Inhibition of MELK Protooncogene as an Innovative Treatment for Intrahepatic Cholangiocarcinoma

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Tissue Samples
2.2. Histology and Immunohistochemistry
2.3. Quantitative Reverse Transcription Real-Time Polymerase Chain Reaction (qRT-PCR)
2.4. In Vitro Experiments
2.5. Statistical and Bioinformatic Analysis
3. Results
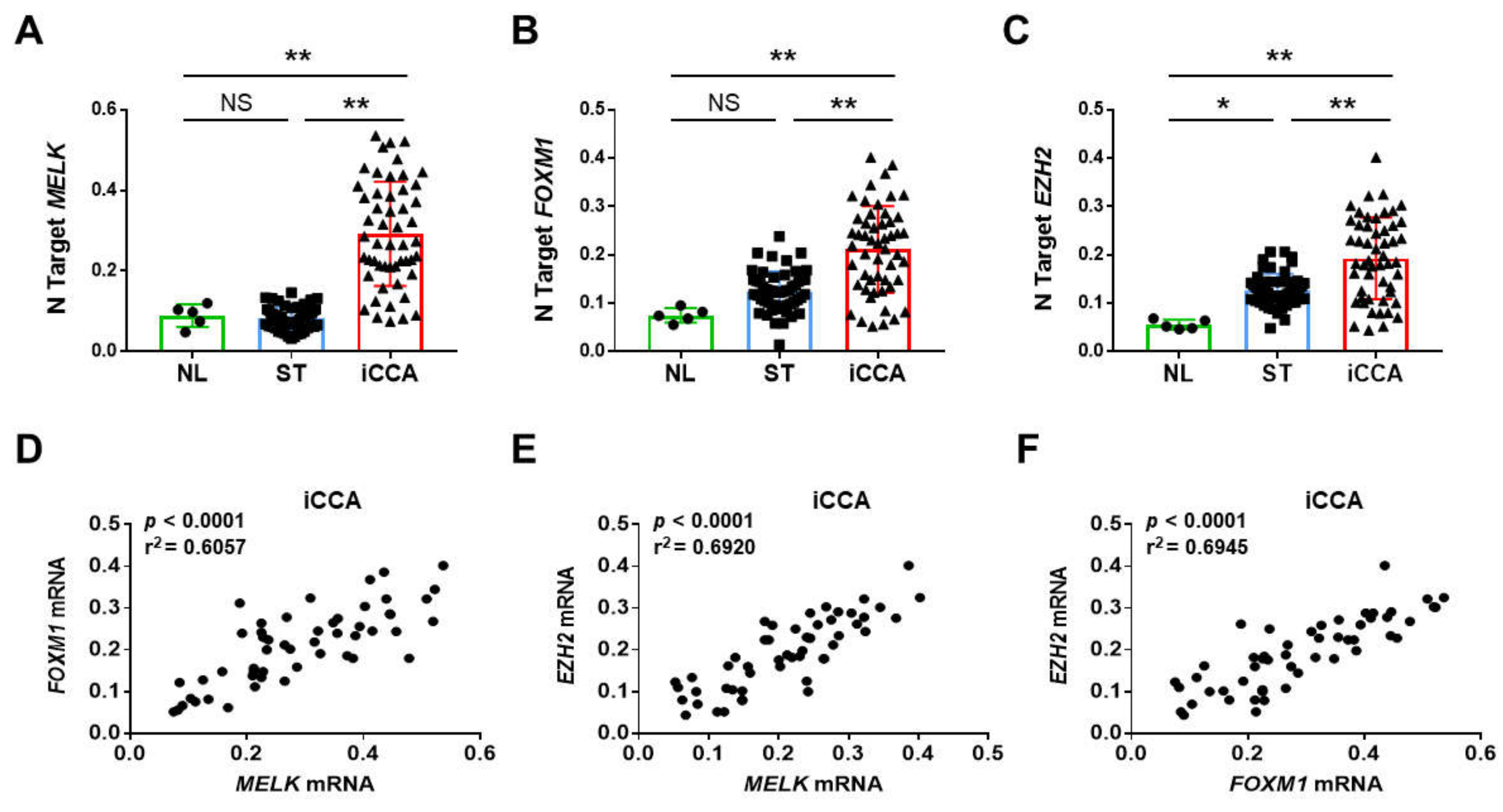
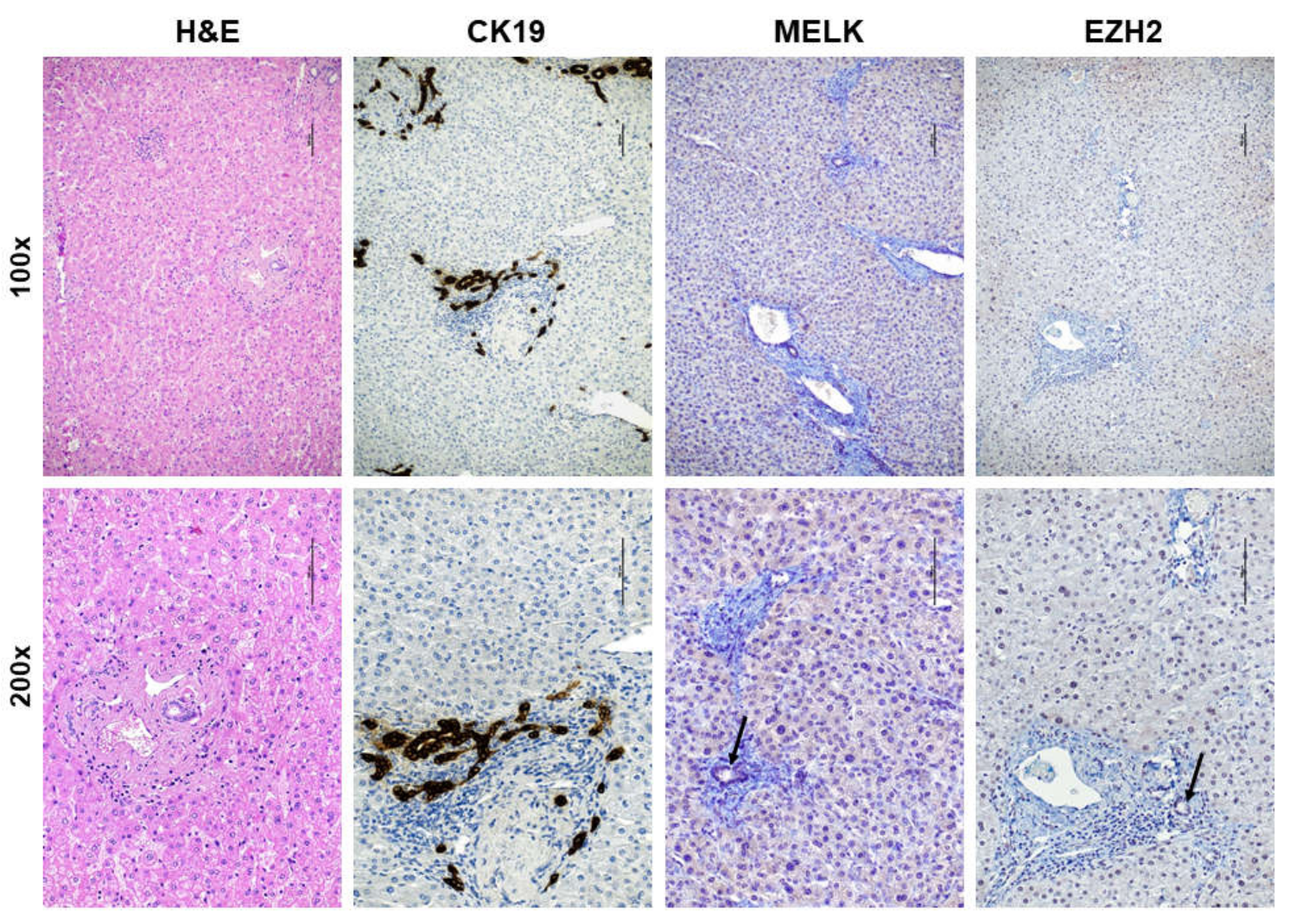
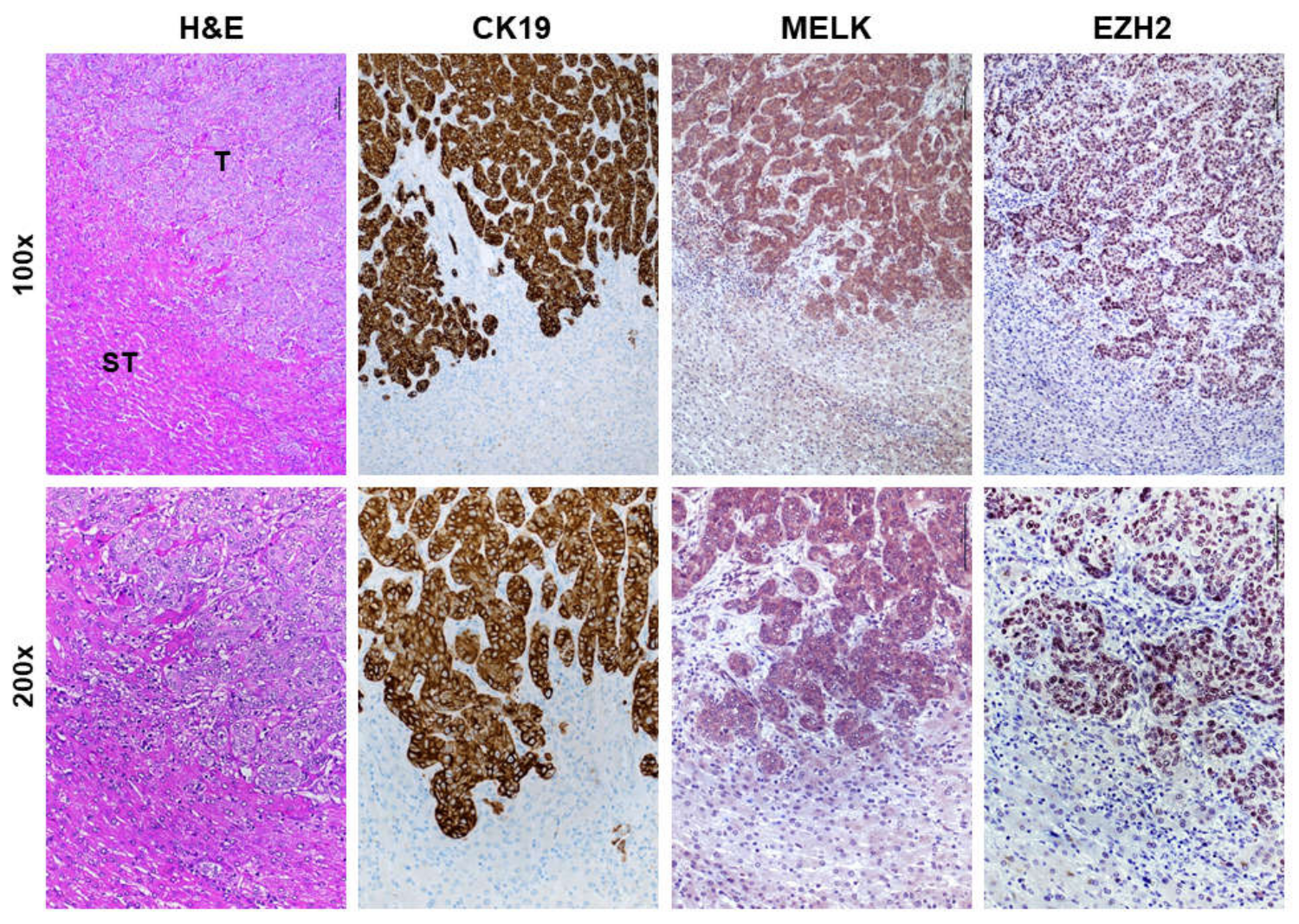
3.1. Overexpression of MELK in Human Intrahepatic Cholangiocarcinoma
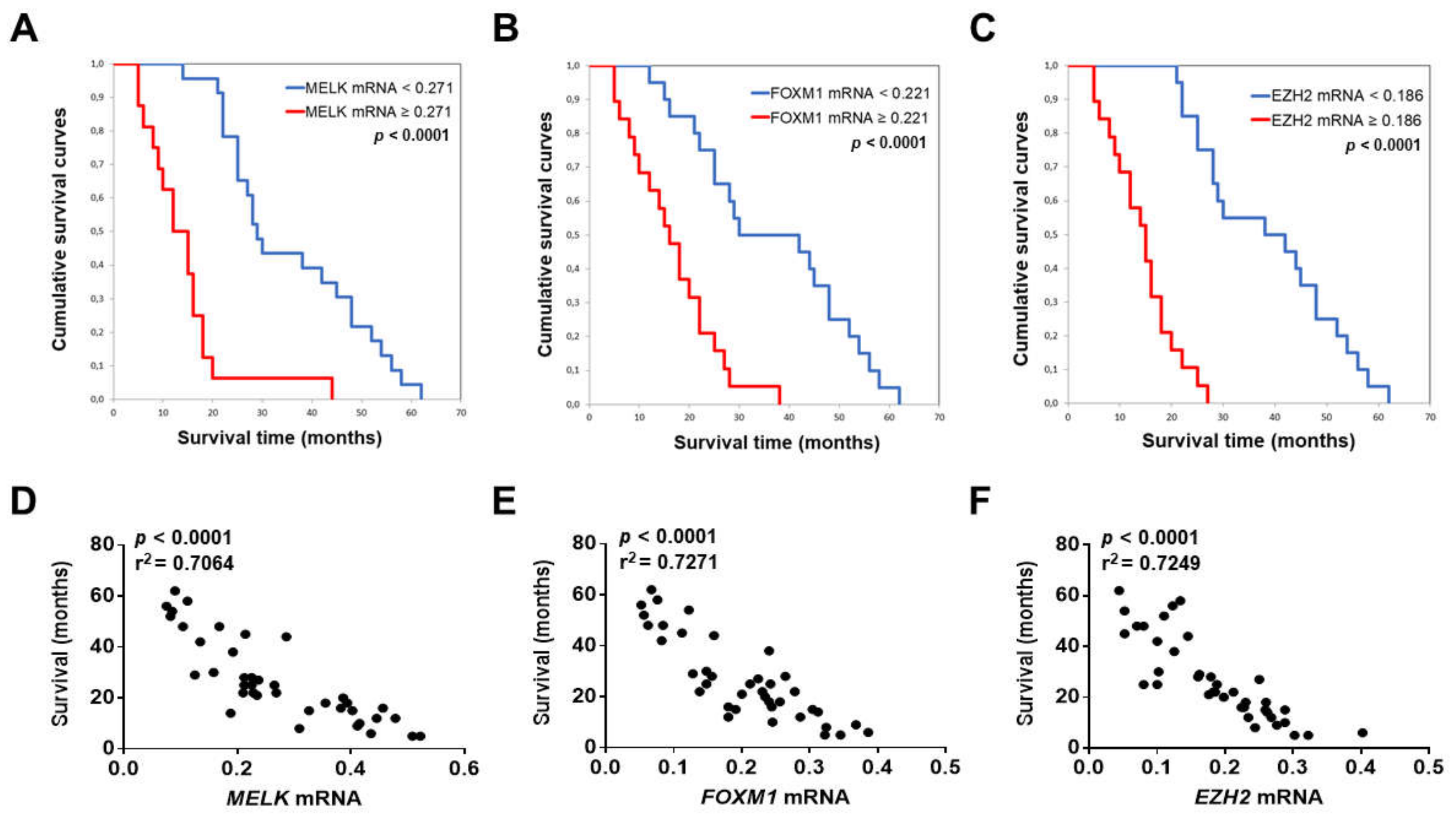
3.2. MELK is a Negative Prognostic Indicator in Intrahepatic Cholangiocarcinoma
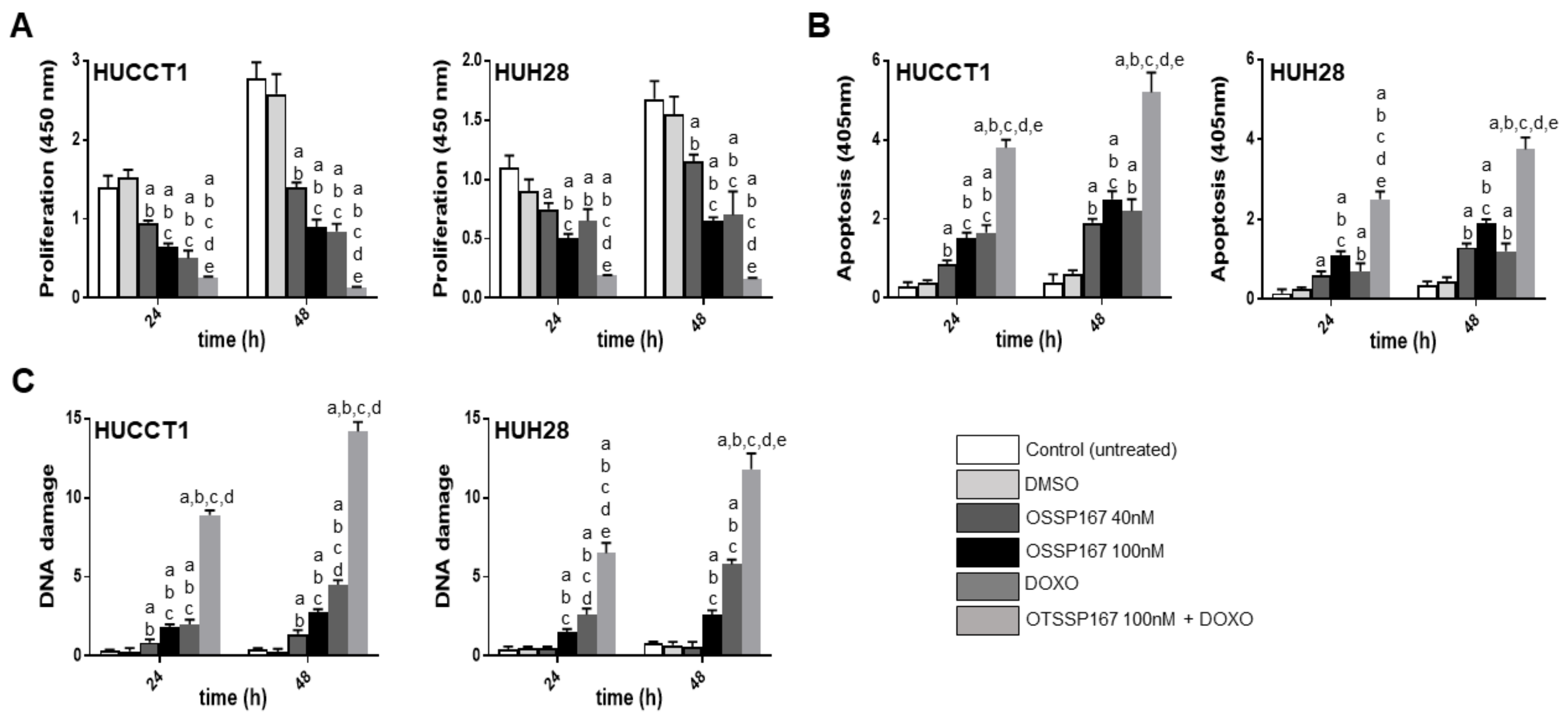
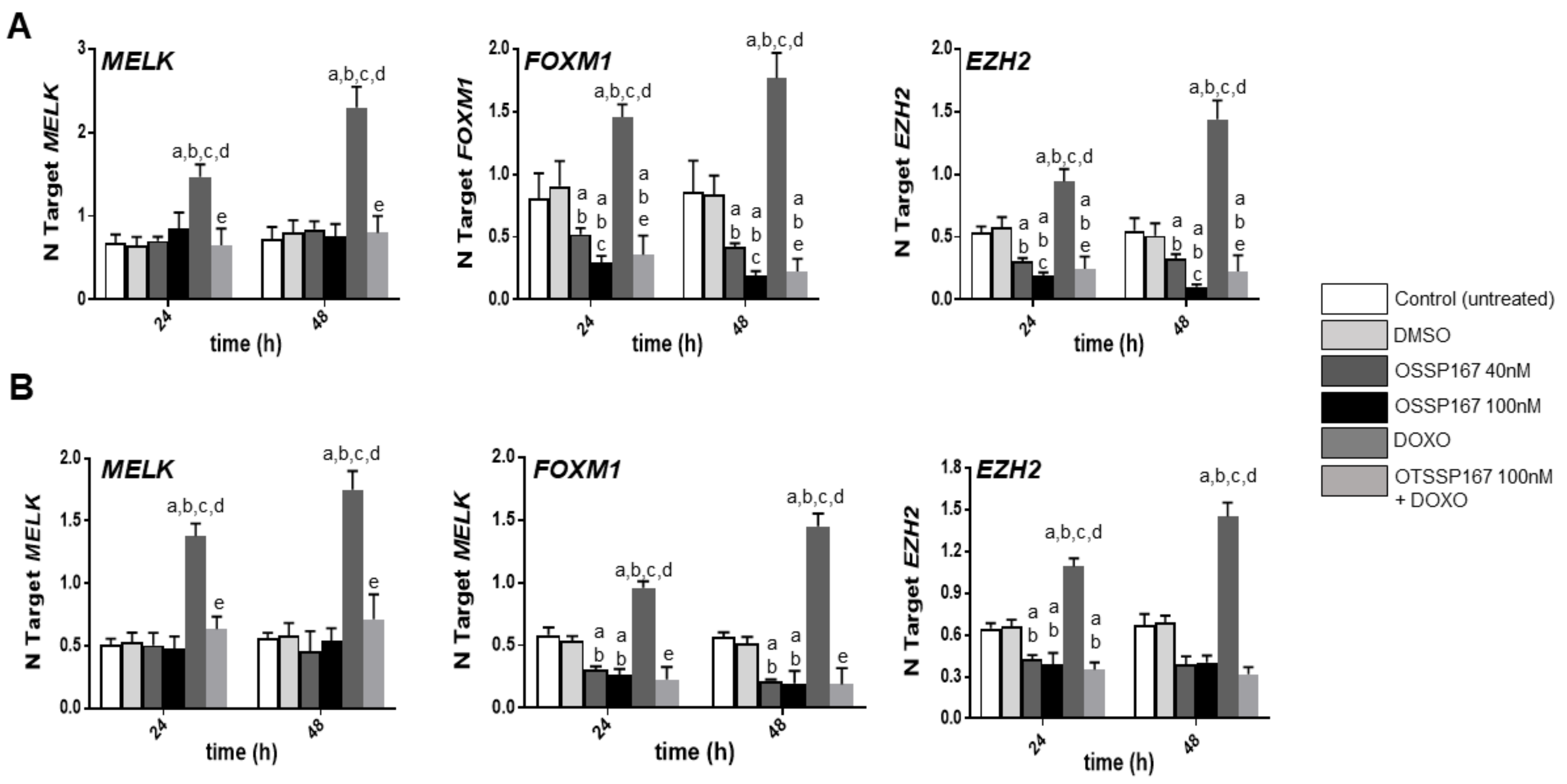
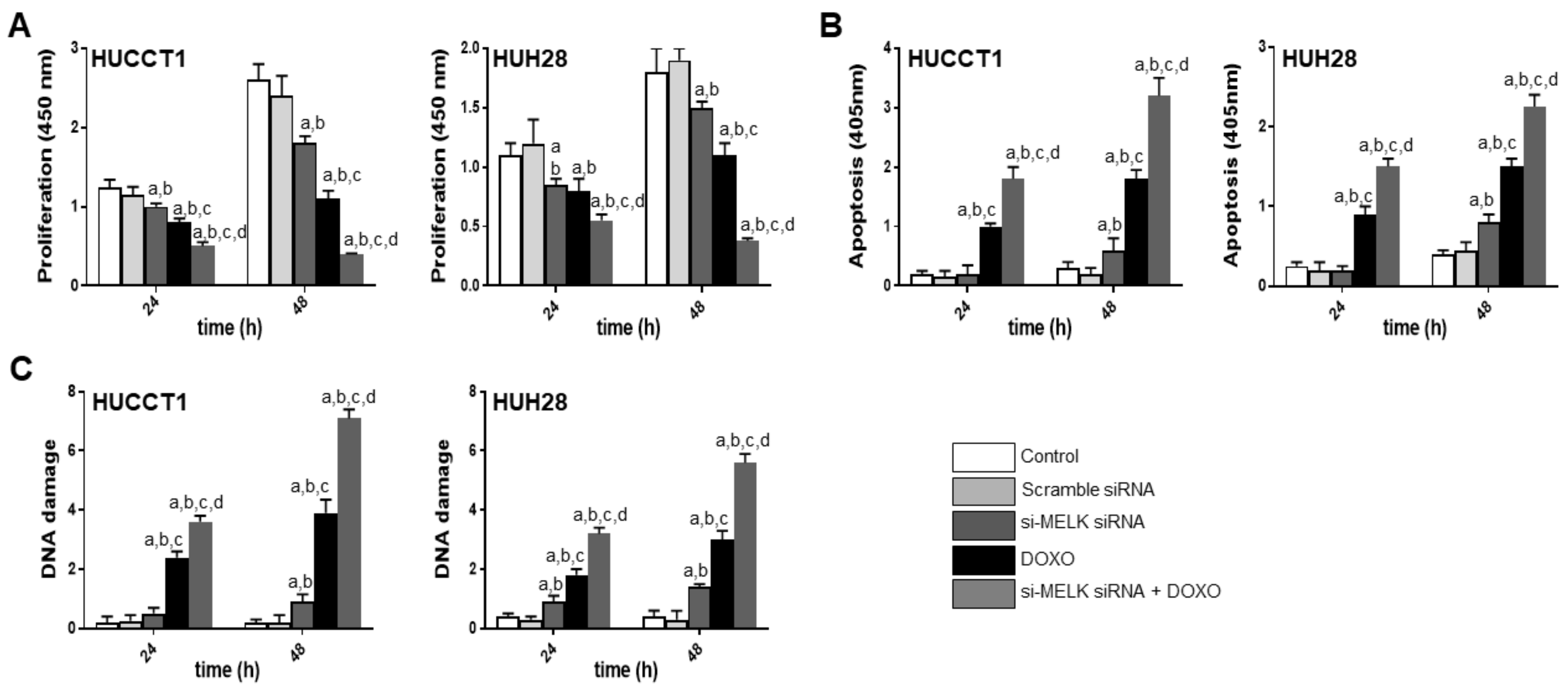
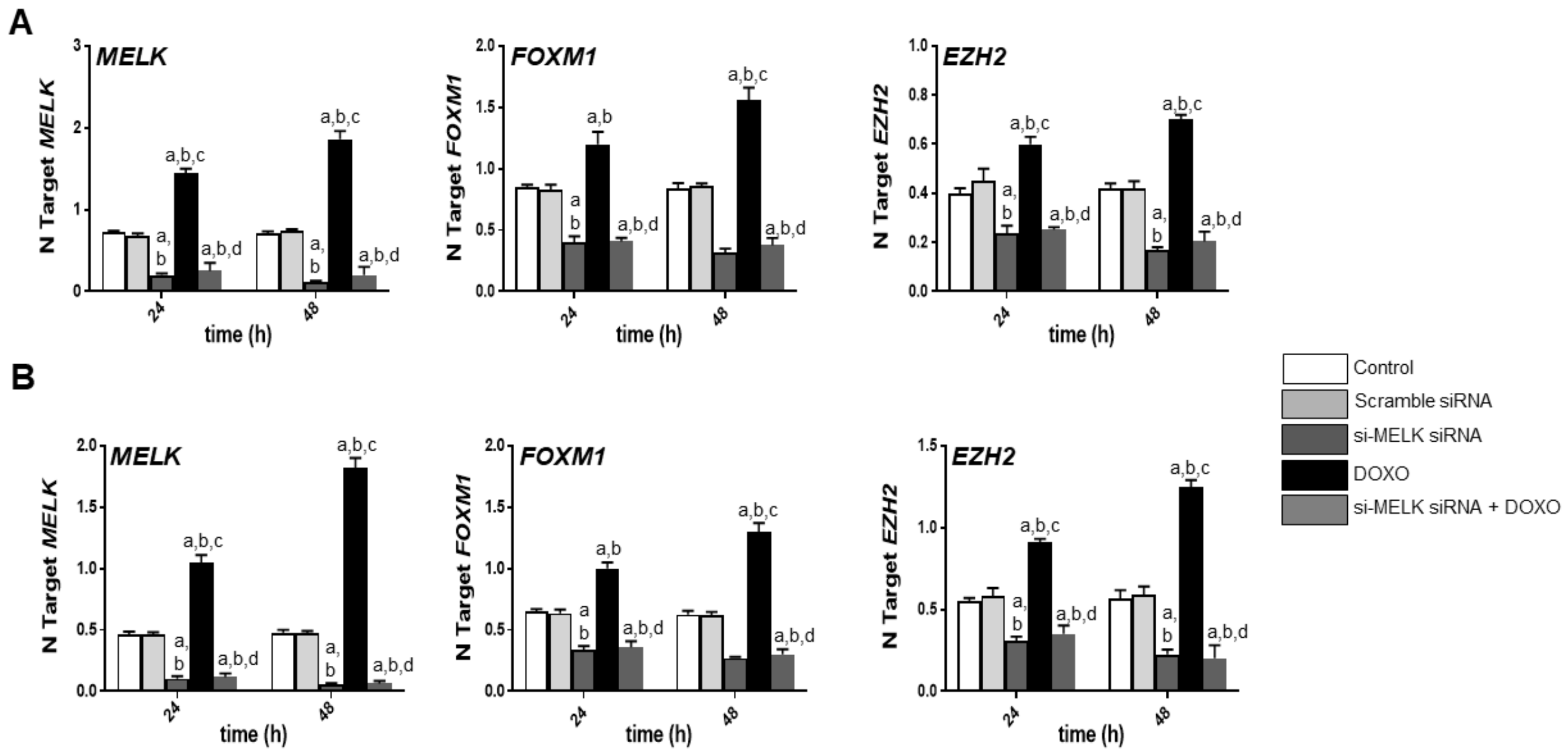
3.3. Suppression of MELK is Highly Detrimental for the Growth of Human iCCA Cell Lines
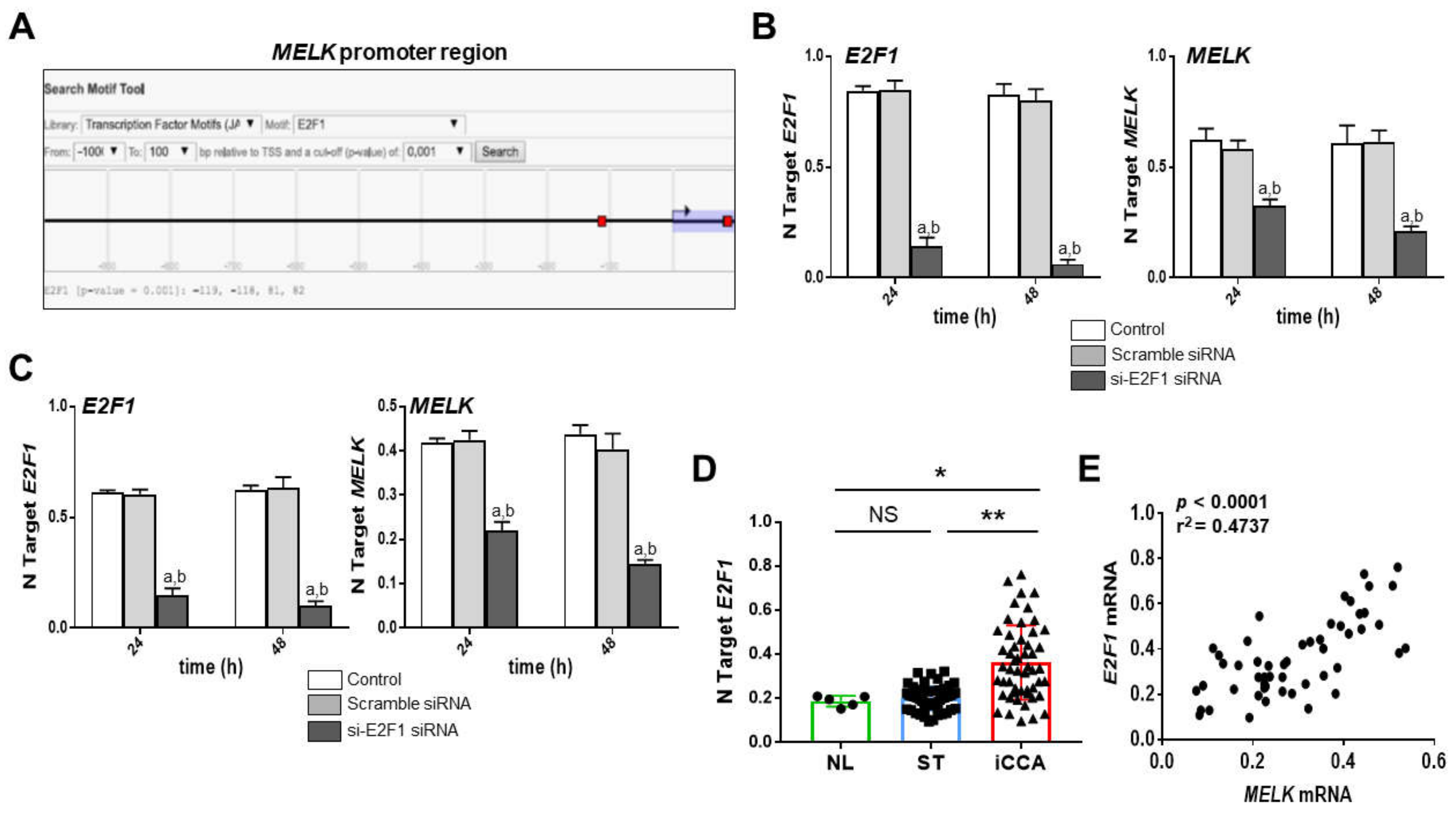
3.4. MELK Is a Transcriptional Target of the E2F1 Protooncogene in Intrahepatic Cholangiocarcinoma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangiocarcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef] [PubMed]
- Groopman, J.D.; Zhu, A.X.; Sirica, A.E.; Strazzabosco, M.; Wang, X.W.; Selaru, F.M.; Gores, G.J. Intrahepatic Cholangiocarcinoma: Continuing Challenges and Translational Advances. Hepatology 2019, 69, 1803–1815. [Google Scholar]
- Bertuccio, P.; Malvezzi, M.; Carioli, G.; Hashim, D.; Boffetta, P.; El-Serag, H.B.; La Vecchia, C.; Negri, E. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J. Hepatol. 2019, 71, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Davidson, B.R.; Goldin, R.D.; Heaton, N.; Karani, J.; Pereira, S.P.; Rosenberg, W.M.C.; Tait, P.; Taylor-Robinson, S.D.; Thillainayagam, A.V.; et al. Guidelines for the diagnosis and treatment of cholangiocarcinoma: An update. Gut 2012, 61, 1657–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simile, M.M.; Bagella, P.; Vidili, G.; Spanu, A.; Manetti, R.; Seddaiu, M.A.; Babudieri, S.; Madeddu, G.; Serra, P.A.; Altana, M.; et al. Targeted Therapies in Cholangiocarcinoma: Emerging Evidence from Clinical Trials. Medicina 2019, 55, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohyeldin, A.; Kornblum, H.I.; Thiel, J.; Nakano, I.; Ganguly, R.; Beullens, M. MELK—A conserved kinase: Functions, signaling, cancer, and controversy. Clin. Transl. Med. 2015, 4, 11. [Google Scholar]
- Jiang, P.; Zhang, D. Maternal embryonic leucine zipper kinase (MELK): A novel regulator in cell cycle control, embryonic development, and cancer. Int. J. Mol. Sci. 2013, 14, 21551–21560. [Google Scholar] [CrossRef]
- Gray, D.; Jubb, A.M.; Hogue, D.; Dowd, P.; Kljavin, N.; Yi, S.; Bai, W.; Frantz, G.; Zhang, Z.; Koeppen, H.; et al. Maternal embryonic leucine zipper kinase/murine protein serine-threonine kinase 38 is a promising therapeutic target for multiple cancers. Cancer Res. 2005, 65, 9751–9761. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, Y.; Shen, F.; Xu, Y.; Zhang, Y.; Zou, X.; Zhou, J.; Chen, Y. Maternal embryonic leucine zipper kinase: A novel biomarker and a potential therapeutic target of cervical cancer. Cancer Med. 2018, 7, 5665–5678. [Google Scholar] [CrossRef] [Green Version]
- Davezac, N.; Baldin, V.; Blot, J.; Ducommun, B.; Tassan, J.P. Human pEg3 kinase associates with and phosphorylates CDC25B phosphatase: A potential role for pEg3 in cell cycle regulation. Oncogene 2002, 21, 7630–7641. [Google Scholar] [CrossRef] [Green Version]
- Badouel, C.; Körner, R.; Frank-Vaillant, M.; Couturier, A.; Nigg, E.A.; Tassan, J.P. M-phase MELK activity is regulated by MPF and MAPK. Cell Cycle 2006, 5, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.L.; Park, J.H.; Nishidate, T.; Nakamura, Y.; Katagiri, T. Involvement of maternal embryonic leucine zipper kinase (MELK) in mammary carcinogenesis through interaction with Bcl-G, a pro-apoptotic member of the Bcl-2 family. Breast Cancer Res. 2007, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seong, H.A.; Jung, H.; Ha, H. Murine protein serine/threonine kinase 38 stimulates TGF-β signaling in a kinase-dependent manner via direct phosphorylation of smad proteins. J. Biol. Chem. 2010, 285, 30959–30970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seong, H.A.; Ha, H. Murine protein serine-threonine kinase 38 activates p53 function through Ser 15 phosphorylation. J. Biol. Chem. 2012, 287, 20797–20810. [Google Scholar] [CrossRef] [Green Version]
- Pitner, M.K.; Taliaferro, J.M.; Dalby, K.N.; Bartholomeusz, C. MELK: A potential novel therapeutic target for TNBC and other aggressive malignancies. Expert Opin. Ther. Targets 2017, 21, 849–859. [Google Scholar] [CrossRef]
- Gu, C.; Banasavadi-Siddegowda, Y.K.; Joshi, K.; Nakamura, Y.; Kurt, H.; Gupta, S.; Nakano, I. Tumor-specific activation of the C-JUN/MELK pathway regulates glioma stem cell growth in a p53-dependent manner. Stem Cells 2013, 31, 870–881. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Joshi, K.; Ezhilarasan, R.; Myers, T.R.; Siu, J.; Gu, C.; Nakano-Okuno, M.; Taylor, D.; Minata, M.; Sulman, E.P.; et al. EZH2 protects Glioma stem cells from radiation-induced cell death in a MELK/FOXM1-dependent manner. Stem Cell Rep. 2015, 4, 226–238. [Google Scholar] [CrossRef] [Green Version]
- Pickard, M.R.; Green, A.R.; Ellis, I.O.; Caldas, C.; Hedge, V.L.; Mourtada-Maarabouni, M.; Williams, G.T. Dysregulated expression of Fau and MELK is associated with poor prognosis in breast cancer. Breast Cancer Res. 2009, 11, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Nakano, I.; Masterman-Smith, M.; Saigusa, K.; Paucar, A.A.; Horvath, S.; Shoemaker, L.; Watanabe, M.; Negro, A.; Bajpai, R.; Howes, A.; et al. Maternal embryonic leucine zipper kinase is a key regulator of the proliferation of malignant brain tumors, including brain tumor stem cells. J. Neurosci. Res. 2008, 86, 48–60. [Google Scholar] [CrossRef]
- Saito, R.; Nakauchi, H.; Watanabe, S. Serine/threonine kinase, Melk, regulates proliferation and glial differentiation of retinal progenitor cells. Cancer Sci. 2012, 103, 42–49. [Google Scholar] [CrossRef]
- Shi, M.; Ooi, L.L.; Rajasekaran, M.; Kong, S.N.; Seshachalam, V.P.; Goh, B.K.P.; Hui, K.M.; Sekar, K.; Xia, H.; Chen, J. MELK is an oncogenic kinase essential for early hepatocellular carcinoma recurrence. Cancer Lett. 2016, 383, 85–93. [Google Scholar]
- Inoue, H.; Kato, T.; Olugbile, S.; Tamura, K.; Chung, S.; Miyamoto, T.; Matsuo, Y.; Salgia, R.; Nakamura, Y.; Park, J.-H. Effective growth-suppressive activity of maternal embryonic leucine-zipper kinase (MELK) inhibitor against small cell lung cancer. Oncotarget 2016, 7, 13621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, Y.; Chen, Y.; Xie, Q.; Dong, N.; Gao, Y.; Deng, H.; Lu, C.; Wang, S. MicroRNA-214-3p inhibits proliferation and cell cycle progression by targeting MELK in hepatocellular carcinoma and correlates cancer prognosis. Cancer Cell Int. 2017, 17, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Speers, C.; Zhao, S.G.; Kothari, V.; Santola, A.; Liu, M.; Wilder-Romans, K.; Evans, J.; Batra, N.; Bartelink, H.; Hayes, D.F.; et al. Maternal embryonic leucine zipper kinase (MELK) as a novel mediator and biomarker of radioresistance in human breast cancer. Clin. Cancer Res. 2016, 22, 5864–5875. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Inoue, H.; Imoto, S.; Tamada, Y.; Miyamoto, T.; Matsuo, Y.; Nakamura, Y.; Park, J.-H. Oncogenic roles of TOPK and MELK, and effective growth suppression by small molecular inhibitors in kidney cancer cells. Oncotarget 2016, 7, 17652. [Google Scholar] [CrossRef] [Green Version]
- Calcagno, D.Q.; Takeno, S.S.; Gigek, C.O.; Leal, M.F.; Wisnieski, F.; Chen, E.S.; Araújo, T.M.T.; Lima, E.M.; Melaragno, M.I.; Demachki, S.; et al. Identification of IL11RA and MELK amplification in gastric cancer by comprehensive genomic profiling of gastric cancer cell lines. World J. Gastroenterol. 2016, 22, 9506–9514. [Google Scholar] [CrossRef]
- Janostiak, R.; Rauniyar, N.; Lam, T.T.; Ou, J.; Zhu, L.J.; Green, M.R.; Wajapeyee, N. MELK Promotes Melanoma Growth by Stimulating the NF-κB Pathway. Cell Rep. 2017, 21, 2829–2841. [Google Scholar] [CrossRef]
- Alachkar, H.; Mutonga, M.; Chung, S.; Matsuo, Y.; Stock, W.; Nakamura, Y. Abstract 952: Preclinical efficacy of maternal embryonic leucine-zipper kinase (MELK) inhibition in acute myeloid leukemia. Cancer Res. 2015, 74, 952. [Google Scholar]
- Kohler, R.S.; Kettelhack, H.; Knipprath-Mészaros, A.M.; Fedier, A.; Schoetzau, A.; Jacob, F.; Heinzelmann-Schwarz, V. MELK expression in ovarian cancer correlates with poor outcome and its inhibition by OTSSP167 abrogates proliferation and viability of ovarian cancer cells. Gynecol. Oncol. 2017, 145, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Guan, S.; Lu, J.; Zhao, Y.; Yu, Y.; Li, H.; Chen, Z.; Shi, Z.; Liang, H.; Wang, M.; Guo, K.; et al. MELK is a novel therapeutic target in high-risk neuroblastoma. Oncotarget 2017, 9, 2591–2602. [Google Scholar] [CrossRef] [Green Version]
- Bolomsky, A.; Heusschen, R.; Schlangen, K.; Stangelberger, K.; Muller, J.; Schreiner, W.; Zojer, N.; Caers, J.; Ludwig, H. Maternal embryonic leucine zipper kinase is a novel target for proliferation-associated high-risk myeloma. Haematologica 2018, 103, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, Z.; Guo, T.; Xing, X.-F.; Cheng, X.; Du, H.; Wen, X.-Z.; Ji, J.-F. Maternal embryonic leucine zipper kinase serves as a poor prognosis marker and therapeutic target in gastric cancer. Oncotarget 2015, 7, 14–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waclaw, R.; Kig, C.; Sobol, R.W.; Mo, X.; Kornblum, H.I.; Chow, L.M.L.; Joshi, K.; Kim, S.-H.; Nardini, D.; Nakano, I.; et al. MELK-Dependent FOXM1 Phosphorylation is Essential for Proliferation of Glioma Stem Cells. Stem Cells 2013, 31, 1051–1063. [Google Scholar]
- Kurahara, H.; Yonemori, K.; Nishizono, Y.; Mataki, Y.; Iino, S.; Ueno, S.; Minami, K.; Maemura, K.; Hiwatashi, K.; Sakoda, M.; et al. Expression of Maternal Embryonic Leucine Zipper Kinase (MELK) Correlates to Malignant Potentials in Hepatocellular Carcinoma. Anticancer Res. 2016, 36, 5183–5188. [Google Scholar]
- Yuan, H.; Wang, H.; Yu, C.; Sun, Q.; Gu, C.; Liu, H.; Zhang, J.; Sun, Y.; Zhang, M.; Pang, S.; et al. MELK and EZH2 Cooperate to Regulate Medulloblastoma Cancer Stem-like Cell Proliferation and Differentiation. Mol. Cancer Res. 2017, 15, 1275–1286. [Google Scholar]
- Chlenski, A.; Dobratic, M.; Wilkinson, E.; Budke, B.; Cohn, S.L.; Miller, R.; Applebaum, M.A.; Park, J.-H.; Park, C.; Nakamura, Y.; et al. Maternal Embryonic Leucine Zipper Kinase (MELK), a Potential Therapeutic Target for Neuroblastoma. Mol. Cancer Ther. 2019, 18, 507–516. [Google Scholar] [CrossRef] [Green Version]
- Marie, S.K.N.; Oba-Shinjo, S.M.; da Silva, R.; Gimenez, M.; Nunes Reis, G.; Tassan, J.P.; Rosa, J.C.; Uno, M. Stathmin involvement in the maternal embryonic leucine zipper kinase pathway in glioblastoma. Proteome Sci. 2016, 14, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.-T.; Zhao, J.J.; Li, Q.; Mitchison, T.J.; Eck, M.J.; Gray, N.S.; Lako, A.; Wang, Y.; Cantley, L.C.; Begley, M. Mitotic MELK-eIF4B signaling controls protein synthesis and tumor cell survival. Proc. Natl. Acad. Sci. USA 2016, 113, 9810–9815. [Google Scholar]
- Hallstrom, T.C.; Mori, S.; Nevins, J.R. An E2F1-dependent gene expression program that determines the balance between proliferation and cell death. Cancer Cell. 2008, 13, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Verlinden, L.; Eelen, G.; Beullens, I.; Van Camp, M.; Van Hummelen, P.; Engelen, K.; Van Hellemont, R.; Marchal, K.; De Moor, B.; Foijer, F.; et al. Characterization of the condensin component Cnap1 and protein kinase Melk as novel E2F target genes down-regulated by 1,25-dihydroxyvitamin D3. J. Biol. Chem. 2005, 280, 37319–37330. [Google Scholar] [CrossRef] [Green Version]
- Poppy Roworth, A.; Ghari, F.; La Thangue, N.B. To live or let die—Complexity within the E2F1 pathway. Mol. Cell Oncol. 2015, 2, e970480. [Google Scholar] [CrossRef] [PubMed]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Denechaud, P.D.; Fajas, L.; Giralt, A. E2F1, a Novel Regulator of Metabolism. Front. Endocrinol. 2017, 8, 311. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.; Nakamural, Y. MELK inhibitor, novel molecular targeted therapeutics for human cancer stem cells. Cell Cycle 2013, 12, 1655–1656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, S.; Cohen-Solal, M.; Bolomsky, A.; Lejeune, M.; Muller, J.; Duray, E.; Hempel, U.; Stangelberger, K.; Beguin, Y.; Heusschen, R.; et al. Maternal embryonic leucine zipper kinase inhibitor OTSSP167 has preclinical activity in multiple myeloma bone disease. Haematologica 2018, 103, 1359–1368. [Google Scholar]
- Stefka, A.T.; Park, J.-H.; Matsuo, Y.; Chung, S.; Nakamura, Y.; Jakubowiak, A.J.; Rosebeck, S. Anti-myeloma activity of MELK inhibitor OTS167: Effects on drug-resistant myeloma cells and putative myeloma stem cell replenishment of malignant plasma cells. Blood Cancer J. 2016, 6, e460. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.; Deng, B.; Saloura, V.; Park, J.H.; Nakamura, Y. MELK inhibition targets cancer stem cells through downregulation of SOX2 expression in head and neck cancer cells. Oncol. Rep. 2019, 41, 2540–2548. [Google Scholar] [CrossRef]
- McDonald, I.M.; Gilbert, T.S.K.; Arend, K.C.; Moorman, N.J.; Lazear, E.; Graves, L.M.; Rashid, N.; Lenarcic, E.M.; Vincent, H.A.; Johnson, G.L.; et al. Kinome Profiling Identifies Druggable Targets for Novel Human Cytomegalovirus (HCMV) Antivirals. Mol. Cell. Proteom. 2017, 16, S263–S276. [Google Scholar]
- Reinecke, M.; Greif, P.A.; Schlegl, J.; Feuchtinger, A.; Ruprecht, B.; Helm, D.; Wu, Z.; Médard, G.; Vick, B.; Ruland, J.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, eaan4368. [Google Scholar]
- Lin, A.; Giuliano, C.J.; Sayles, N.M.; Sheltzer, J.M. CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on-going clinical trials. Elife 2017, 6, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Dreos, R.; Ambrosini, G.; Cavin Périer, R.; Bucher, P. EPD and EPDnew, high-quality promoter resources in the next-generation sequencing era. Nucleic Acids Res. 2013, 41, D157–D164. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Codony-Servat, C.; Codony-Servat, J.; Lligé, D.; Chaib, I.; Sun, X.; Miao, J.; Sun, R.; Cai, X.; Verlicchi, A.; et al. Targeting PKCι-PAK1 signaling pathways in EGFR and KRAS mutant adenocarcinoma and lung squamous cell carcinoma. Cell Commun. Signal. 2019, 17, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, W.; Arnst, C.; Tipton, A.R.; Bekier, M.E., 2nd; Taylor, W.R.; Yen, T.J.; Liu, S.T. OTSSP167 Abrogates Mitotic Checkpoint through Inhibiting Multiple Mitotic Kinases. PLoS ONE 2016, 11, e0153518. [Google Scholar] [CrossRef] [PubMed]
- Beke, L.; Kig, C.; Linders, J.T.; Boens, S.; Boeckx, A.; van Heerde, E.; Parade, M.; De Bondt, A.; Van den Wyngaert, I.; Bashir, T.; et al. MELK-T1, a small-molecule inhibitor of protein kinase MELK, decreases DNA-damage tolerance in proliferating cancer cells. Biosci. Rep. 2015, 35, e00267. [Google Scholar] [CrossRef] [Green Version]
- Black, E.P.; Hallstrom, T.; Dressman, H.K.; West, M.; Nevins, J.R. Distinctions in the specificity of E2F function revealed by gene expression signatures. Proc. Natl. Acad. Sci. USA 2005, 102, 15948–15953. [Google Scholar] [CrossRef] [Green Version]
- Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef] [Green Version]
- Frolov, M.V.; Dyson, N.J. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J. Cell Sci. 2004, 117, 2173–2181. [Google Scholar] [CrossRef] [Green Version]
- Polager, S.; Kalma, Y.; Berkovich, E.; Ginsberg, D. E2Fs up-regulate expression of genes involved in DNA replication, DNA repair and mitosis. Oncogene 2002, 21, 437–446. [Google Scholar] [CrossRef] [Green Version]
- La Thangue, N.B. DP and E2F proteins: Components of a heterodimeric transcription factor implicated in cell cycle control. Curr. Opin. Cell Biol. 1994, 6, 443–450. [Google Scholar] [CrossRef]
- Song, X.; Liu, X.; Wang, H.; Wang, J.; Qiao, Y.; Cigliano, A.; Utpatel, K.; Ribback, S.; Pilo, M.G.; Serra, M.; et al. Combined CDK4/6 and Pan-mTOR Inhibition Is Synergistic Against Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2019, 25, 403–413. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | |
|---|---|
| No. of patients; male; female | 52; 31; 21 |
| Age (years); <60; >60 | 14; 38 |
| Etiology; HBV; HCV; hepatolithiasis; PSC; NA | 12; 7; 12; 3; 18 |
| Liver cirrhosis; yes; no | 20; 32 |
| Tumor differentiation; well; moderately; poorly | 24; 20; 8 |
| Tumor size (cm); <5; >5 | 36; 16 |
| Tumor number; single; multiple | 38; 14 |
| Lymph node metastases; yes; no; NA | 19; 21; 12 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cigliano, A.; Pilo, M.G.; Mela, M.; Ribback, S.; Dombrowski, F.; Pes, G.M.; Cossu, A.; Evert, M.; Calvisi, D.F.; Utpatel, K. Inhibition of MELK Protooncogene as an Innovative Treatment for Intrahepatic Cholangiocarcinoma. Medicina 2020, 56, 1. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina56010001
Cigliano A, Pilo MG, Mela M, Ribback S, Dombrowski F, Pes GM, Cossu A, Evert M, Calvisi DF, Utpatel K. Inhibition of MELK Protooncogene as an Innovative Treatment for Intrahepatic Cholangiocarcinoma. Medicina. 2020; 56(1):1. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina56010001
Chicago/Turabian StyleCigliano, Antonio, Maria Giulia Pilo, Marta Mela, Silvia Ribback, Frank Dombrowski, Giovanni Mario Pes, Antonio Cossu, Matthias Evert, Diego Francesco Calvisi, and Kirsten Utpatel. 2020. "Inhibition of MELK Protooncogene as an Innovative Treatment for Intrahepatic Cholangiocarcinoma" Medicina 56, no. 1: 1. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina56010001