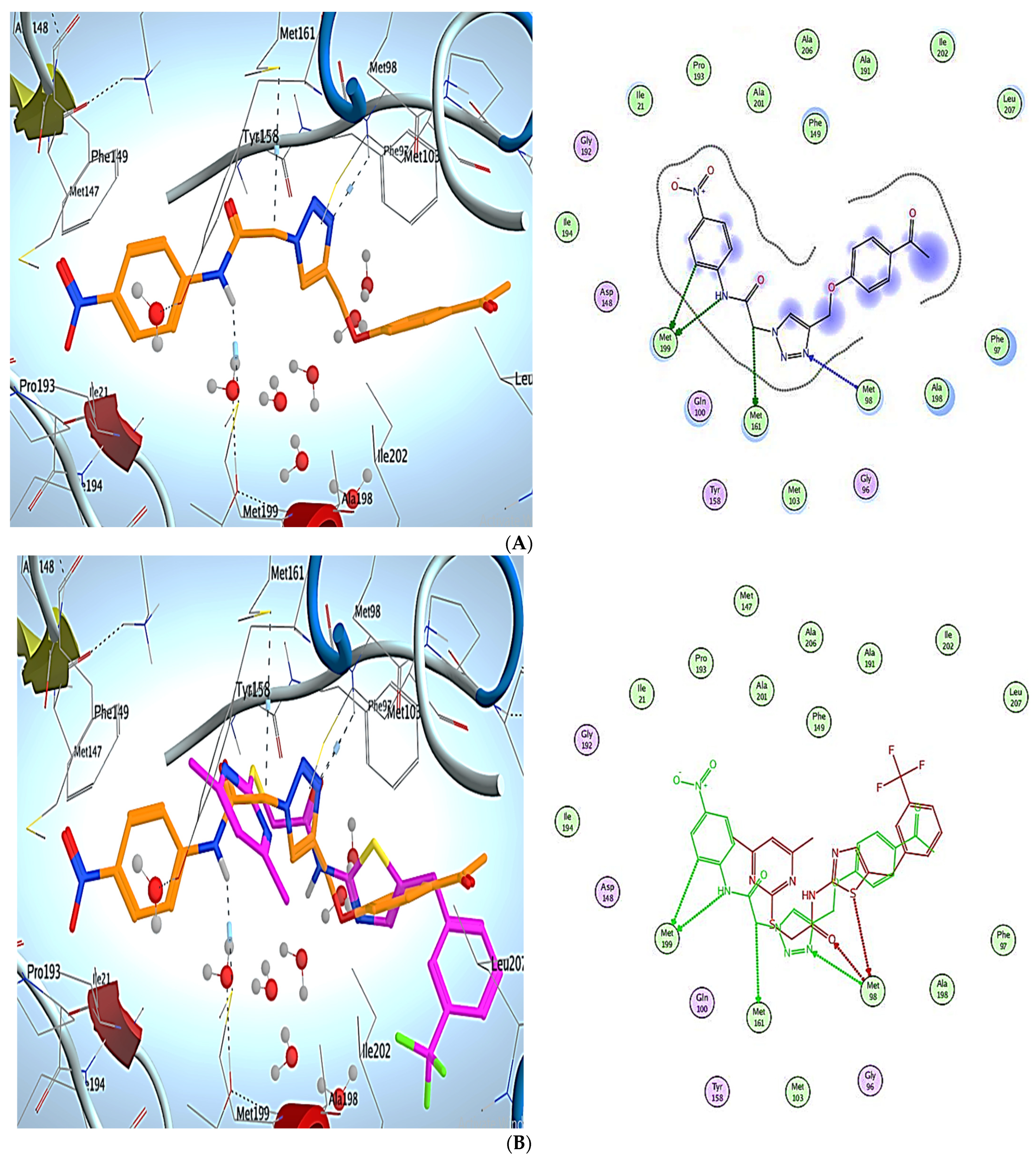
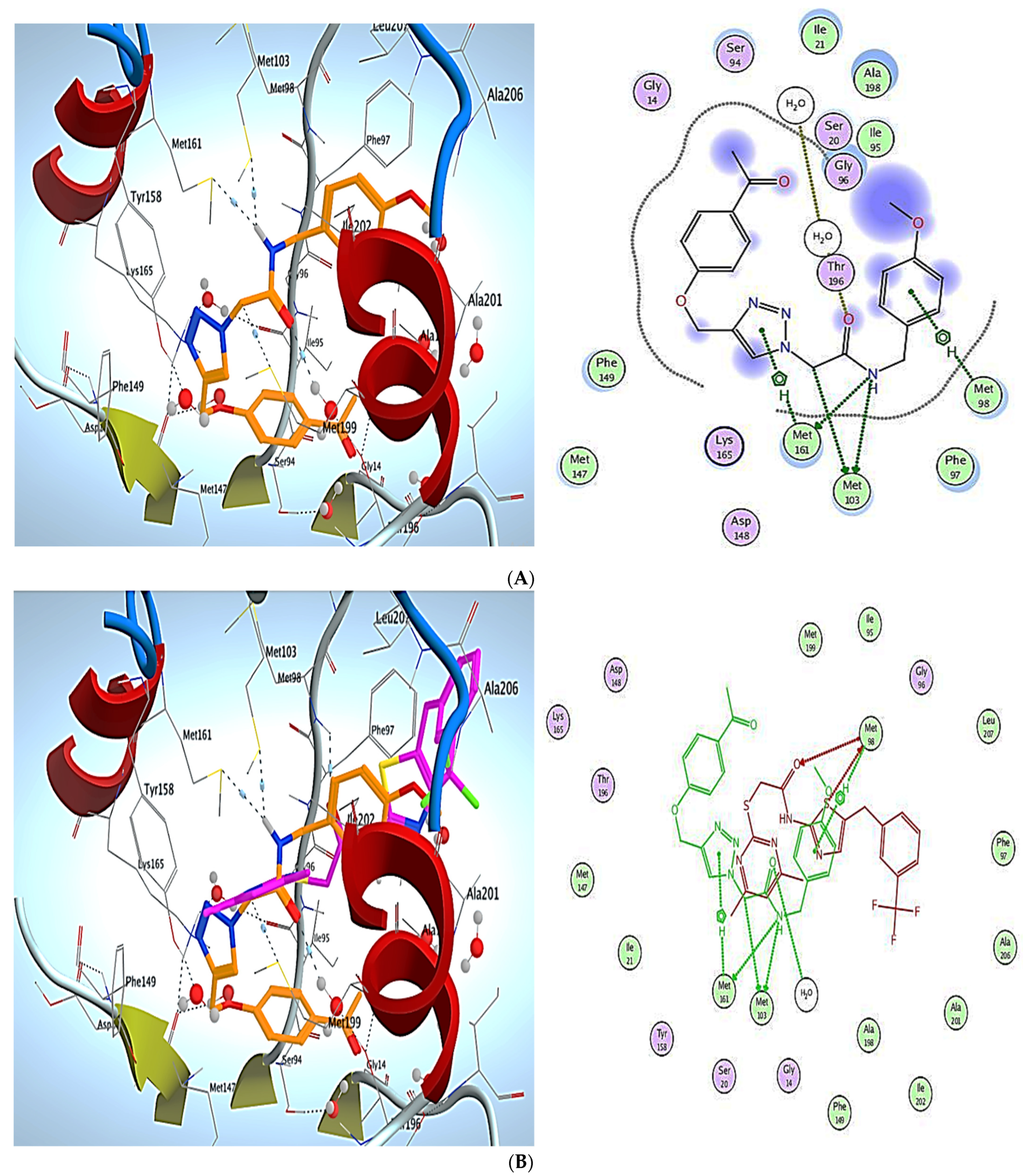
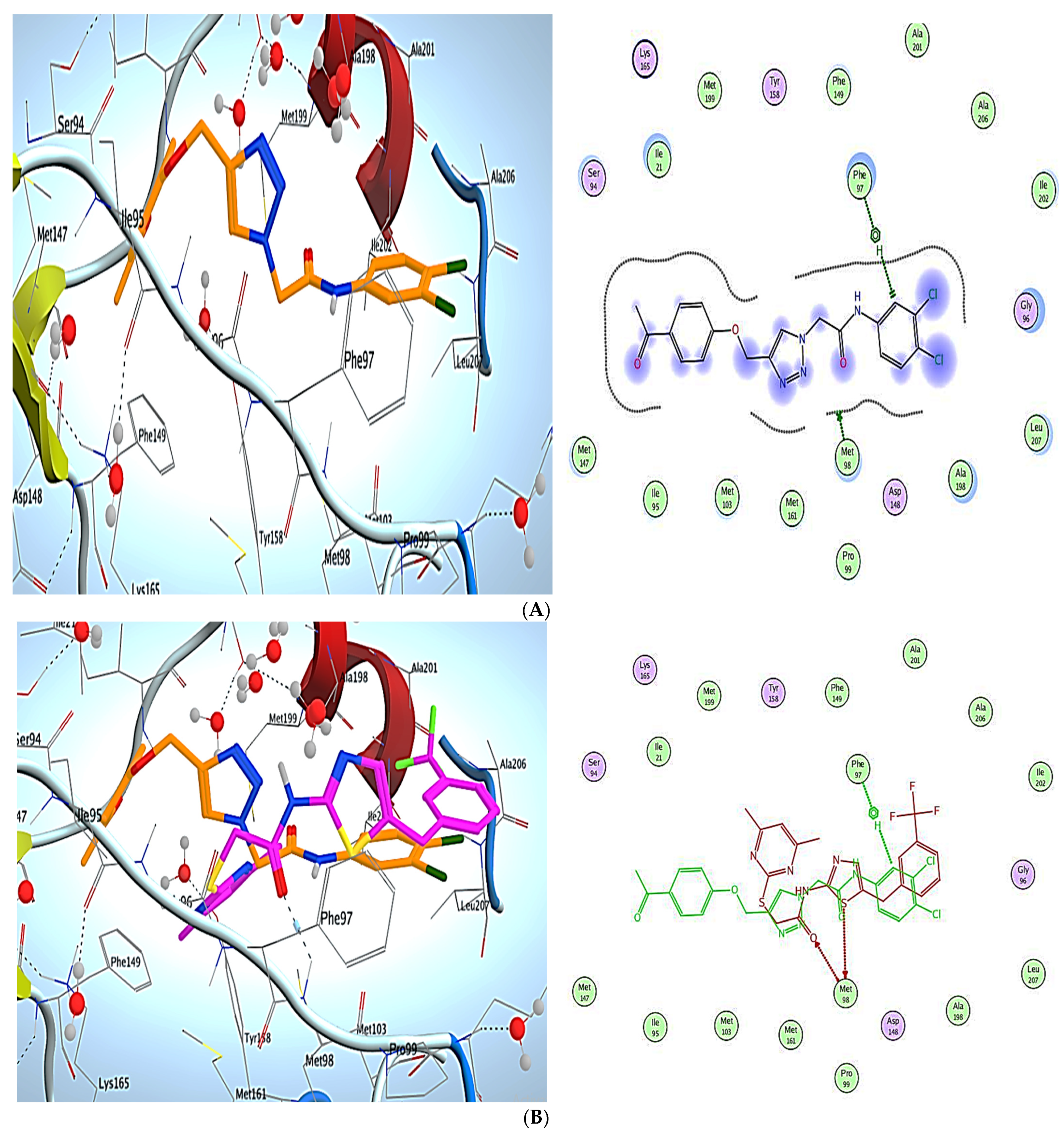
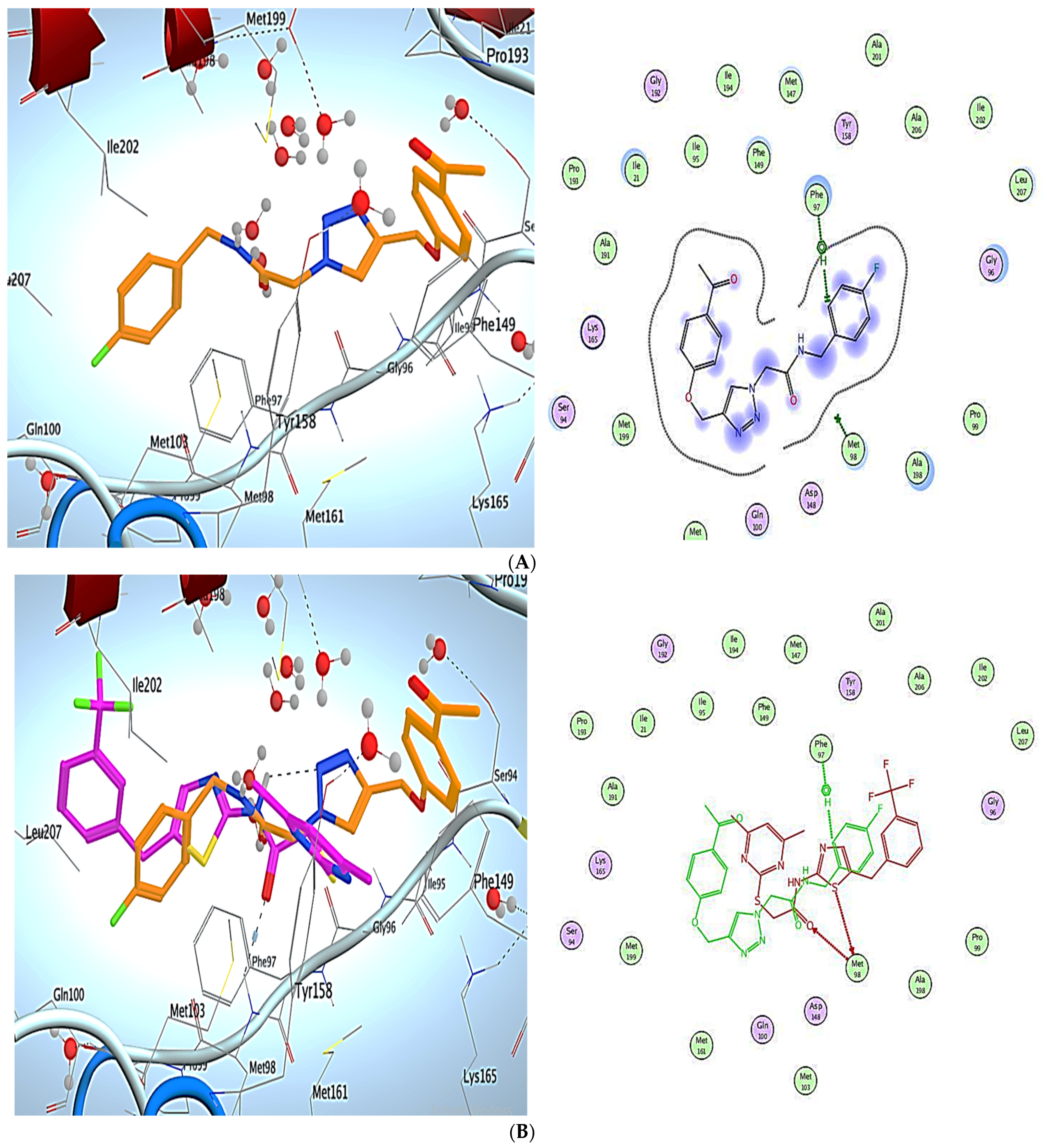
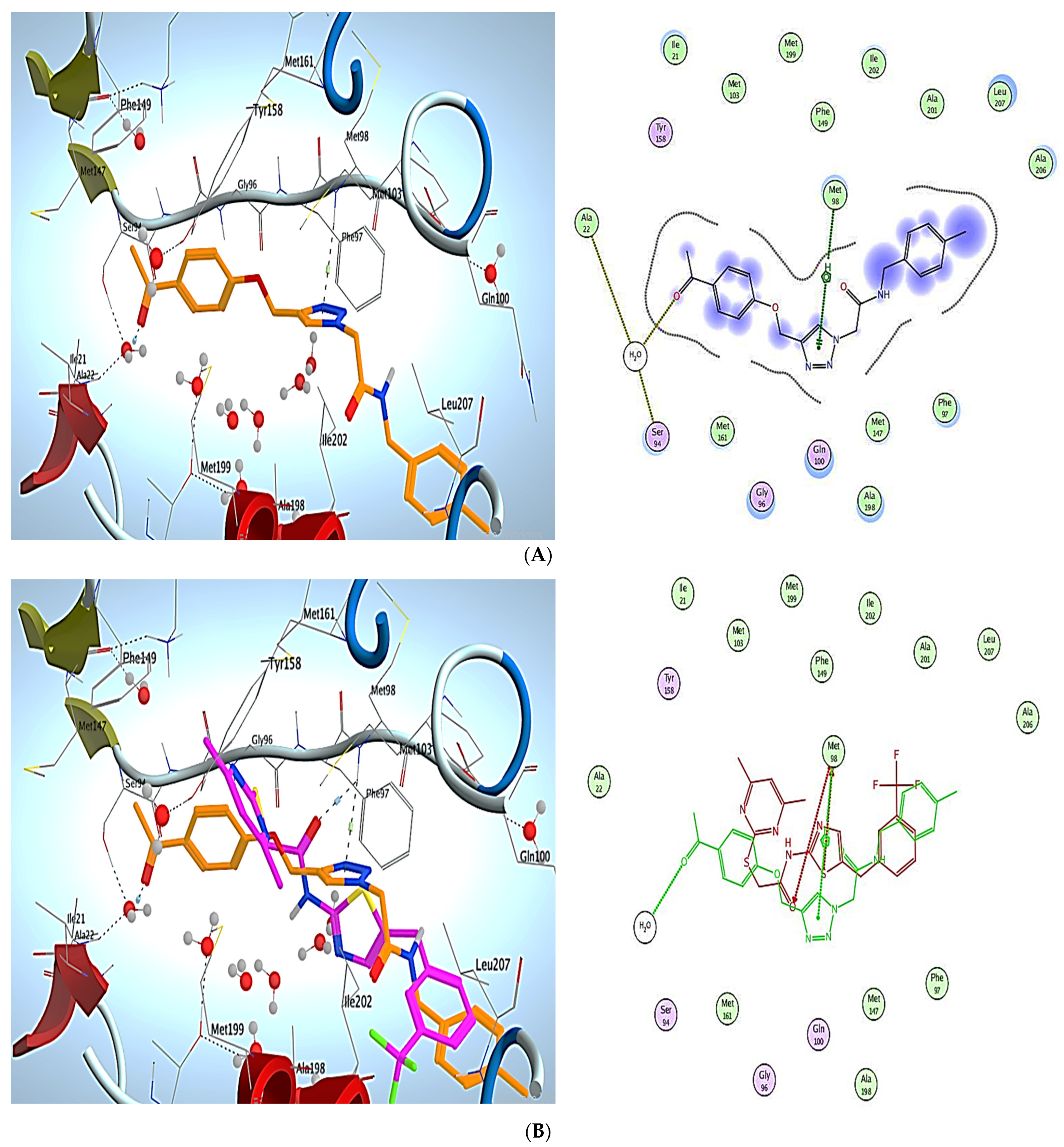
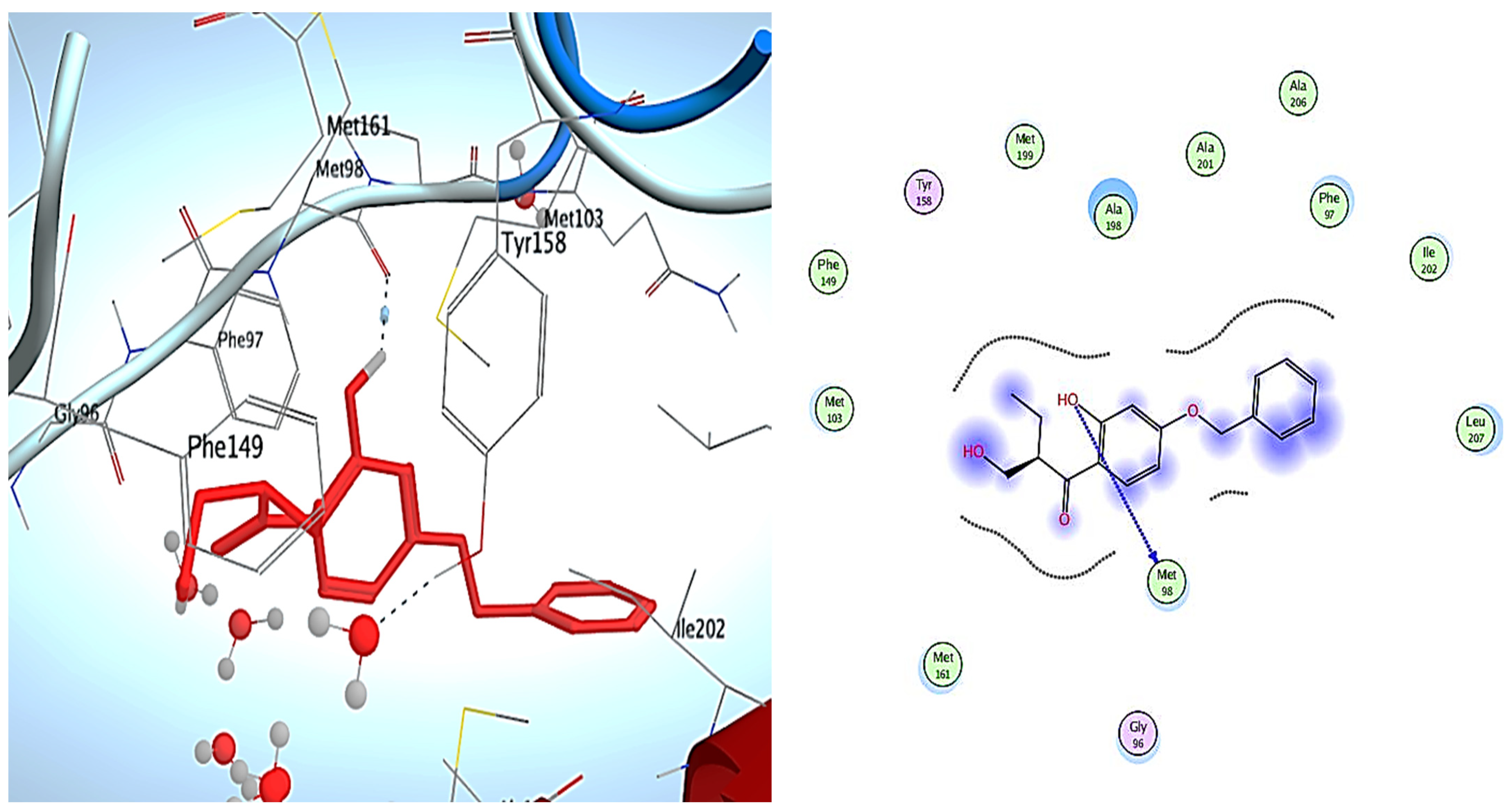
3. Molecular Modeling
Docking Simulations
4. Experimental Methods
4.1. Chemistry
4.1.1. General Information
4.1.2. Synthesis Andof 1-(4-(Prop-2-Yn-1-yloxy) phenyl) ethan-1-one (2)
2-(4-((4-Acetylphenoxy)methyl)-1H-1,2,3-triazol-1-Yl)-N-(2-fluorophenyl)acetamide (9)
2-(4-((4-Acetylphenoxy)methyl)-1H-1,2,3-triazol-1-Yl)-N-(4-nitrophenyl)acetamide (10)
2-(4-((4-Acetylphenoxy)methyl)-1H-1,2,3-triazol-1-Yl)-N-(3,4-dichlorophenyl)acetamide (11)
2-(4-((4-Acetylphenoxy)methyl)-1H-1,2,3-triazol-1-Yl)-N-(4-fluorobenzyl)acetamide (12)
2-(4-((4-Acetylphenoxy)methyl)-1H-1,2,3-triazol-1-Yl)-N-(4-methylbenzyl)acetamide (13)
2-(4-((4-Acetylphenoxy)methyl)-1H-1,2,3-triazol-1-Yl)-N-(4-methoxybenzyl)acetamide (14)
4.2. Enzymatic Inhibition Experiments
4.3. Docking Study
4.3.1. Preparation of the Protein Crystal Structures
4.3.2. Preparation of the Selected Compounds for Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Juríček, M.; Kouwer, P.H.; Rowan, A.E. Triazole: A unique building block for the construction of functional materials. Chem. Commun. 2011, 47, 8740–8749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordão, A.K.; Ferreira, V.F.; Souza, T.M.; de Souza Faria, G.G.; Machado, V.; Abrantes, J.L.; de Souza, M.C.; Cunha, A. Synthesis and anti-HSV-1 activity of new 1, 2, 3-triazole derivatives. Bioorganic Med. Chem. 2011, 19, 1860–1865. [Google Scholar] [CrossRef] [Green Version]
- Pereira, D.; Fernandes, P. Synthesis and antibacterial activity of novel 4-aryl-[1, 2, 3]-triazole containing macrolides. Bioorganic Med. Chem. Lett. 2011, 21, 510–513. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Gu, J.; Wang, C.; Wang, S.; Liu, N.; Jiang, Y.; Dong, G.; Wang, Y.; Liu, Y.; Yao, J. Design, synthesis and antifungal activity of novel triazole derivatives containing substituted 1, 2, 3-triazole-piperdine side chains. Eur. J. Med. Chem. 2014, 82, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.S.; Zheng, M.J.; Zhao, F.; Liu, D. 1, 2, 3-Triazole hybrids with anti-HIV-1 activity. Arch. Der Pharm. 2021, 354, 2000163. [Google Scholar] [CrossRef]
- Al-Blewi, F.F.; Almehmadi, M.A.; Aouad, M.R.; Bardaweel, S.K.; Sahu, P.K.; Messali, M.; Rezki, N.; El Ashry, E.S.H. Design, synthesis, ADME prediction and pharmacological evaluation of novel benzimidazole-1, 2, 3-triazole-sulfonamide hybrids as antimicrobial and antiproliferative agents. Chem. Central J. 2018, 12, 1–14. [Google Scholar] [CrossRef]
- El Ashry, E.; Elshatanofy, M.; Badawy, M.; Kandeel, K.; Elhady, O.; Abdel-Sayed, M. Synthesis and Evaluation of Antioxidant, Antibacterial, and Target Protein-Molecular Docking of Novel 5-Phenyl-2, 4-dihydro-3H-1, 2, 4-triazole Derivatives Hybridized with 1, 2, 3-Triazole via the Flexible SCH2-Bonding. Russ. J. Gen. Chem. 2020, 90, 2419–2434. [Google Scholar] [CrossRef]
- Lopes, F.V.; Stroppa, P.H.F.; Marinho, J.A.; Soares, R.R.; de Azevedo Alves, L.; Goliatt, P.V.Z.C.; Abramo, C.; da Silva, A.D. 1, 2, 3-Triazole derivatives: Synthesis, docking, cytotoxicity analysis and in vivo antimalarial activity. Chem. Interact. 2021, 350, 109688. [Google Scholar] [CrossRef]
- Junqueira, G.G.; Carvalho, M.R.; de Andrade, P.; Lopes, C.D.; Carneiro, Z.A.; Sesti-Costa, R.; Silva, J.S.; Carvalho, I. Synthesis and in vitro evaluation of novel galactosyl-triazolo-benzenesulfonamides against Trypanosoma cruzi. J. Braz. Chem. Soc. 2014, 25, 1872–1884. [Google Scholar]
- Xu, Z.; Zhao, S.-J.; Liu, Y. 1, 2, 3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. [Google Scholar] [CrossRef]
- Saad, H.A.; Osman, N.A.; Moustafa, A.H. Synthesis and analgesic activity of some new pyrazoles and triazoles bearing a 6, 8-dibromo-2-methylquinazoline moiety. Molecules 2011, 16, 10187–10201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, M.K.; Kumar, K.S.; Sreenivas, P.; Krupadanam, G.D.; Reddy, K.J. Synthesis of novel 1, 4-disubstituted-1, 2, 3-triazole semi synthetic analogues of forskolin by click reaction. Tetrahedron Lett. 2011, 52, 6537–6540. [Google Scholar] [CrossRef]
- Aouad, M.R.; Almehmadi, M.A.; Rezki, N.; Al-blewi, F.F.; Messali, M.; Ali, I. Design, click synthesis, anticancer screening and docking studies of novel benzothiazole-1, 2, 3-triazoles appended with some bioactive benzofused heterocycles. J. Mol. Struct. 2019, 1188, 153–164. [Google Scholar] [CrossRef]
- Rezki, N.; Almehmadi, M.A.; Ihmaid, S.; Shehata, A.M.; Omar, A.M.; Ahmed, H.E.; Aouad, M.R. Novel scaffold hopping of potent benzothiazole and isatin analogues linked to 1, 2, 3-triazole fragment that mimic quinazoline epidermal growth factor receptor inhibitors: Synthesis, antitumor and mechanistic analyses. Bioorganic Chem. 2020, 103, 104133. [Google Scholar] [CrossRef] [PubMed]
- Almehmadi, M.A.; Aljuhani, A.; Alraqa, S.Y.; Ali, I.; Rezki, N.; Aouad, M.R.; Hagar, M. Design, synthesis, DNA binding, modeling, anticancer studies and DFT calculations of Schiff bases tethering benzothiazole-1, 2, 3-triazole conjugates. J. Mol. Struct. 2021, 1225, 129148. [Google Scholar] [CrossRef]
- Alraqa, S.Y.; Alharbi, K.; Aljuhani, A.; Rezki, N.; Aouad, M.R.; Ali, I. Design, click conventional and microwave syntheses, DNA binding, docking and anticancer studies of benzotriazole-1, 2, 3-triazole molecular hybrids with different pharmacophores. J. Mol. Struct. 2021, 1225, 129192. [Google Scholar] [CrossRef]
- Ihmaid, S.K.; Alraqa, S.Y.; Aouad, M.R.; Aljuhani, A.; Elbadawy, H.M.; Salama, S.A.; Rezki, N.; Ahmed, H.E. Design of molecular hybrids of phthalimide-triazole agents with potent selective MCF-7/HepG2 cytotoxicity: Synthesis, EGFR inhibitory effect, and metabolic stability. Bioorganic Chem. 2021, 111, 104835. [Google Scholar] [CrossRef]
- Albelwi, F.F.; Teleb, M.; Abu-Serie, M.M.; Moaty, M.N.A.A.; Alsubaie, M.S.; Zakaria, M.A.; El Kilany, Y.; Aouad, M.R.; Hagar, M.; Rezki, N. Halting Tumor Progression via Novel Non-Hydroxamate Triazole-Based Mannich Bases MMP-2/9 Inhibitors; Design, Microwave-Assisted Synthesis, and Biological Evaluation. Int. J. Mol. Sci. 2021, 22, 10324. [Google Scholar] [CrossRef]
- Karakurt, A.; Aytemir, M.D.; Stables, J.P.; Özalp, M.; Betül Kaynak, F.; Özbey, S.; Dalkara, S. Synthesis of Some Oxime Ether Derivatives of 1-(2-Naphthyl)-2-(1, 2, 4-triazol-1-yl) ethanone and Their Anticonvulsant and Antimicrobial Activities. Arch. Der Pharm. 2006, 339, 513–520. [Google Scholar] [CrossRef]
- Kumudha, D.; Reddy, R.; Kalavathi, T. Synthesis and evaluation of some 1, 3, 4-thiadiazoles having substituted 1, 2, 4-triazole moiety for anticonvulsant and CNS depressant activity. World J. Pharm. Pharm. Sci. 2014, 3, 728–740. [Google Scholar]
- Kumar, S.; Sharma, B.; Mehra, V.; Kumar, V. Recent accomplishments on the synthetic/biological facets of pharmacologically active 1H-1, 2, 3-triazoles. Eur. J. Med. Chem. 2021, 212, 113069. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; He, Y.; Zhang, X.; Xu, J.; Luo, Y.; Wang, Y.; Franzblau, S.G.; Yang, Z.; Chan, R.J.; Liu, Y. Targeting mycobacterium protein tyrosine phosphatase B for antituberculosis agents. Proc. Natl. Acad. Sci. USA 2010, 107, 4573–4578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Sawy, M.A.; Elshatanofy, M.M.; El Kilany, Y.; Kandeel, K.; Elwakil, B.H.; Hagar, M.; Aouad, M.R.; Albelwi, F.F.; Rezki, N.; Jaremko, M. Novel Hybrid 1, 2, 4-and 1, 2, 3-Triazoles Targeting Mycobacterium Tuberculosis Enoyl Acyl Carrier Protein Reductase (InhA): Design, Synthesis, and Molecular Docking. Int. J. Mol. Sci. 2022, 23, 4706. [Google Scholar] [CrossRef]
- Massarotti, A.; Aprile, S.; Mercalli, V.; Del Grosso, E.; Grosa, G.; Sorba, G.; Tron, G.C. Are 1, 4- and 1, 5-Disubstituted 1, 2, 3-Triazoles Good Pharmacophoric Groups? ChemMedChem 2014, 9, 2497–2508. [Google Scholar] [CrossRef] [PubMed]
- Surineni, G.; Yogeeswari, P.; Sriram, D.; Kantevari, S. Rational design, synthesis and evaluation of novel-substituted 1, 2, 3-triazolylmethyl carbazoles as potent inhibitors of Mycobacterium tuberculosis. Med. Chem. Res. 2015, 24, 1298–1309. [Google Scholar] [CrossRef]
- Kamal, A.; Hussaini, S.M.A.; Faazil, S.; Poornachandra, Y.; Reddy, G.N.; Kumar, C.G.; Rajput, V.S.; Rani, C.; Sharma, R.; Khan, I.A.; et al. Anti-tubercular agents. Part 8: Synthesis, antibacterial and antitubercular activity of 5-nitrofuran based 1, 2, 3-triazoles. Bioorganic Med. Chem. Lett. 2013, 23, 6842–6846. [Google Scholar] [CrossRef]
- Patil, P.S.; Kasare, S.L.; Haval, N.B.; Khedkar, V.M.; Dixit, P.P.; Rekha, E.M.; Sriram, D.; Haval, K.P. Novel isoniazid embedded triazole derivatives: Synthesis, antitubercular and antimicrobial activity evaluation. Bioorganic Med. Chem. Lett. 2020, 30, 127434. [Google Scholar] [CrossRef]
- Shaikh, M.H.; Subhedar, D.D.; Nawale, L.; Sarkar, D.; Khan, F.A.K.; Sangshetti, J.N.; Shingate, B.B. 1, 2, 3-Triazole derivatives as antitubercular agents: Synthesis, biological evaluation and molecular docking study. MedChemComm 2015, 6, 1104–1116. [Google Scholar] [CrossRef]
- Aouad, M.R.; Khan, D.J.; Said, M.A.; Al-Kaff, N.S.; Rezki, N.; Ali, A.A.; Bouqellah, N.; Hagar, M. Novel 1, 2, 3-Triazole Derivatives as Potential Inhibitors against Covid-19 Main Protease: Synthesis, Characterization, Molecular Docking and DFT Studies. ChemistrySelect 2021, 6, 3468–3486. [Google Scholar] [CrossRef]
- Alzahrani, A.Y.; Shaaban, M.M.; Elwakil, B.H.; Hamed, M.T.; Rezki, N.; Aouad, M.R.; Zakaria, M.A.; Hagar, M. Anti-COVID-19 activity of some benzofused 1, 2, 3-triazolesulfonamide hybrids using in silico and in vitro analyses. Chemom. Intell. Lab. Syst. 2021, 217, 104421. [Google Scholar] [CrossRef]
- Said, M.A.; Khan, D.J.; Al-Blewi, F.F.; Al-Kaff, N.S.; Ali, A.A.; Rezki, N.; Aouad, M.R.; Hagar, M. New 1, 2, 3-Triazole Scaffold Schiff Bases as Potential Anti-COVID-19: Design, Synthesis, DFT-Molecular Docking, and Cytotoxicity Aspects. Vaccines 2021, 9, 1012. [Google Scholar] [CrossRef] [PubMed]
- Kamsri, P.; Hanwarinroj, C.; Phusi, N.; Pornprom, T.; Chayajarus, K.; Punkvang, A.; Suttipanta, N.; Srimanote, P.; Suttisintong, K.; Songsiriritthigul, C.; et al. Discovery of new and potent InhA inhibitors as antituberculosis agents: Structure-based virtual screening validated by biological assays and X-ray crystallography. J. Chem. Inf. Modeling 2019, 60, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Phatak, P.S.; Bakale, R.D.; Kulkarni, R.S.; Dhumal, S.T.; Dixit, P.P.; Krishna, V.S.; Sriram, D.; Khedkar, V.M.; Haval, K.P. Design and synthesis of new indanol-1, 2, 3-triazole derivatives as potent antitubercular and antimicrobial agents. Bioorganic Med. Chem. Lett. 2020, 30, 127579. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, C.; Pahwa, A.; Singh, D.; Kumar, K.; Luxmi, R. Efficient synthesis, antitubercular and antimicrobial evaluation of 1, 4-disubstituted 1, 2, 3-triazoles with amide functionality. Mon. Für Chem.-Chem. Mon. 2019, 150, 1127–1136. [Google Scholar] [CrossRef]
- Rozwarski, D.A.; Vilcheze, C.; Sugantino, M.; Bittman, R.; Sacchettini, J.C. Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J. Biol. Chem. 1999, 274, 15582–15589. [Google Scholar] [CrossRef] [Green Version]
- Naik, S.K.; Mohanty, S.; Padhi, A.; Pati, R.; Sonawane, A. Evaluation of antibacterial and cytotoxic activity of Artemisia nilagirica and Murraya koenigii leaf extracts against mycobacteria and macrophages. Altern. Med. 2014, 14, 87. [Google Scholar] [CrossRef]
- Devender, N.; Gunjan, S.; Tripathi, R.; Tripathi, R.P. Synthesis and antiplasmodial activity of novel indoleamide derivatives bearing sulfonamide and triazole pharmacophores. Eur. J. Med. Chem. 2017, 131, 171–184. [Google Scholar] [CrossRef]
- Wang, G.; Peng, Z.; Wang, J.; Li, X.; Li, J. Synthesis, in vitro evaluation and molecular docking studies of novel triazine-triazole derivatives as potential α-glucosidase inhibitors. Eur. J. Med. Chem. 2017, 125, 423–429. [Google Scholar] [CrossRef]
- Minvielle, M.J.; Bunders, C.A.; Melander, C. Indole–triazole conjugates are selective inhibitors and inducers of bacterial biofilms. MedChemComm 2013, 4, 916–919. [Google Scholar] [CrossRef]
- Rezki, Z.; Aouad, M.R. Green ultrasound-assisted three-component click synthesis of novel 1H-1, 2, 3-triazole carrying benzothiazoles and fluorinated-1, 2, 4-triazole conjugates and their antimicrobial evaluation. Acta Pharm. 2017, 67, 309–324. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, F.; Saffon, N.; Sammartino, J.C.; Degiacomi, G.; Pasca, M.R.; Lherbet, C. First triclosan-based macrocyclic inhibitors of InhA enzyme. Bioorganic Chem. 2020, 95, 103498. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Structure | IC50 (µM) |
|---|---|---|
| Rifampicin |  | 8.50 |
| Isoniazide |  | 0.054 |
| 9 |  | 0.005 |
| 10 |  | 0.008 |
| 11 |  | 0.043 |
| 12 |  | 0.041 |
| 13 |  | 0.084 |
| 14 |  | 0.002 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Albelwi, F.F.; Abdu Mansour, H.M.; Elshatanofy, M.M.; El Kilany, Y.; Kandeel, K.; Elwakil, B.H.; Hagar, M.; Aouad, M.R.; El Ashry, E.S.H.; Rezki, N.; et al. Design, Synthesis and Molecular Docking of Novel Acetophenone-1,2,3-Triazoles Containing Compounds as Potent Enoyl-Acyl Carrier Protein Reductase (InhA) Inhibitors. Pharmaceuticals 2022, 15, 799. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15070799
Albelwi FF, Abdu Mansour HM, Elshatanofy MM, El Kilany Y, Kandeel K, Elwakil BH, Hagar M, Aouad MR, El Ashry ESH, Rezki N, et al. Design, Synthesis and Molecular Docking of Novel Acetophenone-1,2,3-Triazoles Containing Compounds as Potent Enoyl-Acyl Carrier Protein Reductase (InhA) Inhibitors. Pharmaceuticals. 2022; 15(7):799. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15070799
Chicago/Turabian StyleAlbelwi, Fawzia Faleh, Hanaa M. Abdu Mansour, Maram M. Elshatanofy, Yeldez El Kilany, Kamal Kandeel, Bassma H. Elwakil, Mohamed Hagar, Mohamed Reda Aouad, El Sayed H. El Ashry, Nadjet Rezki, and et al. 2022. "Design, Synthesis and Molecular Docking of Novel Acetophenone-1,2,3-Triazoles Containing Compounds as Potent Enoyl-Acyl Carrier Protein Reductase (InhA) Inhibitors" Pharmaceuticals 15, no. 7: 799. https://0-doi-org.brum.beds.ac.uk/10.3390/ph15070799