Targeted Therapies in Cholangiocarcinoma: Emerging Evidence from Clinical Trials

 , , ,
, , , 
Abstract
:1. Introduction
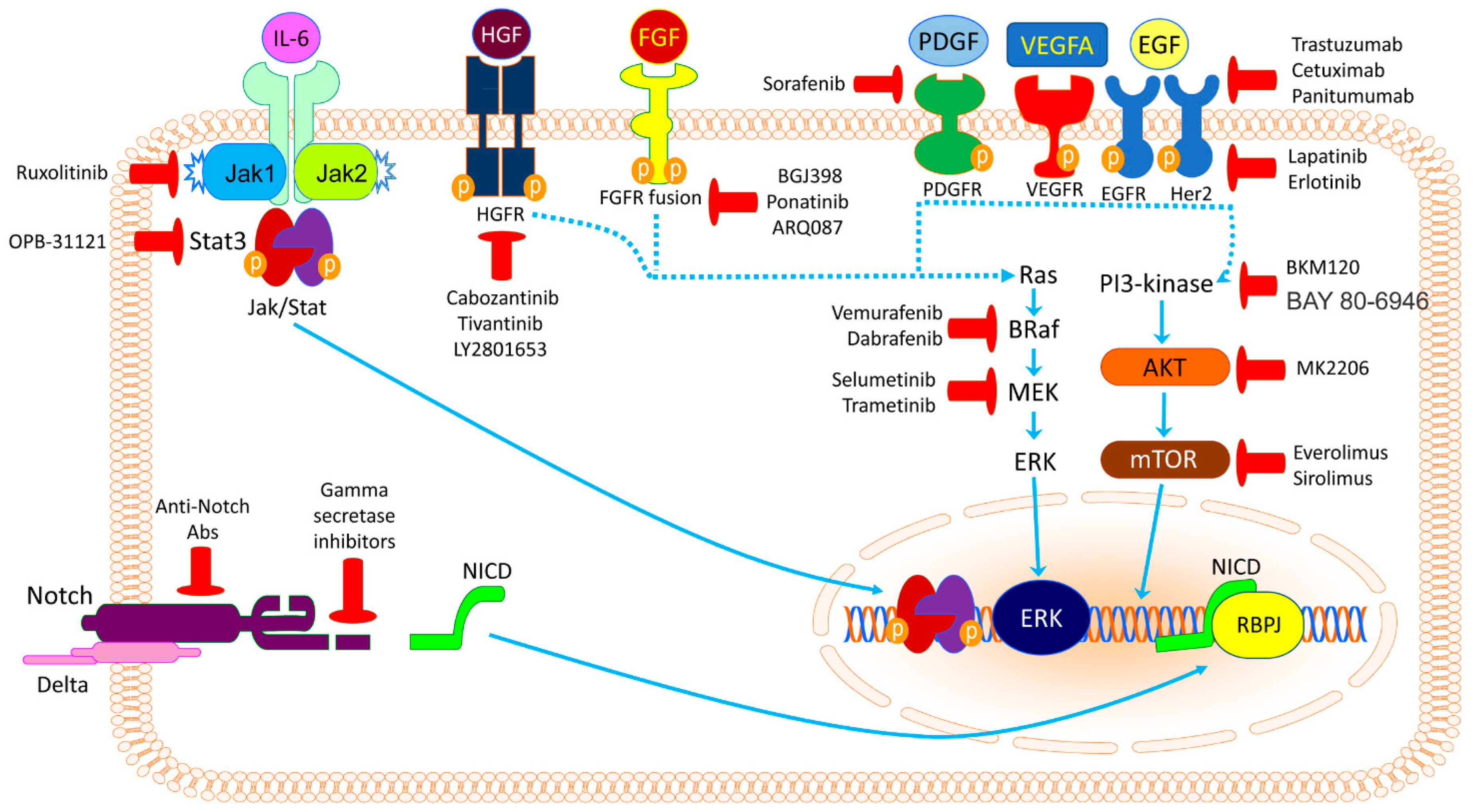
2. Potential Targetable Genes and Molecular Pathways
2.1. Tyrosine Kinase Inhibitors
2.1.1. ErbB Inhibitors
2.1.2. VEGFR and PDGFR Inhibitors
2.1.3. FGFR Inhibitors
2.1.4. MET Inhibitors
2.1.5. ROS1 (ALK) Inhibitors
2.2. RAS/RAF/MEK/ERK Signaling Pathway Inhibitors
2.3. PI3K/AKT/mTOR Signaling Pathway Inhibitors
2.4. Glucose Metabolism Enzyme Inhibitors
2.5. Promising Targets and Inhibitors
2.6. Rising Hopes from CCA Immunotherapy
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bergquist, A.; von Seth, E. Epidemiology of cholangiocarcinoma. Best. Pract. Res. Clin. Gastroenterol. 2015, 29, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Fan, B.; Malato, Y.; Calvisi, D.F.; Naqvi, S.; Razumilava, N.; Ribback, S.; Gores, G.J.; Dombrowski, F.; Evert, M.; Chen, X.; et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Invest. 2012, 122, 2911–2915. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, S.; Suzuki, A. Intrahepatic cholangiocarcinoma can arise from Notch-mediated conversion of hepatocytes. J. Clin. Invest. 2012, 122, 3914–3918. [Google Scholar] [CrossRef] [PubMed]
- Blechacz, B.; Gores, G.J. Cholangiocarcinoma: Advances in pathogenesis, diagnosis, and treatment. Hepatology 2008, 48, 308–321. [Google Scholar] [CrossRef] [PubMed]
- Patel, T. Cholangiocarcinoma—Controversies and challenges. Nat. Rev. Gastroenterol. Hepatoogy. 2011, 8, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Kirstein, M.M.; Vogel, A. Epidemiology and Risk Factors of Cholangiocarcinoma. Visc. Med. 2016, 32, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, S.; Beretta, G.D.; Labianca, R.; Zampino, M.G.; Gatta, G.; Heinemann, V. Cholangiocarcinoma. Crit. Rev. Oncol. Hematol. 2009, 69, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Patel, T. Worldwide trends in mortality from biliary tract malignancies. BMC Cancer 2002, 2, 10. [Google Scholar] [CrossRef]
- Shaib, Y.H.; Davila, J.A.; McGlynn, K.; El-Serag, H.B. Rising incidence of intrahepatic cholangiocarcinoma in the United States: A true increase? J. Hepatol. 2004, 40, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Farley, D.R.; Weaver, A.L.; Nagorney, D.M. “Natural history” of unresected cholangiocarcinoma: Patient outcome after noncurative intervention. Mayo Clin. Proc. 1995, 70, 425–429. [Google Scholar] [CrossRef]
- Ustundag, Y.; Bayraktar, Y. Cholangiocarcinoma: A compact review of the literature. World J. Gastroenterol. 2008, 14, 6458–6466. [Google Scholar] [CrossRef]
- Khan, S.A.; Davidson, B.R.; Goldin, R.D.; Heaton, N.; Karani, J.; Pereira, S.P.; Rosenberg, W.M.; Tait, P.; Taylor-Robinson, S.D.; Thillainayagam, A.V.; et al. British Society of Gastroenterology. Guidelines for the diagnosis and treatment of cholangiocarcinoma: An update. Gut 2012, 61, 1657–1669. [Google Scholar] [CrossRef] [PubMed]
- Everhart, J.E.; Ruhl, C.E. Burden of digestive diseases in the United States Part III: Liver, biliary tract, and pancreas. Gastroenterology 2009, 136, 1134–1144. [Google Scholar] [CrossRef]
- Friman, S. Cholangiocarcinoma—Current treatment options. Scand. J. Surg. 2011, 100, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, D.V.; Zhang, S.; Chen, X.; Calvisi, D.F.; Andersen, J.B. Molecular profiling of intrahepatic cholangiocarcinoma: The search for new therapeutic targets. Expert. Rev. Gastroenterol. Hepatol. 2017, 11, 349–356. [Google Scholar] [CrossRef]
- Sia, D.; Tovar, V.; Moeini, A.; Llovet, J.M. Intrahepatic cholangiocarcinoma: Pathogenesis and rationale for molecular therapies. Oncogene 2013, 32, 4861–4870. [Google Scholar] [CrossRef]
- Sirica, A.E. Role of ErbB family receptor tyrosine kinases in intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2008, 14, 7033–7058. [Google Scholar] [CrossRef]
- Yoshikawa, D.; Ojima, H.; Iwasaki, M.; Hiraoka, N.; Kosuge, T.; Kasai, S.; Hirohashi, S.; Shibata, T. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br. J. Cancer 2008, 98, 418–425. [Google Scholar] [CrossRef]
- Rizvi, S.; Borad, M.J.; Patel, T.; Gores, G.J. Cholangiocarcinoma: Molecular pathways and therapeutic opportunities. Semin. Liver Dis. 2014, 34, 456–464. [Google Scholar] [CrossRef]
- Walter, D.; Hartmann, S.; Waidmann, O. Update on cholangiocarcinoma: Potential impact of genomic studies on clinical management. Z. Gastroenterol. 2017, 55, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Borad, M. The rise of the FGFR inhibitor in advanced biliary cancer: The next cover of time magazine? J. Gastrointest. Oncol. 2016, 7, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Moeini, A.; Sia, D.; Bardeesy, N.; Mazzaferro, V.; Llovet, J.M. Molecular Pathogenesis and Targeted Therapies for Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2016, 22, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, M.; Ojima, H.; Iwasaki, M.; Shimizu, H.; Kokubu, A.; Hiraoka, N.; Kosuge, T.; Yoshikawa, D.; Kono, T.; Furukawa, H.; et al. Prognostic significance of overexpression of c-Met oncoprotein in cholangiocarcinoma. Br. J. Cancer 2011, 105, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Terada, T.; Nakanuma, Y.; Sirica, A.E. Immunohistochemical demonstration of MET overexpression in human intrahepatic cholangiocarcinoma and in hepatolithiasis. Hum. Pathol. 1998, 29, 175–180. [Google Scholar] [CrossRef]
- Rahnemai-Azar, A.A.; Weisbrod, A.B.; Dillhoff, M.; Schmidt, C.; Pawlik, T.M. Intrahepatic cholangiocarcinoma: Current management and emerging therapies. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef]
- Paliogiannis, P.; Attene, F.; Cossu, A.; Defraia, E.; Porcu, G.; Carta, A.; Sotgiu, M.I.; Pazzola, A.; Cordero, L.; Capelli, F.; et al. Impact of tissue type and content of neoplastic cells of samples on the quality of epidermal growth factor receptor mutation analysis among patients with lung adenocarcinoma. Mol. Med. Rep. 2015, 12, 187–191. [Google Scholar] [CrossRef]
- Han, W.; Lo, H.W. Landscape of EGFR signaling network in human cancers: Biology and therapeutic response in relation to receptor subcellular locations. Cancer Lett. 2012, 318, 124–134. [Google Scholar] [CrossRef]
- Sharip, A.; Abdukhakimova, D.; Wang, X.; Kim, A.; Kim, Y.; Sharip, A.; Orakov, A.; Miao, L.; Sun, Q.; Chen, Y.; et al. Analysis of origin and protein-protein interaction maps suggests distinct oncogenic role of nuclear EGFR during cancer evolution. J. Cancer 2017, 8, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Matsuoka, Y.; Funahashi, A.; Kitano, H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol. 2005, 1. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Oyesanya, R.A.; Campbell, D.J.; Almenara, J.A.; Dewitt, J.L.; Sirica, A.E. Preclinical assessment of simultaneous targeting of epidermal growth factor receptor (ErbB1) and ErbB2 as a strategy for cholangiocarcinoma therapy. Hepatology 2010, 52, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Philip, P.A.; Mahoney, M.R.; Allmer, C.; Thomas, J.; Pitot, H.C.; Kim, G.; Donehower, R.C.; Fitch, T.; Picus, J.; Erlichman, C. Phase II study of erlotinib in patients with advanced biliary cancer. J. Clin. Oncol. 2006, 24, 3069–3074. [Google Scholar] [CrossRef] [PubMed]
- Gruenberger, B.; Schueller, J.; Heubrandtner, U.; Wrba, F.; Tamandl, D.; Kaczirek, K.; Roka, R.; Freimann-Pircher, S.; Gruenberger, T. Cetuximab, gemcitabine, and oxaliplatin in patients with unresectable advanced or metastatic biliary tract cancer: A phase 2 study. Lancet Oncol. 2010, 11, 1142–1148. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; Belani, C.P.; Singh, D.A.; Tanaka, M.; Lenz, H.J.; Yen, Y.; Kindler, H.L.; Iqbal, S.; Longmate, J.; Mack, P.C.; et al. A phase II study of lapatinib in patients with advanced biliary tree and hepatocellular cancer. Cancer Chemother. Pharmacol. 2009, 64, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Peck, J.; Wei, L.; Zalupski, M.; O’Neil, B.; Villalona Calero, M.; Bekaii-Saab, T. HER2/neu may not be an interesting target in biliary cancers: Results of an early phase II study with lapatinib. Oncology 2012, 82, 175–179. [Google Scholar] [CrossRef]
- Jensen, L.H.; Lindebjerg, J.; Ploen, J.; Hansen, T.F.; Jakobsen, A. Phase II marker-driven trial of panitumumab and chemotherapy in KRAS wild-type biliary tract cancer. Ann. Oncol. 2012, 23, 2341–2346. [Google Scholar] [CrossRef]
- Leone, F.; Marino, D.; Cereda, S.; Filippi, R.; Belli, C.; Spadi, R.; Nasti, G.; Montano, M.; Amatu, A.; Aprile, G.; et al. Panitumumab in combination with gemcitabine and oxaliplatin does not prolong survival in wild-type KRAS advanced biliary tract cancer: A randomized phase 2 trial (Vecti-BIL study). Cancer 2016, 122, 574–581. [Google Scholar] [CrossRef]
- Ole Larsen, F.; Taksony Solyom Hoegdall, D.; Hoegdall, E.; Nielsen, D. Gemcitabine, capecitabine and oxaliplatin with or without cetuximab in advanced biliary tract carcinoma. Acta Oncol. 2016, 55, 382–385. [Google Scholar] [CrossRef]
- Lee, J.; Park, S.; Chang, H.M.; Kim, J.S.; Choi, H.J.; Lee, M.A.; Jang, J.S.; Jeung, H.C.; Kang, J.H.; Lee, H.W.; et al. Gemcitabine and oxaliplatin with or without erlotinib in advanced biliary-tract cancer: A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2012, 13, 181–188. [Google Scholar] [CrossRef]
- Javle, M.; Churi, C.; Kang, H.C.; Shroff, R.; Janku, F.; Surapaneni, R.; Zuo, M.; Barrera, C.; Alshamsi, H.; Krishnan, S.; et al. HER2/neu-directed therapy for biliary tract cancer. J. Hematol. Oncol. 2015, 8, 58. [Google Scholar] [CrossRef]
- Kessler, E.R.; Eckhardt, S.G.; Pitts, T.M.; Bradshaw-Pierce, E.L.; O’byrant, C.L.; Messersmith, W.A.; Nallapreddy, S.; Weekes, C.; Spratlin, J.; Lieu, C.H.; et al. Phase I trial of vandetanib in combination with gemcitabine and capecitabine in patients with advanced solid tumors with an expanded cohort in pancreatic and biliary cancers. Invest. New Drugs 2016, 34, 176–183. [Google Scholar] [CrossRef]
- Santoro, A.; Gebbia, V.; Pressiani, T.; Testa, A.; Personeni, N.; Arrivas Bajardi, E.; Foa, P.; Buonadonna, A.; Bencardino, K.; Barone, C.; et al. A randomized, multicenter, phase II study of vandetanib monotherapy versus vandetanib in combination with gemcitabine versus gemcitabine plus placebo in subjects with advanced biliary tract cancer: The VanGogh study. Ann. Oncol. 2015, 26, 542–547. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis. A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Tang, D.; Nagano, H.; Yamamoto, H.; Wada, H.; Nakamura, M.; Kondo, M.; Ota, H.; Yoshioka, S.; Kato, H.; Damdinsuren, B.; et al. Angiogenesis in cholangiocellular carcinoma: Expression of vascular endothelial growth factor, angiopoietin-1/2, thrombospondin-1 and clinicopathological significance. Oncol. Rep. 2006, 15, 525–532. [Google Scholar] [CrossRef]
- Guedj, N.; Zhan, Q.; Perigny, M.; Rautou, P.E.; Degos, F.; Belghiti, J.; Farges, O.; Bedossa, P.; Paradis, V. Comparative protein expression profiles of hilar and peripheral hepatic cholangiocarcinomas. J. Hepatol. 2009, 51, 93–101. [Google Scholar] [CrossRef]
- Boonjaraspinyo, S.; Boonmars, T.; Wu, Z.; Loilome, W.; Sithithaworn, P.; Nagano, I.; Pinlaor, S.; Yongvanit, P.; Nielsen, P.S.; Pairojkul, C.; et al. Platelet-derived growth factor may be a potential diagnostic and prognostic marker for cholangiocarcinoma. Tumour Biol. 2012, 33, 1785–1802. [Google Scholar] [CrossRef]
- Vaeteewoottacharn, K.; Kariya, R.; Dana, P.; Fujikawa, S.; Matsuda, K.; Ohkuma, K.; Kudo, E.; Kraiklang, R.; Wongkham, C.; Wongkham, S.; et al. Inhibition of carbonic anhydrase potentiates bevacizumab treatment in cholangiocarcinoma. Tumour Biol. 2016, 37, 9023–9035. [Google Scholar] [CrossRef]
- Zhu, A.X.; Meyerhardt, J.A.; Blaszkowsky, L.S.; Kambadakone, A.R.; Muzikansky, A.; Zheng, H.; Clark, J.W.; Abrams, T.A.; Chan, J.A.; Enzinger, P.C.; et al. Efficacy and safety of gemcitabine, oxaliplatin, and bevacizumab in advanced biliary-tract cancers and correlation of changes in 18-fluorodeoxyglucose PET with clinical outcome: A phase 2 study. Lancet Oncol. 2010, 11, 48–54. [Google Scholar] [CrossRef]
- Lubner, S.J.; Mahoney, M.R.; Kolesar, J.L.; Loconte, N.K.; Kim, G.P.; Pitot, H.C.; Philip, P.A.; Picus, J.; Yong, W.P.; Horvath, L.; et al. Report of a multicenter phase II trial testing a combination of biweekly bevacizumab and daily erlotinib in patients with unresectable biliary cancer: A phase II Consortium study. J. Clin. Oncol. 2010, 28, 3491–3497. [Google Scholar] [CrossRef]
- Guion-Dusserre, J.F.; Lorgis, V.; Vincent, J.; Bengrine, L.; Ghiringhelli, F. FOLFIRI plus bevacizumab as a second-line therapy for metastatic intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2015, 21, 2096–2101. [Google Scholar] [CrossRef]
- Bengala, C.; Bertolini, F.; Malavasi, N.; Boni, C.; Aitini, E.; Dealis, C.; Zironi, S.; Depenni, R.; Fontana, A.; Del Giovane, C.; et al. Sorafenib in patients with advanced biliary tract carcinoma: A phase II trial. Br. J. Cancer 2010, 102, 68–72. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Rankin, C.J.; Ben-Josef, E.; Lenz, H.J.; Gold, P.J.; Hamilton, R.D.; Govindarajan, R.; Eng, C.; Blanke, C.D. SWOG 0514: A phase II study of sorafenib in patients with unresectable or metastatic gallbladder carcinoma and cholangiocarcinoma. Invest. New Drugs 2012, 30, 1646–1651. [Google Scholar] [CrossRef]
- Moehler, M.; Maderer, A.; Schimanski, C.; Kanzler, S.; Denzer, U.; Kolligs, F.T.; Ebert, M.P.; Distelrath, A.; Geissler, M.; Trojan, J.; et al. Gemcitabine plus sorafenib versus gemcitabine alone in advanced biliary tract cancer: A double-blind placebo-controlled multicentre phase II AIO study with biomarker and serum programme. Eur. J. Cancer 2014, 50, 3125–3135. [Google Scholar] [CrossRef]
- Lee, J.K.; Capanu, M.; O’Reilly, E.M.; Ma, J.; Chou, J.F.; Shia, J.; Katz, S.S.; Gansukh, B.; Reidy-Lagunes, D.; Segal, N.H.; et al. A phase II study of gemcitabine and cisplatin plus sorafenib in patients with advanced biliary adenocarcinomas. Br. J. Cancer 2013, 109, 915–919. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Rankin, C.; Siegel, A.B.; Iqbal, S.; Gong, I.Y.; Micetich, K.C.; Kayaleh, O.R.; Lenz, H.J.; Blanke, C.D. S0941: A phase 2 SWOG study of sorafenib and erlotinib in patients with advanced gallbladder carcinoma or cholangiocarcinoma. Br. J. Cancer 2014, 110, 882–887. [Google Scholar] [CrossRef]
- Valle, J.W.; Wasan, H.; Lopes, A.; Backen, A.C.; Palmer, D.H.; Morris, K.; Duggan, M.; Cunningham, D.; Anthoney, D.A.; Corrie, P.; et al. Cediranib or placebo in combination with cisplatin and gemcitabine chemotherapy for patients with advanced biliary tract cancer (ABC-03): A randomised phase 2 trial. Lancet Oncol. 2015, 16, 967–978. [Google Scholar] [CrossRef]
- Sleeman, M.; Fraser, J.; McDonald, M.; Yuan, S.; White, D.; Grandison, P.; Kumble, K.; Watson, J.D.; Murison, J.G. Identification of a new fibroblast growth factor receptor, FGFR5. Gene 2001, 271, 171–182. [Google Scholar] [CrossRef]
- Duchesne, L.; Tissot, B.; Rudd, T.R.; Dell, A.; Fernig, D.G. N-glycosylation of fibroblast growth factor receptor 1 regulates ligand and heparan sulfate co-receptor binding. J. Biol. Chem. 2006, 281, 27178–27189. [Google Scholar] [CrossRef]
- Coutts, J.C.; Gallagher, J.T. Receptors for fibroblast growth factors. Immunol. Cell. Biol. 1995, 73, 584–589. [Google Scholar] [CrossRef]
- Belov, A.A.; Mohammadi, M. Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb. Perspect. Biol. 2013, 5, 6. [Google Scholar] [CrossRef]
- Rizvi, S.; Yamada, D.; Hirsova, P.; Bronk, S.F.; Werneburg, N.W.; Krishnan, A.; Salim, W.; Zhang, L.; Trushina, E.; Truty, M.J.; et al. A Hippo and Fibroblast Growth Factor Receptor Autocrine Pathway in Cholangiocarcinoma. J. Biol. Chem. 2016, 291, 8031–8047. [Google Scholar] [CrossRef]
- Cooper, C.S. The met oncogene: From detection by transfection to transmembrane receptor for hepatocyte growth factor. Oncogene 1992, 7, 3–7. [Google Scholar]
- Goyal, L.; Yurgelun, M.B.; Abrams, T.A.; Kwak, E.L.; Cleary, J.M.; Knowles, M.; Regan, E.; Gisondi, A.; Sheehan, S.; Zheng, H.; et al. A phase II trial of cabozantinib (XL-184) in patients with advanced cholangiocarcinoma. J. Clin. Oncol. 2015, 33. [Google Scholar] [CrossRef]
- Pant, S.; Saleh, M.; Bendell, J.; Infante, J.R.; Jones, S.; Kurkjian, C.D.; Moore, K.M.; Kazakin, J.; Abbadessa, G.; Wang, Y.; et al. A phase I dose escalation study of oral c-MET inhibitor tivantinib (ARQ 197) in combination with gemcitabine in patients with solid tumors. Ann. Oncol. 2014, 25, 1416–1421. [Google Scholar] [CrossRef]
- Davies, K.D.; Le, A.T.; Theodoro, M.F.; Skokan, M.C.; Aisner, D.L.; Berge, E.M.; Terracciano, L.M.; Cappuzzo, F.; Incarbone, M.; Roncalli, M.; et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin. Cancer Res. 2012, 18, 4570–4579. [Google Scholar] [CrossRef]
- Morgan, P.; Woolacott, N.; Biswas, M.; Mebrahtu, T.; Harden, M.; Hodgson, R. Crizotinib for untreated anaplastic lymphoma kinase-positive non-small-cell lung cancer: An evidence review group perspective of a NICE single technology appraisal. Pharmacoeconomics 2017, 35, 909–919. [Google Scholar] [CrossRef]
- Palomba, G.; Doneddu, V.; Cossu, A.; Paliogiannis, P.; Manca, A.; Casula, M.; Colombino, M.; Lanzillo, A.; Defraia, E.; Pazzola, A.; et al. Prognostic impact of KRAS, NRAS, BRAF, and PIK3CA mutations in primary colorectal carcinomas: A population-based study. J. Transl. Med. 2016, 14, 292. [Google Scholar] [CrossRef]
- Palmieri, G.; Ombra, M.; Colombino, M.; Casula, M.; Sini, M.; Manca, A.; Paliogiannis, P.; Ascierto, P.A.; Cossu, A. Multiple Molecular Pathways in Melanomagenesis: Characterization of Therapeutic Targets. Front. Oncol. 2015, 5, 183. [Google Scholar] [CrossRef]
- Silkin, S.V.; Startsev, S.S.; Krasnova, M.E.; Raskin, G.A.; Mitiushkina, N.V.; Iyevleva, A.G.; Sokolenko, A.P.; Imyanitov, E.N. Complete Clinical Response of BRAF-Mutated Cholangiocarcinoma to Vemurafenib, Panitumumab, and Irinotecan. J. Gastrointest. Cancer 2016, 47, 502–505. [Google Scholar] [CrossRef]
- Ioka, T.; Ikeda, M.; Fukutomi, A.; Morizane, C.; Kasuga, A.; Takada, R.; Takahashi, H.; Todaka, A.; Okusaka, T.; Creasy, C.; et al. A proof-of-concept study of MEK inhibitor trametinib monotherapy in patients with biliary tract cancers. Eur. J. Cancer 2015, 51, S464. [Google Scholar] [CrossRef]
- King, D.; Yeomanson, D.; Bryant, H.E. PI3King the lock: Targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma. J. Pediatr. Hematol. Oncol. 2015, 37, 245–251. [Google Scholar] [CrossRef]
- McRee, A.J.; Sanoff, H.K.; Carlson, C.; Ivanova, A.; O’Neil, B.H. A phase I trial of mFOLFOX6 combined with the oral PI3K inhibitor BKM120 in patients with advanced refractory solid tumors. Invest. New Drugs 2015, 33, 1225–1231. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov (accessed on 23 January 2019).
- Corpas, F.J.; Barroso, J.B.; Sandalio, L.M.; Palma, J.M.; Lupiáñez, J.A.; del Río, L.A. Peroxisomal NADP-Dependent Isocitrate Dehydrogenase. Characterization and Activity Regulation during Natural Senescence. Plant Physiol. 1999, 121, 921–928. [Google Scholar] [CrossRef]
- Lowery, M.A.; Abou-Alfa, G.K.; Burris, H.A.; Janku, F.; Shroff, R.T.; Cleary, J.M.; Azad, N.S.; Goyal, L.; Maher, E.A.; Gore, L.; et al. Phase I study of AG-120, an IDH1 mutant enzyme inhibitor: Results from the cholangiocarcinoma dose escalation and expansion cohorts. J. Clin. Oncol. 2017, 35, 4015. [Google Scholar] [CrossRef]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don’t know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef]
- Yang, J.; Farren, M.R.; Ahn, D.; Bekaii-Saab, T.; Lesinski, G.B. Signaling pathways as therapeutic targets in biliary tract cancer. Expert. Opin. Ther. Targets 2017, 21, 485–498. [Google Scholar] [CrossRef]
- Okusaka, T.; Ueno, H.; Ikeda, M.; Mitsunaga, S.; Ozaka, M.; Ishii, H.; Yokosuka, O.; Ooka, Y.; Yoshimoto, R.; Yanagihara, Y.; et al. Phase 1 and pharmacological trial of OPB-31121, a signal transducer and activator of transcription-3 inhibitor, in patients with advanced hepatocellular carcinoma. Hepatol. Res. 2015, 45, 1283–1291. [Google Scholar] [CrossRef]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef]
- Morell, C.M.; Strazzabosco, M. Notch signaling and new therapeutic options in liver disease. J. Hepatol. 2014, 60, 885–890. [Google Scholar] [CrossRef]
- Cigliano, A.; Wang, J.; Chen, X.; Calvisi, D.F. Role of the Notch signaling in cholangiocarcinoma. Expert. Opin. Ther. Targets 2017, 21, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Lum, L.; Beachy, P.A. The Hedgehog response network: Sensors, switches, and routers. Science 2004, 304, 1755–1759. [Google Scholar] [CrossRef]
- Riedlinger, D.; Bahra, M.; Boas-Knoop, S.; Lippert, S.; Bradtmöller, M.; Guse, K.; Seehofer, D.; Bova, R.; Sauer, I.M.; Neuhaus, P.; et al. Hedgehog pathway as a potential treatment target in human cholangiocarcinoma. J. Hepatobiliary Pancreat. Sci. 2014, 21, 607–615. [Google Scholar] [CrossRef]
- El Khatib, M.; Kalnytska, A.; Palagani, V.; Kossatz, U.; Manns, M.P.; Malek, N.P.; Wilkens, L.; Plentz, R.R. Inhibition of hedgehog signaling attenuates carcinogenesis in vitro and increases necrosis of cholangiocellular carcinoma. Hepatology 2013, 57, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gradilone, S.A.; Smoot, R.L.; Mertens, J.C.; Bronk, S.F.; Sirica, A.E.; Gores, G.J. Non-canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. J. Hepatol. 2014, 60, 599–605. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Sirica, A.E.; Gores, G.J. Desmoplastic stroma and cholangiocarcinoma: Clinical implications and therapeutic targeting. Hepatology 2014, 59, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Gores, G.J. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 2013, 145, 1215–1229. [Google Scholar] [CrossRef]
- DeClerck, Y.A. Desmoplasia: A response or a niche? Cancer Discov. 2012, 2, 772–774. [Google Scholar] [CrossRef]
- Ling, H.; Roux, E.; Hempel, D.; Tao, J.; Smith, M.; Lonning, S. Transforming growth factor β neutralization ameliorates pre-existing hepatic fibrosis and reduces cholangiocarcinoma in thioacetamide-treated rats. PLoS ONE 2013, 8, e54499. [Google Scholar] [CrossRef] [PubMed]
- Pinlaor, S.; Prakobwong, S.; Hiraku, Y.; Pinlaor, P.; Laothong, U.; Yongvanit, P. Reduction of periductal fibrosis in liver fluke-infected hamsters after long-term curcumin treatment. Eur. J. Pharmacol. 2010, 638, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Zhou, L.; Xie, X.; Jiang, G.; Xie, H.; Zheng, S. Interaction of B7-H1 on intrahepatic cholangiocarcinoma cells with PD-1 on tumor-infiltrating T cells as a mechanism of immune evasion. J. Surg. Oncol. 2009, 100, 500–504. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene | % | Comment |
|---|---|---|
| EGFR | 1.6 | 2.2–20% in other series [17,18,19,20,21]. Overexpression of EGFR and/or HER2/neu has been documented in 10–32% of iCCAs [17,18,19]. |
| VEGF | 0.7 | VEGF overexpression has been reported in about 54% of iCCAs [19]. |
| KRAS | 23 | More common in eCCAs [20,21]. |
| NRAS | 4 | Similar distribution between iCCAs and eCCAs [20,21]. |
| BRAF | 4 | There were no BRAF mutations in 137 eCCA cases reviewed by Walter et al. They were detected in 3.3% of 723 iCCAs [20,21]. |
| FGFR2 | 2.1 | FGFR2 fusions were observed in approximately 3–50% of iCCAS [22,23,24]. |
| MET | 0.7 | MET has been found overexpressed in 12–58% of iCCAs [25,26]. |
| ROS1 | 0.7 | In other reports, the frequency of ROS1 alterations varies between 1.1% and 8.8% [27]. |
| PIK3CA | 7 | Slightly more frequent in eCCAs in accordance with Walter et al. [21]. |
| PTEN | 3.3 | Similar distribution between iCCAs and eCCAs [20,21]. |
| IDH1 | 9 | Rare in eCCAs [20,21]. |
| IDH2 | 3 | Not found in eCCAs [20,21]. |
| JAK1/2, STAT3 | 0.6–1% | JAK/STAT signaling pathway activated in 50–70% of iCCAS [28]. |
| ARID1A | 13 | Similar distribution between iCCAs and eCCAs [20,21]. |
| PBRM1 | 6 | More common in iCCAs [20,21]. |
| BAP1 | 9 | Rare in eCCAs [20,21]. |
| Treatment | Target(s) | Phase | Identifier |
|---|---|---|---|
| Gemcitabine + oxaliplatin + capecitabine vs. gemcitabine + oxaliplatin + panitumumab + capecitabine | EGFR | II | NCT00779454 |
| Gemcitabine + oxaliplatin + capecitabine + panitumumab or bevacizumab | EGFR − VEGFR | II | NCT01206049 |
| Trastuzumab + tipifarnib | HER2/neu + FTI | I | NCT00005842 |
| Varlitinib | EGFR | II | NCT02609958 |
| Gemcitabine + oxaliplatin + cetuximab + trastuzumab + gefitinib + lapatinib + sorafenib + crizotinib | Multiple targets | I/II | NCT02836847 |
| CART-EGFR | EGFR | I/II | NCT01869166 |
| Afatinib + capecitabine | EGFR | I | NCT02451553 |
| LY2801653 + cetuximab or cisplatin or gemcitabine or ramucirumab | Multiple targets | I | NCT01438554 |
| Pazopanib + GSK1120212 | Multiple targets | I | NCT01438554 |
| Sunitinib | Multiple targets | II | NCT01718327 |
| Gemcitabine + pazopanib | Multiple targets | II | NCT01855724 |
| Regorafenib | Multiple targets | II | NCT02053376 NCT02162914 |
| Gemcitabine + oxaliplatin + regorafenib | Multiple targets | II | NCT02386397 |
| Ramucirumab | VEGFR | II | NCT02520141 |
| Ramucirumab + pembrolizumab | Multiple targets | I | NCT02443324 |
| Cediranib + AZD0530 | Multiple targets | I | NCT00475956 |
| Oxaliplatin + leucovorin calcium + fluorouracil + cediranib | Multiple targets | II | NCT01229111 |
| Sorafenib | VEGFR − PDGFR − BRAF | II | NCT00238212 |
| Sorafenib + oxaliplatin/capecitabine | VEGFR − PDGFR − BRAF | I/II | NCT00634751 |
| Ponatinib | FGFR | II | NCT02265341 NCT02272998 |
| JNJ-42756493 | FGFR | I | NCT01703481 |
| BGJ398 | FGFR2 | II | NCT02150967 |
| ARQ087 | FGFR2 | II | NCT01752920 |
| FRA144 | FGFR2b | I | NCT02318329 |
| Ceritinib | ROS1 − ALK | II | NCT02638909 |
| Entrectinib | ROS1 − ALK | II | NCT02568267 |
| LDK378 (Ceritinib) | ROS1 − ALK | II | NCT02374489 |
| PLX8394 | BRAF | I/II | NCT02428712 NCT02012231 |
| Gemcitabine + selumetinib vs gemcitabine | MEK | II | NCT02151084 |
| Refametinib | MEK | II | NCT02346032 |
| Trametinib vs 5-fluoruracil or capecitabine | MEK | II | NCT02042443 |
| Gemcitabine + MEK162 | MEK | II | NCT01828034 |
| Everolimus + gemcitabine | mTOR | I | NCT00949949 |
| Sirolimus + gemcitabine | mTOR | I | NCT01888302 |
| Copanlisib + gemcitabine | PI3K | II | NCT02631590 |
| AG-881 | IDH | I | NCT02481154 |
| AG-120 | IDH | I | NCT02073994 |
| AG-120 | IDH | III | NCT02989857 |
| Rucaparib + nivolumab | PARP | II | NCT03639935 |
| BBI-503 | STAT3 | II | NCT02232633 |
| Pembrolizumab + GM − CSF | PD1 | II | NCT02703714 |
| Pembrolizumab | PD1 | II | NCT02628067 |
| Pembrolizumab | PD1 | I/II | NCT02268825 |
| Nivolumab or Ipilimumab | PD1 − CTL4 | II | NCT02834013 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simile, M.M.; Bagella, P.; Vidili, G.; Spanu, A.; Manetti, R.; Seddaiu, M.A.; Babudieri, S.; Madeddu, G.; Serra, P.A.; Altana, M.; et al. Targeted Therapies in Cholangiocarcinoma: Emerging Evidence from Clinical Trials. Medicina 2019, 55, 42. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina55020042
Simile MM, Bagella P, Vidili G, Spanu A, Manetti R, Seddaiu MA, Babudieri S, Madeddu G, Serra PA, Altana M, et al. Targeted Therapies in Cholangiocarcinoma: Emerging Evidence from Clinical Trials. Medicina. 2019; 55(2):42. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina55020042
Chicago/Turabian StyleSimile, Maria Maddalena, Paola Bagella, Gianpaolo Vidili, Angela Spanu, Roberto Manetti, Maria Antonietta Seddaiu, Sergio Babudieri, Giordano Madeddu, Pier Andrea Serra, Matteo Altana, and et al. 2019. "Targeted Therapies in Cholangiocarcinoma: Emerging Evidence from Clinical Trials" Medicina 55, no. 2: 42. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina55020042