1. Introduction
The accurate prediction of drug-target binding affinity (DTA) plays an essential role in the discovery of new drugs [
1], as well as drug repositioning [
2,
3,
4]. Proteins usually act as targets to interact with small molecules to regulate important biological functions in drug discovery. Although wet-lab experimental methods have been developed to screen and characterize chemical molecules, it is time-consuming and labor-intensive to identify potential compounds on a large scale. To relieve this bottleneck, People have proposed many computational methods to identify drug-target binding affinity.
Traditional methods such as molecular docking [
5,
6] and molecular dynamics simulation [
7] have been used in the virtual screening of compounds. Although these methods are very explanatory and even uncover the potential binding posture, their practical applications are limited, The reason is that these methods rely heavily on the existing high-quality 3D structure of the protein of interest. Besides, these methods consumes lots of computational resources.
Many methods applied machine learning to predict drug-target interactions, which was regarded as a binary classification task [
8,
9,
10,
11,
12]. However, the binding affinities between drugs and targets are actually real-valued continuous variables, and some weak drug-target interactions also play important functions. So, there have been some methods proposed to predict the quantitative binding affinity representing the strength of protein-drug interactions, usually in terms of the dissociation constant (
), the inhibition constant (
), or half of the maximum inhibitory concentration (IC50). In principle, a low IC50 value (low
value or high
value) indicates high binding affinity. The use of continuous values to measure the binding strength is more informative. For example, Pahikkala et al. used the least-squares algorithm called KronRLS [
13], which is based on the similarity of the drug-target pair calculated by Smith-Waterman (S-W) algorithm [
14]. SimBoost [
15] calculates drug and target ontological features and network features and then inputs them into the gradient boosting machines [
16] to predict the binding affinity. CGKronRLS [
17] is one of the best performers in the recent binding affinity prediction challenges of protein kinases. It uses 2D structure-based compound-compound similarity and normalized Smith-Waterman alignment scores to obtain protein-protein similarity, and then inputs the pre-calculated similarity into Kronecker kernel to calculate the binding affinity between the compounds and proteins.Pred-binding [
18] method utilizes protein sequences and molecular structures combined with support vector machines [
19] and random forests [
20] to predict the binding affinity between proteins and compounds.
In recent years, deep learning has advanced in image processing [
21], natural language processing [
22], speech recognition [
23], and other fields. Some studies have been inspired to develop deep learning-based methods to predict drug-target binding affinity. For example, Ozturk et al. proposed a deep convolutional neural network (CNN) method called DeepDTA [
24], which uses drug SMILES [
25] and sequences representation of protein as input of convolution Neural network to extract features for binding affinity prediction. WideDTA [
26] used a text-based method to encode the drug SMILES and protein sequences, including four different textual pieces of information. GANsDTA [
27] proposed a novel semi-supervised model based on generative adversarial networks (GANs) [
28] to predict binding affinity via drug SMILES and protein sequences. DeepGS [
29] is another method that takes the drug SMILES descriptors and the protein sequences to predict binding affinity. These methods have shown that deep networks can better capture the essential features than traditional machine learning algorithms. In addition to CNN, Deep-Affnity [
30] combined CNN and long and short-term memory networks (LSTM) to extract sequence features, as LSTM often better captures long-distance dependency in the sequence. DeepAffnity shows that the binding affinity can be effectively predicted using only the original sequences without relying on feature engineering. Besides, some models use extended connectivity fingerprints (ECFP) [
31] and graph convolutional networks [
32,
33,
34,
35] to extract drug information. However, due to the black-box nature of deep learning, the deep learning-based methods achieved remarkable performance, but these methods have limited interpretability.
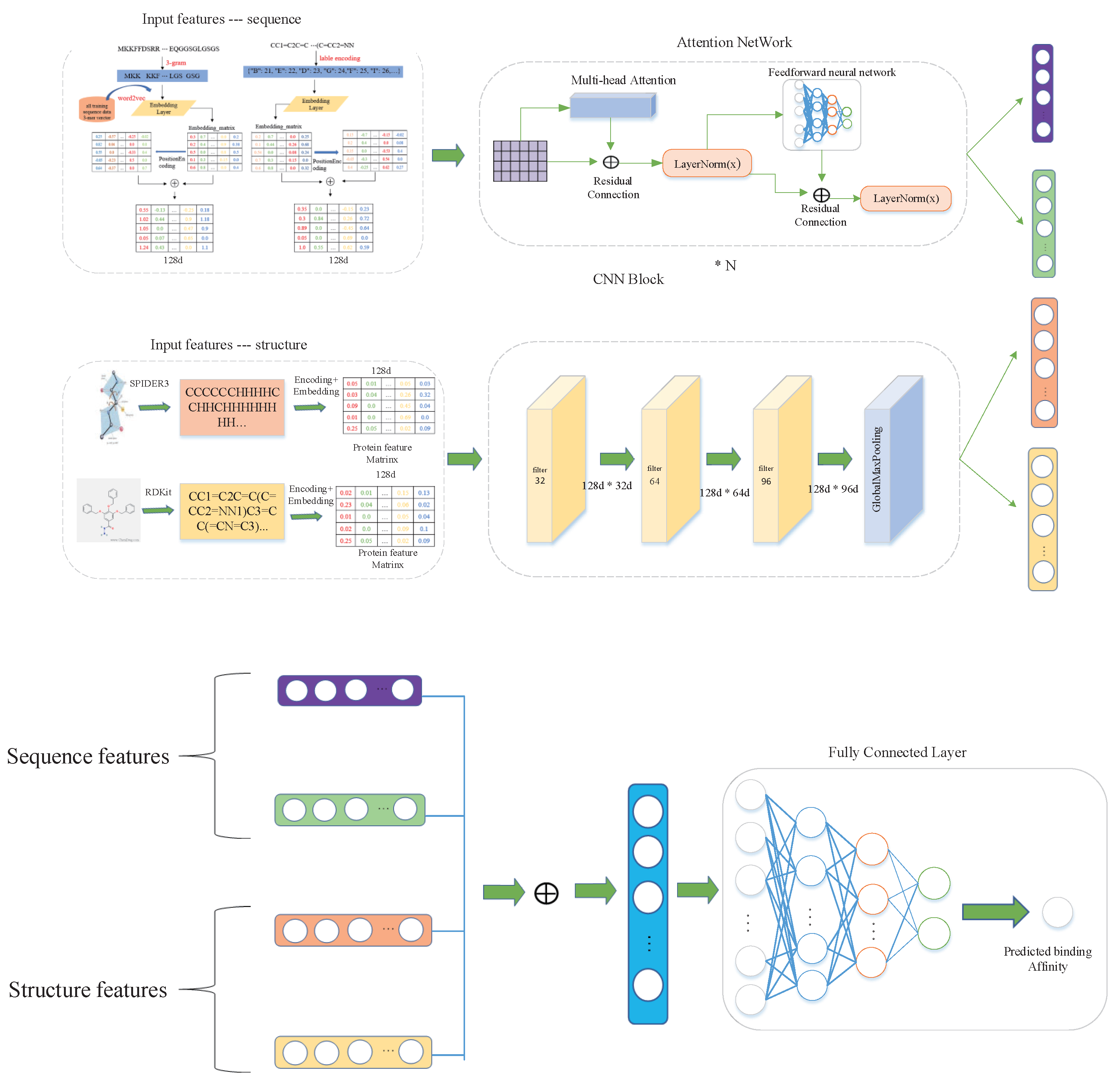
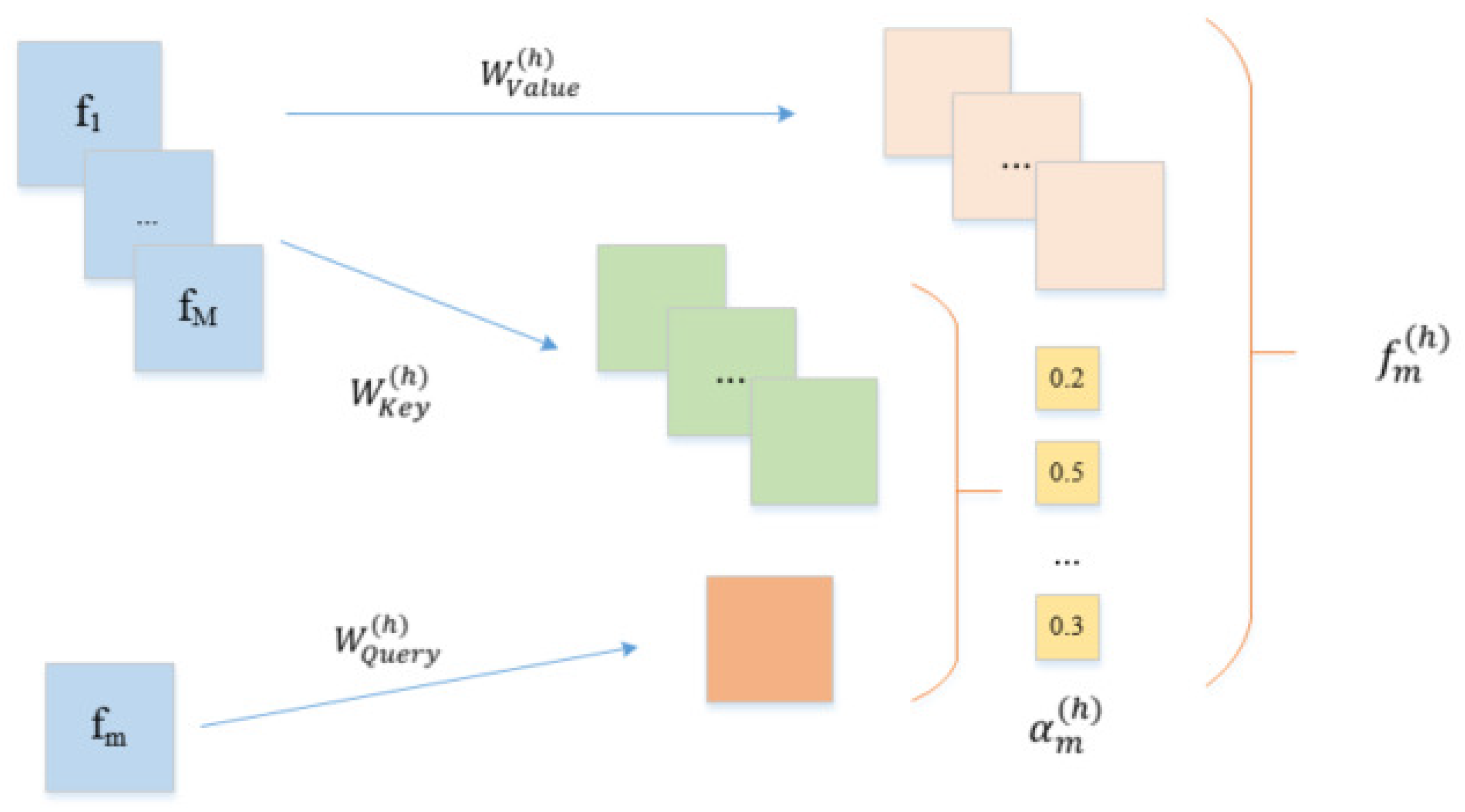
In this paper, we propose an end-to-end deep learning method to predict the binding affinity of proteins and drugs. First, a multi-head attention mechanism layer is introduced to promote the associations between different features so as to identify high-order semantic features automatically. The multi-head attention mechanism can map the original features to multiple subspaces, so that our model can capture different feature associations. Second, the residual network is applied to the feature extraction layers, which allows the combination of features in a different layer. Finally, the features of the protein and drug are concatenated and fed into a fully connected layer for prediction. For performance evaluation, the Davis kinase binding affinity dataset and KIBA large-scale kinase inhibitors dataset are used to calibrate our method. We compare our method with several current state-of-the-art methods and report four performance measures, including CI, AUPR, and MSE. The experimental results show that our method is significantly better than other methods on these two datasets.
2. Results and Discussion
2.1. Evaluation Metrics
As we formulate the prediction of drug-target binding affinity as a regression problem, four metrics are used to evaluate our model: (1) the consistency index (CI); (2) the mean square error (MSE); (3) coefficient; (4) PR curve area (AUPR). The first three indicators are often used to evaluate the continuous output value of the model, and the fourth metric is used to evaluate the binary output of the model.
The Concordance Index (CI) is a model evaluation method proposed by Gönen and Heller [
36]. It measures the probability of agreement between actual and predicted values. Let
and
denote the ground truth and predicted value of the i-th sample, respectively. Therefore, the metric is defined as follows:
and
represent the measured value and predicted value of the
i-th sample, respectively.
Z is the normalization constant and b is the step function. The definition of
is as follows:
This metric measures whether the predicted binding affinity values of two random drug-target pairs are predicted in the same order as their true values. The range of this value is 0–1, the closer the value is to 1, the better the model is.
The mean square error (MSE) is calculated as follows:
in which,
n represents the total number of samples in the data set,
represents the predicted value of the
i-th sample, and
represents the true value of the
i-th sample.
The mean regression coefficient
, which is proposed by previous paper [
37], is calculated using the following formulation:
The area under the precision-recall curve (AUPR) evaluates the binary classification model by averaging the precision of all recall values. We choose the AUPR metric because the PR curve is more suitable than the ROC curve in the case of unbalanced data. To calculate the AUPR value, we introduce different binding affinity thresholds to convert continuous predicted values into binary values. Similar to DeepDTA, we choose threshold value 7 for Davis dataset and 12.1 for KIBA dataset.
The hyperparameters of the model are tuned through five-fold cross-validation and evaluated on an independent test set. We use the mean square error as the loss function and the Adam optimizer to minimize the loss function. Tensorflow is used to build the model and the model is trained on a workstation with two GPUs. For the Davis dataset, we use dropout and regularization to prevent overfitting. For the KIBA dataset, which is four times the size of the Davis dataset, we use only regularization to prevent overfitting.
2.2. Hyperparameter Analysis
For model hyperparameters, we use grid search to tune their values. The filter sizes of the three convolutional layers are set to 32, 64, and 96, respectively. For other hyperparameters, such as learning rate, batch size, and regularization, we conducted parameter tuning experiments and the final hyperparameter values are shown in
Table 1. For performance evaluation on the independent test set, we run 500 epoches for training and then used for prediction.
2.3. Competitive Methods
To verify the superiority of our method, we compare it with six baseline methods, including both machine learning and deep learning methods. They are briefly introduced as below:
KronRLS: KronRLS [
13] is implemented based on the Kronecker Regularized Least Square, which uses Kronecker product algebraic properties to perform predictions on the whole drug-target space, without the explicit calculation of the pairwise kernels.
SimBoost: SimBoost [
15] obtains three types of features through feature engineering, and then uses gradient boosting trees trained on the extracted features to predict the binding affinity of targets and drugs.
DeepCPI: DeepCPI [
38] uses graph neural network and CNN to extract features from the SMILES of the compound and the sequence of the protein respectively. We transfer it to the regression model by modifying the neurons in the final fully connected layer to output real-value binding affinity.
DeepDTA: DeepDTA [
24] uses drug SMILES and the protein sequence as the input into three-layer CNN to learn protein and drug features, and fed into fully connected layer to predict binding affinity.
GANsDTA: GANsDTA [
27] proposes a semi-supervised generative adversarial network to predict the binding affinity of drugs and targets. This semi-supervised learning mechanism allows the method to work on unlabeled data.
DeepGS: DeepGS [
29] takes the SMILES string of the drug and the sequence information of the protein as input, and uses Prot2Vec and Smi2Vec to obtain a two-dimensional feature matrix representation of amino acids and atom. Meanwhile, graph attention network is used to extract the topological information of drugs.
2.4. Performance Comparison
We first conduct a performance comparison on the Davis dataset.
Table 2 shows the average CI value, mean square error, AUPR and
comparison. Note that DeepMHADTA1 means that only regularization is used, while DeepMHADTA2 means that both regularization and dropout are used.
It can be seen from
Table 2 that DeepMHADTA2 obtained the best performance, and achieve 0.895, 0.701, 0.766 and 0.244 for CI,
, AUPR and MSE metric, respectively. In particular, our method performs markedly better on
and AUPR. We also noticed that the traditional machine learning methods, KronRLS and SimBoost, perform slightly weak compared to deep learning-based methods. The reason lies in that traditional machine learning relies on manually curated features. As KronRLS and SimBoost use protein and drug similarity as input, while the deep learning-based methods are end-to-end learning frameworks and automatically capture the features from input data.
Next, we compared our model with other methods on another independent dataset.
Table 3 shows the performance metric on the KIBA dataset. Our method achieves 0.876, 0.719, 0.806 and 0.186 of CI index,
, AUPR and MSE, which outperforms all competitive methods.
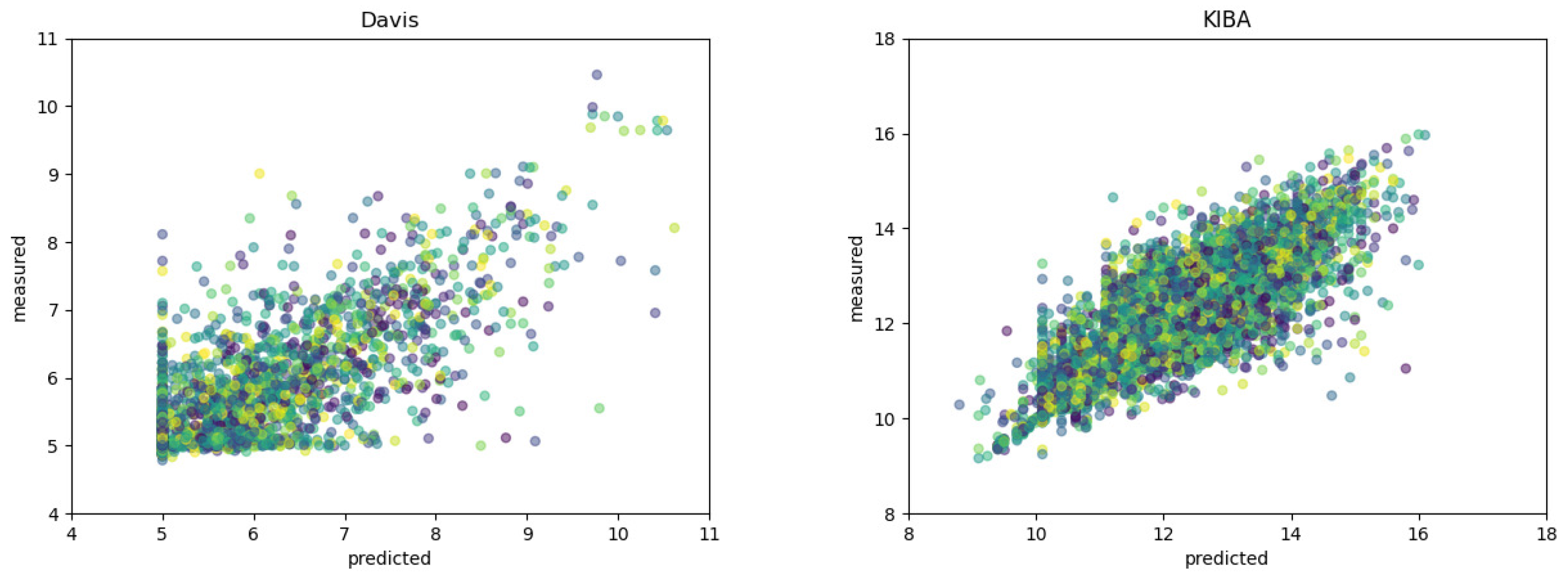
Moreover,
Figure 1 shows the scatter plots of the measured and predicted binding affinities on two datasets. A good model should predict value as close to the true value as possible, namely, the points locate closely to the diagonal line as much as possible. It can be seen from
Figure 1 that our method achieve superior performance.
To verify the effectiveness of our method, we plot the histogram of the binding affinity values in two datasets. As shown in
Figure 1, binding affinity are mostly distributed in the region from 5 to 6 in the Daivs dataset, while in the KIBA dataset most values fall in the range of 10 to 15.
2.5. Ablation Study
The input of our model contains two parts: (1) The sequence information of proteins and drugs, and (2) the structural information of proteins and drugs. We conducted an ablation study to evaluate the impact of each part on Davis and KIBA datasets.
Table 4 and
Table 5 show the results. We found that on the Davis dataset, the model using only one type of information does not differ largely from the model using both sequence and structure.
However, on the KIBA dataset, the performance of the two ablation models decline significantly. The ablation study shows that the structural information of proteins and drugs plays an important role for improving model performance besides sequence information.
4. Conclusions
In this paper, we propose a new end-to-end deep learning method called DeepMHADTA to predict the binding affinity of proteins and drugs. We use not only the protein sequence and SMILES descriptors of drugs, but also the protein secondary structure and drug fingerprints. For the extraction of sequence features, we used Word2Vec and label encoding to encode of proteins and drugs, respectively. Also, we combine the multi-head self-attention mechanism with the residual network as feature extraction block, and meanwhile we use the CNN to extract structural features, and finally concatenate all the embedding vectors into the fully connected layer to predict the binding affinity value. Our empirical experiments show that our method achieves superior performance on two independent datasets. We have also tried to use only sequence or structure information alone train the model, and found that both structure and sequence provide informative features. The advantage of our method is multiplex: (1) we use the multi-head self-attention mechanism, which make our model pay attention to important features. (2) For the extraction of protein sequence features, we use Word2Vec instead of label-encoding or one-hot encoding, which is a informative and efficient semantic representation than straightforward one-hot encoding. (3) We consider not only the sequence of proteins and drugs, but also their spatial structure.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}