
Mitochondria and Mitochondrial DNA: Key Elements in the Pathogenesis and Exacerbation of the Inflammatory State Caused by COVID-19

 , , ,
, , , 
Abstract
:1. Introduction
2. General Concepts
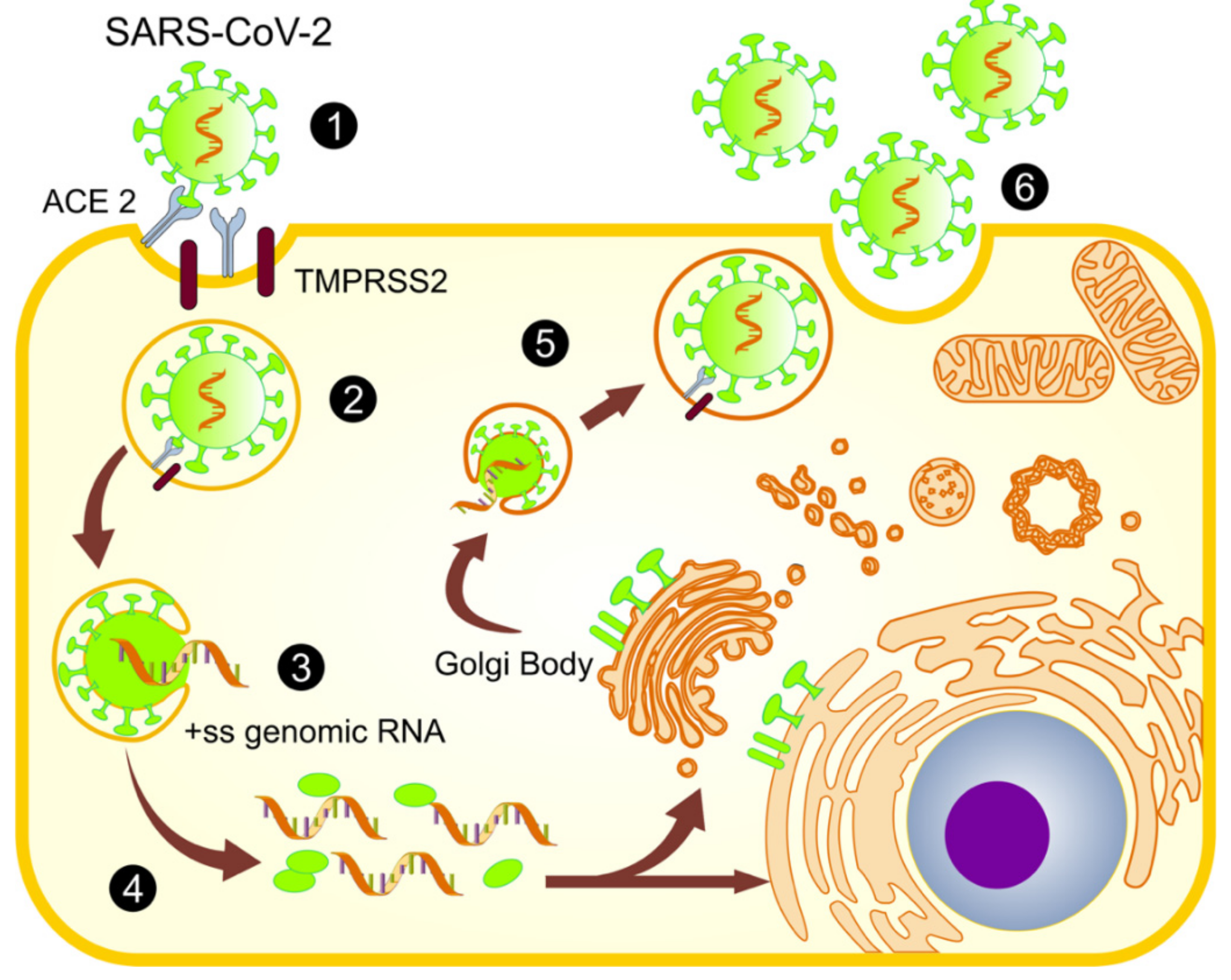
2.1. Infection and Replication of SARS-CoV-2
2.2. Structure and Dynamics of Mitochondria
Mitochondrial DNA (mt-DNA)
2.3. Mitochondrial Functions
2.3.1. ATP Production
2.3.2. Mitochondria and the Production of Reactive Oxygen Species (ROS)
2.3.3. Mitochondria and Cell Death
2.4. Mitochondria and Immune Response
2.4.1. Neutrophils
2.4.2. T lymphocytes
2.4.3. B Lymphocytes
2.4.4. Mitochondria and the Activation of the Immune System
3. Mitochondrial Dysfunction in Viral Infections
4. The Mitochondrial Role in SARS-CoV-2 Replication
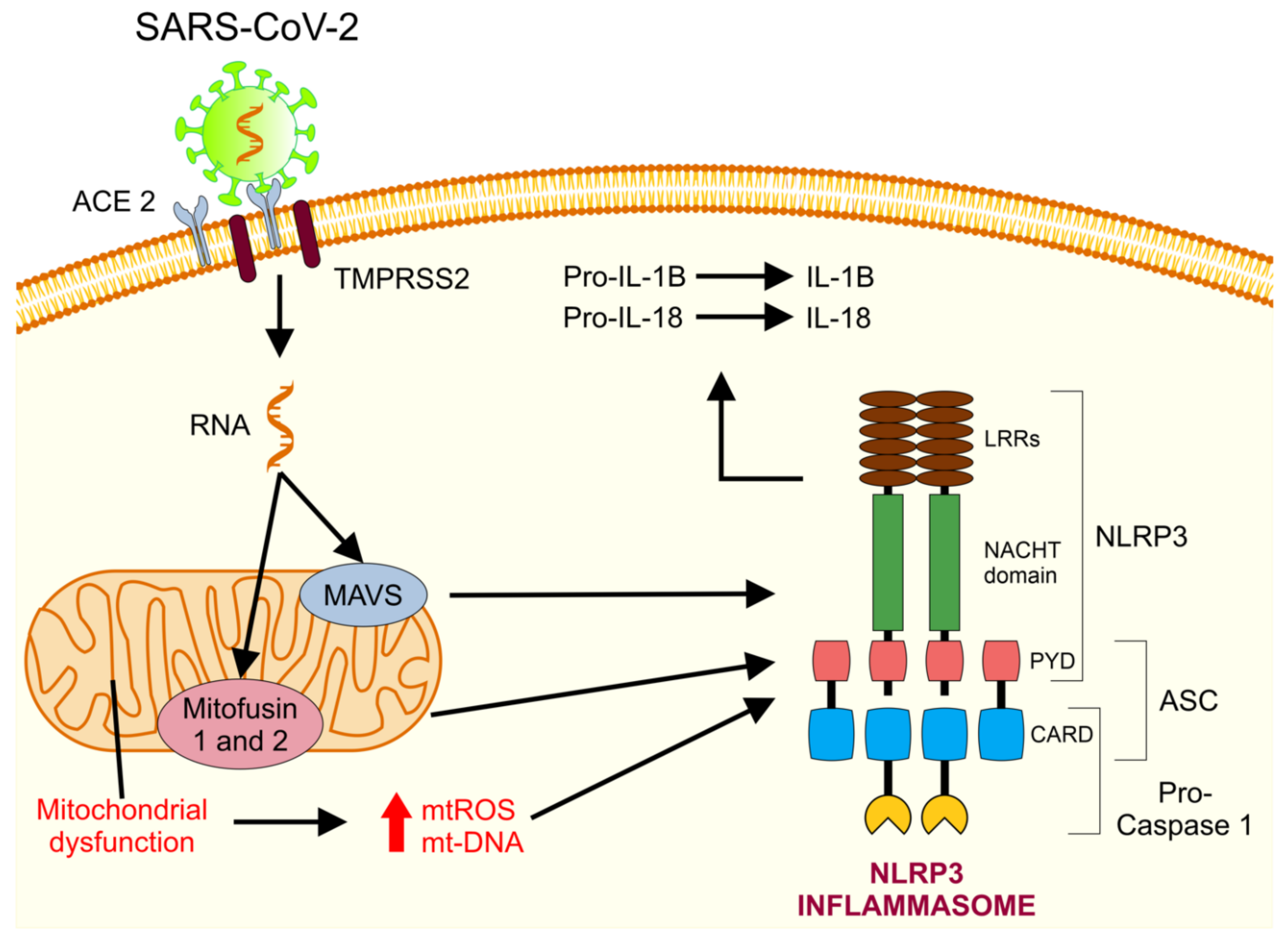
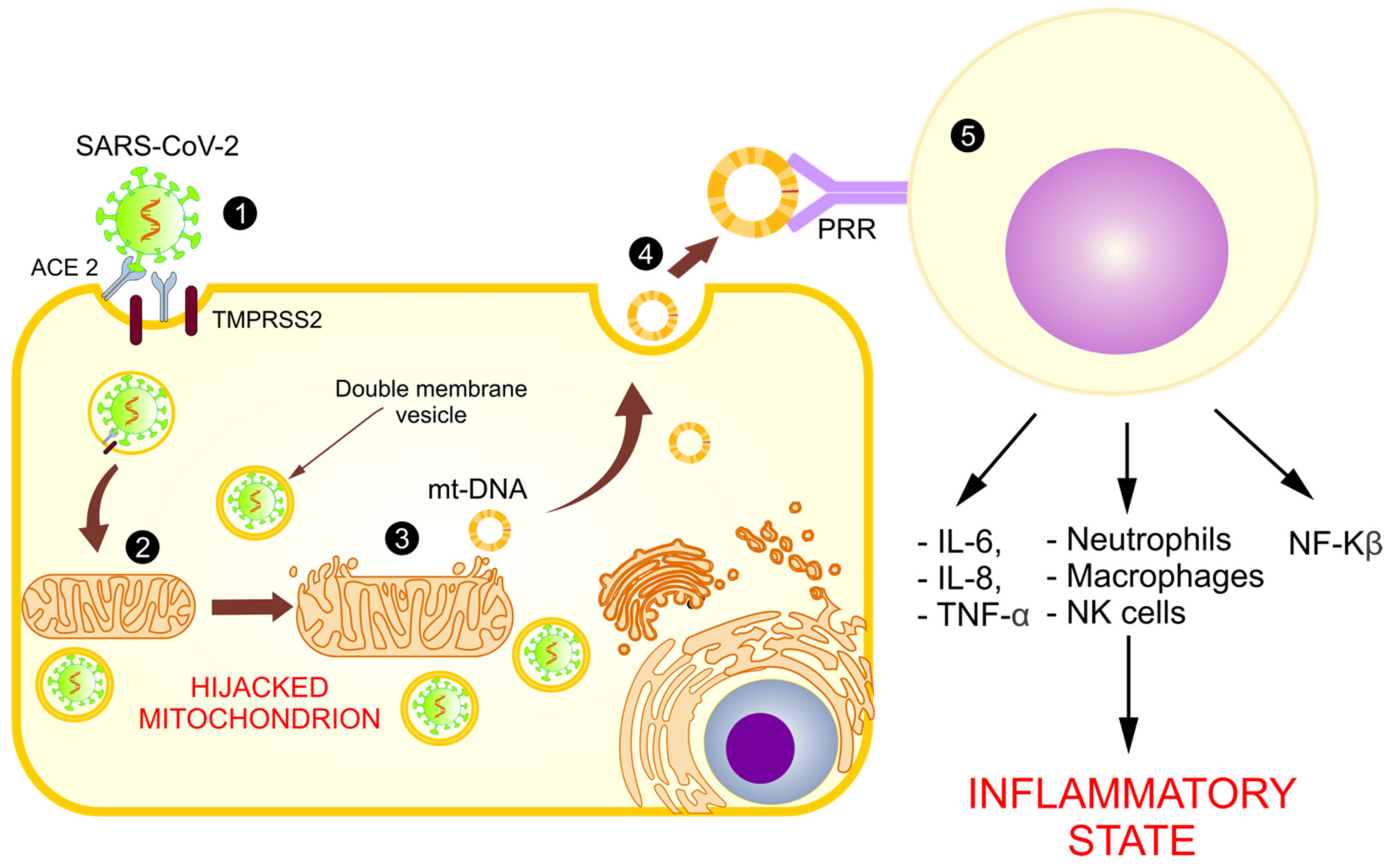
5. Mitochondrial Dysfunction in COVID-19
5.1. Mitochondria and Cytokine Storms
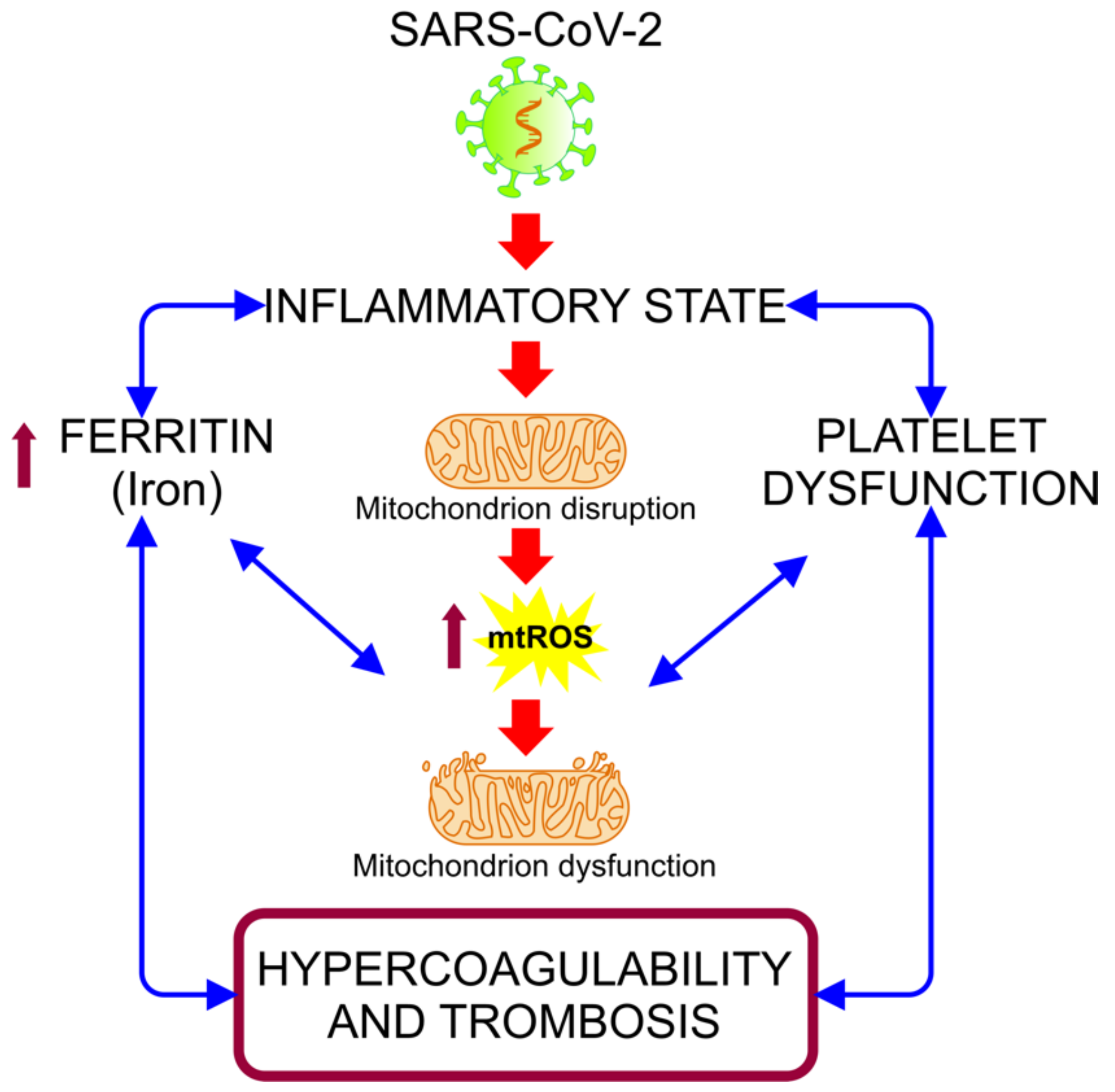
5.2. COVID-19: Disruption in the Mitochondrial Regulation of Iron Homeostasis
5.3. Mitochondrial Disfunction in the Hypercoagulability State Associated with COVID-19 Severity
5.4. Role of the Mitochondrial Dysfunction in Arterial Hypertension and Diabetes: Risk Factors Associated with COVID-19
5.5. Abnormal Functioning of Mitochondria and Release of mt-DNA as DAMPs
6. mt-DNA as a Biomarker in Inflammatory Diseases
mt-DNA: An Early Predictor of Severe Evolution and Mortality in Patients with COVID-19?
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- WHO Coronavirus Disease (COVID 19) Dashboard; World Health Organization: Geneva, Switzerland, 2020.
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- King, A.M.; Lefkowitz, E.; Adams, M.J.; Carstens, E.B. Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: Amsterdam, The Netherlands, 2012; pp. 770–783. [Google Scholar]
- Gallagher, T.M.; Buchmeier, M.J. Coronavirus Spike Proteins in Viral Entry and Pathogenesis. Virology 2001, 279, 371–374. [Google Scholar] [CrossRef] [Green Version]
- Hamming, I.; Van Goor, H.; Turner, A.J.; Rushworth, C.A.; Michaud, A.A.; Corvol, P.; Navis, G. Differential regulation of renal angiotensin-converting enzyme (ACE) and ACE2 during ACE inhibition and dietary sodium restriction in healthy rats. Exp. Physiol. 2008, 93, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Sin, R.; Kubiska, M.; Cmorej, P.C.; Vachalova, J. Clinical and laboratory characteristics of the COVID-19 disease in adult patients. Neuro Endocrinol. Lett. 2020, 41, 223–230. [Google Scholar] [PubMed]
- Santosh, S. Coronavirus (Covid-19) Sepsis: Revisiting Mitochondrial Dysfunction in Pathogenesis, Aging, Inflammation, and Mortality; Springer Nature: Berlin/Heidelberg, Germany, 2020. [Google Scholar]
- Saleh, J.; Peyssonnaux, C.; Singh, K.K.; Edeas, M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion 2020, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Giannis, D.; Ziogas, I.A.; Gianni, P. Coagulation disorders in coronavirus infected patients: COVID-19, SARS-CoV-1, MERS-CoV and lessons from the past. J. Clin. Virol. 2020, 127, 104362. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, G.; Cai, X.P.; Deng, J.W.; Zheng, L.; Zhu, H.H.; Zheng, M.; Yang, B.; Chen, Z. An overview of COVID-19. Biomed. Biotechnol. 2020, 21, 343–360. [Google Scholar] [CrossRef]
- Wax, R.S.; Christian, M. Practical recommendations for critical care and anesthesiology teams caring for novel coronavirus (2019-nCoV) patients. Can. J. Anaesth. 2020, 67, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatti, P.; Ilamathi, H.S.; Todkar, K.; Germain, M. Mitochondria Targeted Viral Replication and Survival Strategies—Prospective on SARS-CoV-2. Front. Pharmacol. 2020, 11, 578599. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2020, 19, 155–170. [Google Scholar] [CrossRef]
- Heras, N.D.L.; Giménez, V.M.M.; Ferder, L.; Manucha, W.; Lahera, V. Implications of Oxidative Stress and Potential Role of Mitochondrial Dysfunction in COVID-19: Therapeutic Effects of Vitamin D. Antioxidants 2020, 9, 897. [Google Scholar] [CrossRef] [PubMed]
- Ledur, P.F.; Karmirian, K.; Pedrosa, C.D.S.G.; Souza, L.R.Q.; Assis-De-Lemos, G.; Martins, T.M.; Ferreira, J.D.C.C.G.; Reis, G.F.D.A.; Silva, E.S.; Silva, D.; et al. Zika virus infection leads to mitochondrial failure, oxidative stress and DNA damage in human iPSC-derived astrocytes. Sci. Rep. 2020, 10, 1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, W.H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Hofmann, H.; Pöhlmann, S. Cellular entry of the SARS coronavirus. Trends Microbiol. 2004, 12, 466–472. [Google Scholar] [CrossRef] [Green Version]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin-converting enzyme 2 (ACE2), SARS-CoV-2 and the pathophysiology of coronavirus disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Zhang, R.; Wu, Y.; Zhao, M.; Liu, C.; Zhou, L.; Shen, S.; Liao, S.; Yang, K.; Li, Q.; Wan, H. Role of HIF-1α in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L631–L640. [Google Scholar] [CrossRef]
- Lely, A.T.; Hamming, I.; van Goor, H.; Navis, G.J. Renal ACE2 expression in human kidney disease. J. Pathol. 2004, 204, 587–593. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Zhang, J.; Yang, H.; Tan, Y.; Wang, H.; Wang, C.; Liu, L.; et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat. Commun. 2021, 12, 2506. [Google Scholar] [CrossRef] [PubMed]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.V.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Jeong, G.U.; Song, H.; Yoon, G.Y.; Kim, D.; Kwon, Y.-C. Therapeutic Strategies against COVID-19 and Structural Characterization of SARS-CoV-2: A Review. Front. Microbiol. 2020, 11, 1723. [Google Scholar] [CrossRef] [PubMed]
- Lebeau, G.; Vagner, D.; Frumence, É.; Ah-Pine, F.; Guillot, X.; Nobécourt, E.; Raffray, L.; Gasque, P. Deciphering SARS-CoV-2 Virologic and Immunologic Features. Int. J. Mol. Sci. 2020, 21, 5932. [Google Scholar] [CrossRef] [PubMed]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and Characterization of MAVS, a Mitochondrial Antiviral Signaling Protein that Activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic Reticulum–Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Drago, I.; Pizzo, P.; Pozzan, T. After half a century mitochondrial calcium in- and efflux machineries reveal themselves. EMBO J. 2011, 30, 4119–4125. [Google Scholar] [CrossRef] [Green Version]
- De Pinto, V.; Palmieri, F. Transmembrane arrangement of mitochondrial porin or voltage-dependent anion channel (VDAC). J. Bioenerg. Biomembr. 1992, 24, 21–26. [Google Scholar] [CrossRef]
- Green, D.R. The Pathophysiology of Mitochondrial Cell Death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Rimessi, A.; Previati, M.; Nigro, F.; Wieckowski, M.R.; Pinton, P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int. J. Biochem. Cell Biol. 2016, 81, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Van Der Bliek, A.M. Mitochondrial Fission, Fusion, and Stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nat. Cell Biol. 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells 2019, 8, 100. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; De Bruijn, M.H.L.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Giles, R.E.; Blanc, H.; Cann, H.M.; Wallace, D.C. Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6715–6719. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, T.; Kang, D. An overview of mammalian mitochondrial DNA replication mechanisms. J. Biochem. 2018, 164, 183–193. [Google Scholar] [CrossRef]
- Sampath, H. Oxidative DNA damage in disease-Insights gained from base excision repair glycosylase-deficient mouse models. Environ. Mol. Mutagen. 2014, 55, 689–703. [Google Scholar] [CrossRef] [Green Version]
- Gorbunova, V.; Seluanov, A.; Mao, Z.; Hine, C. Changes in DNA repair during aging. Nucleic Acids Res. 2007, 35, 7466–7474. [Google Scholar] [CrossRef] [Green Version]
- Crippa, B.L.; Leon, E.; Calhoun, A.; Lowichik, A.; Pasquali, M.; Longo, N. Biochemical abnormalities in Pearson syndrome. Am. J. Med. Genet. Part A 2015, 167, 621–628. [Google Scholar] [CrossRef]
- Sampath, H.; Batra, A.K.; Vartanian, V.; Carmical, J.R.; Prusak, D.; King, I.B.; Lowell, B.; Earley, L.F.; Wood, T.G.; Marks, D.L.; et al. Variable penetrance of metabolic phenotypes and development of high-fat diet-induced adiposity in NEIL1-deficient mice. Am. J. Physiol. Metab. 2011, 300, E724–E734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, M. The role of mitochondrial dysfunction in sepsis-induced multi-organ failure. Virulence 2014, 5, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Bonora, M.; Patergnani, S.; Rimessi, A.; De Marchi, E.; Suski, J.M.; Bononi, A.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. ATP synthesis and storage. Purinergic Signal. 2012, 8, 343–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunt, S.; Heiden, M.G.V. Aerobic Glycolysis: Meeting the Metabolic Requirements of Cell Proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, E.P.; Lehninger, A.L. Oxidation of fatty acids and tricarboxylic acid cycle intermediates by isolated rat liver mitochondria. J. Biol. Chem. 1949, 179, 957–972. [Google Scholar] [CrossRef]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef] [Green Version]
- Mayr, J.A.; Zimmermann, F.A.; Fauth, C.; Bergheim, C.; Meierhofer, D.; Radmayr, D.; Zschocke, J.; Koch, J.; Sperl, W. Lipoic Acid Synthetase Deficiency Causes Neonatal-Onset Epilepsy, Defective Mitochondrial Energy Metabolism, and Glycine Elevation. Am. J. Hum. Genet. 2011, 89, 792–797. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, S. Mitochondrial uncoupling, ROS generation and cardioprotection. Biochim. Biophys. Acta (BBA)-Bioenerg. 2018, 1859, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Starkov, A.A. Mitochondrial metabolism of reactive oxygen species. Biochemistry 2005, 70, 200–214. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.H.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Gurung, P.; Lukens, J.; Kanneganti, T.-D. Mitochondria: Diversity in the regulation of the NLRP3 inflammasome. Trends Mol. Med. 2015, 21, 193–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Cosio, M.G.; Saetta, M.; Agusti, A. Immunologic Aspects of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2009, 360, 2445–2454. [Google Scholar] [CrossRef]
- Yap, J.K.Y.; Moriyama, M.; Iwasaki, A. Inflammasomes and Pyroptosis as Therapeutic Targets for COVID-19. J. Immunol. 2020, 205, 307–312. [Google Scholar] [CrossRef]
- Yang, W.; Ni, H.; Wang, H.; Gu, H. NLRP3 inflammasome is essential for the development of chronic obstructive pulmonary disease. Int. J. Clin. Exp. Pathol. 2015, 8, 13209–13216. [Google Scholar] [PubMed]
- dos Santos, G.; Kutuzov, M.A.; Ridge, K.M. The inflammasome in lung diseases. Am. J. Physiol. Cell. Mol. Physiol. 2012, 303, L627–L633. [Google Scholar] [CrossRef] [Green Version]
- Parkin, J.; Cohen, B. An overview of the immune system. Lancet 2001, 357, 1777–1789. [Google Scholar] [CrossRef]
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Breda, C.N.D.S.; Davanzo, G.; Basso, P.J.; Câmara, N.O.S.; Moraes-Vieira, P.M.M. Mitochondria as central hub of the immune system. Redox Biol. 2019, 26, 101255. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C. Neutrophils and immunity: Challenges and opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Mayadas, T.N.; Cullere, X.; Lowell, C.A. The Multifaceted Functions of Neutrophils. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 181–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefi, S.; Mihalache, C.C.; O Kozlowski, E.; Schmid, I.; Simon, H.-U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.; O’Sullivan, D.; Huang, S.C.-C.; van der Windt, G.J.; Blagih, J.; Qiu, J.; et al. Posttranscriptional Control of T Cell Effector Function by Aerobic Glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- D’Souza, A.D.; Parikh, N.; Kaech, S.M.; Shadel, G.S. Convergence of multiple signaling pathways is required to coordinately up-regulate mtDNA and mitochondrial biogenesis during T cell activation. Mitochondrion 2007, 7, 374–385. [Google Scholar] [CrossRef] [Green Version]
- Boothby, M.R.; Hodges, E.; Thomas, J.W. Molecular regulation of peripheral B cells and their progeny in immunity. Genes Dev. 2019, 33, 26–48. [Google Scholar] [CrossRef]
- Fernández-Ayala, D.J.M.; Navas, P.; López-Lluch, G. Age-related mitochondrial dysfunction as a key factor in COVID-19 disease. Exp. Gerontol. 2020, 142, 111147. [Google Scholar] [CrossRef]
- Ingelsson, B.; Söderberg, D.; Strid, T.; Söderberg, A.; Bergh, A.-C.; Loitto, V.; Lotfi, K.; Segelmark, M.; Spyrou, G.; Rosén, A. Lymphocytes eject interferogenic mitochondrial DNA webs in response to CpG and non-CpG oligodeoxynucleotides of class C. Proc. Natl. Acad. Sci. USA 2018, 115, E478–E487. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Hou, F.; Sun, L.; Zheng, H.; Skaug, B.; Jiang, Q.-X.; Chen, Z.J. MAVS Forms Functional Prion-like Aggregates to Activate and Propagate Antiviral Innate Immune Response. Cell 2011, 146, 448–461. [Google Scholar] [CrossRef] [Green Version]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Pearce, E.L. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol. 2018, 39, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Gong, S.; Singh, P.; Lyu, J.; Bai, Y. The interaction between mitochondria and oncoviruses. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 481–487. [Google Scholar] [CrossRef]
- Medvedev, R.; Ploen, D.; Hildt, E. HCV and Oxidative Stress: Implications for HCV Life Cycle and HCV-Associated Pathogenesis. Oxidative Med. Cell. Longev. 2016, 2016, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.R.; Magalhaes, A.; Camões, F.; Gouveia, A.; Vieira, M.; Kagan, J.C.; Ribeiro, D. Hepatitis C virus NS 3-4A inhibits the peroxisomal MAVS -dependent antiviral signalling response. J. Cell. Mol. Med. 2016, 20, 750–757. [Google Scholar] [CrossRef]
- Huang, S.-H.; Lien, J.-C.; Chen, C.-J.; Liu, Y.-C.; Wang, C.-Y.; Ping, C.-F.; Lin, Y.-F.; Huang, A.-C.; Lin, C.-W. Antiviral Activity of a Novel Compound CW-33 against Japanese Encephalitis Virus through Inhibiting Intracellular Calcium Overload. Int. J. Mol. Sci. 2016, 17, 1386. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.; Wang, M.; Huang, C.; Wu, C.; Hung, L.; Yang, C.; Ke, P.-Y.; Luo, S.; Liu, S.; Ho, L. Infection with the dengue RNA virus activates TLR9 signaling in human dendritic cells. EMBO Rep. 2018, 19, e46182. [Google Scholar] [CrossRef]
- Hottz, E.D.; Oliveira, M.F.; Nunes, P.C.G.; Nogueira, R.M.R.; Valls-De-Souza, R.; Da Poian, A.T.; Weyrich, A.S.; Zimmerman, G.A.; Bozza, P.T.; Bozza, F.A. Dengue induces platelet activation, mitochondrial dysfunction and cell death through mechanisms that involve DC-SIGN and caspases. J. Thromb. Haemost. 2013, 11, 951–962. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-Y.; Liang, J.-J.; Li, J.-K.; Lee, Y.-L.; Chang, B.-L.; Su, C.-I.; Huang, W.-J.; Lai, M.M.C.; Lin, Y.-L. Dengue Virus Impairs Mitochondrial Fusion by Cleaving Mitofusins. PLoS Pathog. 2015, 11, e1005350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatel-Chaix, L.; Cortese, M.; Brey, I.R.; Bender, S.; Neufeldt, C.; Fischl, W.; Scaturro, P.; Schieber, N.; Schwab, Y.; Fischer, B.; et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host Microbe 2016, 20, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Ho, L.-J.; Wang, J.-J.; Shaio, M.-F.; Kao, C.-L.; Chang, D.-M.; Han, S.-W.; Lai, J.-H. Infection of Human Dendritic Cells by Dengue Virus Causes Cell Maturation and Cytokine Production. J. Immunol. 2001, 166, 1499–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinschmidt, M.C.; Michaelis, M.; Ogbomo, H.; Doerr, H.-W.; Cinatl, J. Inhibition of apoptosis prevents West Nile virus induced cell death. BMC Microbiol. 2007, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- Jitobaom, K.; Tongluan, N.; Smith, D.R. Involvement of voltage-dependent anion channel (VDAC) in dengue infection. Sci. Rep. 2016, 6, 35753. [Google Scholar] [CrossRef] [Green Version]
- Kao, Y.-T.; Chang, B.-L.; Liang, J.-J.; Tsai, H.-J.; Lee, Y.-L.; Lin, R.-J.; Lin, Y.-L. Japanese Encephalitis Virus Nonstructural Protein NS5 Interacts with Mitochondrial Trifunctional Protein and Impairs Fatty Acid β-Oxidation. PLoS Pathog. 2015, 11, e1004750. [Google Scholar] [CrossRef] [Green Version]
- El-Bacha, T.; Midlej, V.D.V.P.; da Silva, A.P.P.; da Costa, L.S.; Benchimol, M.; Galina, A.; Da Poian, A. Mitochondrial and bioenergetic dysfunction in human hepatic cells infected with dengue 2 virus. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2007, 1772, 1158–1166. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Bogoyevitch, M.A.; Jans, D.A. Subversion of Host Cell Mitochondria by RSV to Favor Virus Production is Dependent on Inhibition of Mitochondrial Complex I and ROS Generation. Cells 2019, 8, 1417. [Google Scholar] [CrossRef] [Green Version]
- Knoops, K.; Kikkert, M.; Worm, S.H.; Zevenhoven-Dobbe, J.C.; Van Der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, 226. [Google Scholar] [CrossRef]
- Blanchard, E.; Roingeard, P. Virus-induced double-membrane vesicles. Cell. Microbiol. 2015, 17, 45–50. [Google Scholar] [CrossRef]
- Singh, K.K.; Chaubey, G.; Chen, J.Y.; Suravajhala, P. Decoding SARS-CoV-2 hijacking of host mitochondria in COVID-19 pathogenesis. Am. J. Physiol. Physiol. 2020, 319, C258–C267. [Google Scholar] [CrossRef]
- Wu, K.; Zou, J.; Chang, H.Y. RNA-GPS Predicts SARS-CoV-2 RNA Localization to Host Mitochondria and Nucleolus. bioRxiv 2020. [Google Scholar] [CrossRef]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Alfarouk, K.O.; Alhoufie, S.T.S.; Hifny, A.; Schwartz, L.; Alqahtani, A.S.; Ahmed, S.B.M.; Alqahtani, S.S.; Muddathir, A.K.; Ali, H.; Bashir, A.H.H.; et al. Of mitochondrion and COVID-19. J. Enzym. Inhib. Med. Chem. 2021, 36, 1258–1267. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Li, X.; Fang, P.; Mai, J.; Choi, E.T.; Wang, H.; Yang, X.-F. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J. Hematol. Oncol. 2013, 6, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, E.-K.; Kim, J.K.; Shin, D.-M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Chen, Y.C.; Hassanzadeh, S.; Han, K.; Judy, J.T.; Seifuddin, F.; Tunc, I.; Sack, M.N.; Pirooznia, M. Network analysis and transcriptome profiling identify autophagic and mitochondrial dysfunctions in SARS-CoV-2 infection. Front. Genet. 2020, 12, 289. [Google Scholar]
- Zhang, D.; Guo, R.; Lei, L.; Liu, H.; Wang, Y.; Wang, Y.; Qian, H.; Dai, T.; Zhang, T.; Lai, Y.; et al. Frontline Science: COVID-19 infection induces readily detectable morphologic and inflammation-related phenotypic changes in peripheral blood monocytes. J. Leukoc. Biol. 2021, 109, 13–22. [Google Scholar] [CrossRef]
- Hoffmann, R.F.; Jonker, M.R.; Brandenburg, S.M.; De Bruin, H.G.; Hacken, N.H.T.T.; Van Oosterhout, A.J.M.; Heijink, I.H. Mitochondrial dysfunction increases pro-inflammatory cytokine production and impairs repair and corticosteroid responsiveness in lung epithelium. Sci. Rep. 2019, 9, 15047. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.B.; Knutson, M.D.; Paler-Martinez, A.; Lee, S.; Xu, Y.; Viteri, F.E.; Ames, B.N. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc.Natl. Acad. Sci. USA 2002, 99, 2264–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.; Oldford, C.; Mailloux, R.J. Lactate dehydrogenase supports lactate oxidation in mitochondria isolated from different mouse tissues. Redox Biol. 2020, 28, 101339. [Google Scholar] [CrossRef]
- Adriouch, S.; Hubert, S.; Pechberty, S.; Koch-Nolte, F.; Haag, F.; Seman, M. NAD+Released during Inflammation Participates in T Cell Homeostasis by Inducing ART2-Mediated Death of Naive T Cells In Vivo. J. Immunol. 2007, 179, 186–194. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Elalamy, I.; Kastritis, E.; Sergentanis, T.N.; Politou, M.; Psaltopoulou, T.; Gerotziafas, G.; Dimopoulos, M.A. Hematological findings and complications of COVID-19. Am. J. Hematol. 2020, 95, 834–847. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Tanaka, S.; Hori, Y.; Hirayama, F.; Sato, E.F.; Inoue, M. Role of mitochondria in the maintenance of platelet function during in vitro storage. Transfus. Med. 2011, 21, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S. The Failing Heart—An Engine out of Fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamorano-León, J.; Modrego, J.; Mateos-Cáceres, P.; Macaya, C.; Martin-Fernandez, B.; Miana, M.; Heras, N.D.L.; Cachofeiro, V.; Lahera, V.; López-Farre, A. A Proteomic Approach to Determine Changes in Proteins Involved in the Myocardial Metabolism in Left Ventricles of Spontaneously Hypertensive Rats. Cell. Physiol. Biochem. 2010, 25, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Lahera, V.; Heras, N.D.L.; Farre, A.L.; Manucha, W.; Ferder, L. Role of Mitochondrial Dysfunction in Hypertension and Obesity. Curr. Hypertens. Rep. 2017, 19, 11. [Google Scholar] [CrossRef] [PubMed]
- Padmalayam, I. Targeting mitochondrial oxidative stress through lipoic acid synthase: A novel strategy to manage diabetic cardiovascular disease. Cardiovasc. Hematol. Agents Med. Chem. 2012, 10, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Manucha, W.; Ritchie, B.; Ferder, L. Hypertension and Insulin Resistance: Implications of Mitochondrial Dysfunction. Curr. Hypertens. Rep. 2014, 17, 504. [Google Scholar] [CrossRef] [PubMed]
- Baltrusch, S. Mitochondrial network regulation and its potential interference with inflammatory signals in pancreatic beta cells. Diabetologia 2016, 59, 683–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Wakabayashi, N.; Wakabayashi, J.; Tamura, Y.; Song, W.-J.; Sereda, S.; Clerc, P.; Polster, B.M.; Aja, S.M.; Pletnikov, M.V.; et al. The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol. Biol. Cell 2011, 22, 2235–2245. [Google Scholar] [CrossRef]
- Brealey, D.; Brand, M.; Hargreaves, I.; Heales, S.; Land, J.; Smolenski, R.; Davies, N.A.; Cooper, C.E.; Singer, M. Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 2002, 360, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Nakahira, K.; Kyung, S.-Y.; Rogers, A.; Gazourian, L.; Youn, S.; Massaro, A.F.; Quintana, C.; Osorio, J.C.; Wang, Z.; Zhao, Y.; et al. Circulating Mitochondrial DNA in Patients in the ICU as a Marker of Mortality: Derivation and Validation. PLoS Med. 2013, 10, e1001577. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Sun, L.; Chen, Z. Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef]
- Tan, X.; Sun, L.; Chen, J.; Chen, Z.J. Protected, Email Detection of Microbial Infections Through Innate Immune Sensing of Nucleic Acids. Annu. Rev. Microbiol. 2018, 72, 447–478. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-ΚB Activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [Green Version]
- Sursal, T.; Stearns-Kurosawa, D.J.; Itagaki, K.; Oh, S.-Y.; Sun, S.; Kurosawa, S.; Hauser, C.J. Plasma Bacterial and Mitochondrial DNA Distinguish Bacterial Sepsis from Sterile Systemic Inflammatory Response Syndrome and Quantify Inflammatory Tissue Injury in Nonhuman Primates. Shock 2013, 39, 55–62. [Google Scholar] [CrossRef] [Green Version]
- Faas, M.M.; De Vos, P. Mitochondrial function in immune cells in health and disease. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165845. [Google Scholar] [CrossRef] [PubMed]
- Mittra, I.; Nair, N.K.; Mishra, P.K. Nucleic acids in circulation: Are they harmful to the host? J. Biosci. 2012, 37, 301–312. [Google Scholar] [CrossRef]
- Ishii, K.; Suzuki, K.; Coban, C.; Takeshita, F.; Itoh, Y.; Matoba, H.; Kohn, L.D.; Klinman, D.M. Genomic DNA Released by Dying Cells Induces the Maturation of APCs. J. Immunol. 2001, 167, 2602–2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb. Perspect. Med. 2015, 5, a025130. [Google Scholar] [CrossRef] [Green Version]
- Mizumachi, T.; Muskhelishvili, L.; Naito, A.; Furusawa, J.; Fan, C.-Y.; Siegel, E.R.; Kadlubar, F.F.; Kumar, U.; Higuchi, M. Increased distributional variance of mitochondrial DNA content associated with prostate cancer cells as compared with normal prostate cells. Prostate 2008, 68, 408–417. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.-W.; Masayesva, B.; Zahurak, M.; Carvalho, A.; Rosenbaum, E.; Mambo, E.; Zhou, S.; Minhas, K.; Benoit, N.; Westra, W.H.; et al. Increased Mitochondrial DNA Content in Saliva Associated with Head and Neck Cancer. Clin. Cancer Res. 2005, 11, 2486–2491. [Google Scholar] [CrossRef] [Green Version]
- Ellinger, J.; Albers, P.; Müller, S.C.; Von Ruecker, A.; Bastian, P.J. Circulating mitochondrial DNA in the serum of patients with testicular germ cell cancer as a novel noninvasive diagnostic biomarker. BJU Int. 2009, 104, 48–52. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Schaefer, S.T.; Sae-Lee, C.; Byun, H.-M.; Wüllner, U. Elevated serum mitochondrial DNA in females and lack of altered platelet mitochondrial methylation in patients with Parkinson´s disease. Int. J. Neurosci. 2021, 131, 279–282. [Google Scholar] [CrossRef]
- Jauhari, A.; Baranov, S.V.; Suofu, Y.; Kim, J.; Singh, T.; Yablonska, S.; Li, F.; Wang, X.; Oberly, P.; Minnigh, M.B.; et al. Melatonin inhibits cytosolic mitochondrial DNA–induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J. Clin. Investig. 2020, 130, 3124–3136. [Google Scholar] [CrossRef] [Green Version]
- Lowes, H.; Pyle, A.; Santibanez-Koref, M.; Hudson, G. Circulating cell-free mitochondrial DNA levels in Parkinson’s disease are influenced by treatment. Mol. Neurodegener. 2020, 15, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morandi, A.; Zusi, C.; Corradi, M.; Takemoto, K.; Contreas, G.; Olivieri, F.; Fornari, E.; Maffeis, C. Circulating mitochondrial DNA is decreased in children and adolescents with obesity and/or insulin resistance. Acta Diabetol. 2020, 57, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Scozzi, D.; Cano, M.; Ma, L.; Zhou, D.; Zhu, J.H.; O’Halloran, J.A.; Goss, C.W.; Rauseo, A.M.; Liu, Z.; Sahu, S.K.; et al. Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID-19. JCI Insight 2021, 6, e143299. [Google Scholar] [CrossRef]
- Mollo, N.; Cicatiello, R.; Aurilia, M.; Scognamiglio, R.; Genesio, R.; Charalambous, M.; Paladino, S.; Conti, A.; Nitsch, L.; Izzo, A. Targeting Mitochondrial Network Architecture in Down Syndrome and Aging. Int. J. Mol. Sci. 2020, 21, 3134. [Google Scholar] [CrossRef] [PubMed]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus (Family) | Aim and Study Design | Pathological Mechanism Associated with Mitochondria | Ref |
|---|---|---|---|
| Zika (Flaviviridae) | This study analyzed the consequences of ZIKV infection in iPSC-derived astrocytes present in mice and post-mortem infected neonate brains. | The ZKV replicate in the ER. Ca2+ released from the ER to the cytoplasm was taken up by the mitochondria, leading to an increase in ROS production and mitochondrial dysfunction. | [15] |
| Hepatitis C (Flaviviridae) | This review summarizes the mechanism involved in ROS formation in HCV-replicating cells and describes the intervention of HCV with ROS-detoxifying systems. | HCV replication induced ER stress, which released Ca2+ into the cytoplasm, causing an increased Ca2+ influx into the mitochondria via VDACs, thus inducing ROS production. | [81] |
| Japanese encephalitis (Flaviviridae) | Compound CW-33, a synthesized intermediate fluoroquinolone derivative, was investigated to evaluate its antiviral activities against JEV in baby hamster kidneys and human medulloblastoma. | JEV triggered a Ca2+ overload, causing a mitochondrial membrane potential alteration. | [83] |
| Dengue (Flaviviridae) | Freshly isolated platelets from patients infected with dengue were evaluated for the activation of markers and cell death pathways and mitochondrial alterations. | Platelets from dengue-infected patients exhibited classic signs of apoptotic intrinsic pathways that included an increased mitochondrial depolarization, surface phosphatidylserine exposure, and activation of caspase-3 and 9. Mitochondrial dysfunction was followed by the activation of apoptosis in platelets, contributing to thrombocytopenia. | [85] |
| Dengue (Flaviviridae) | To explore if DENV can affect mitochondrial function and dynamics, such as fusion and fission, a mitochondrion intermixing experiment in two stable A549 cell lines infected with DENV was performed. | The mitochondrial fusion was impaired in dengue virus-infected cells because of the cleaving of two mitofusins (Mfn1-Mfn2) by the dengue virus protease, NS2B3. This led to an increased virus replication and disruption of the mitochondrial membrane potential. | [86] |
| Dengue virus (Flaviviridae) | Evaluated the functions of NS4B, a DENV non-structural protein associated with a promising drug target for this virus (in relation to the alteration of mitochondrial proteins and morphology), the NS4B in DENV-infected cells were elucidated. | Dengue’s protein, NS4B, induced mitochondria elongation, compromising the integrity of MAM, which are sites of the ER-mitochondria interface critical for innate immune signaling. This promotes virus replication and the activation of interferon responses. | [87] |
| Hepatitis C Virus (Flaviviridae) | In this study, the ability of HCV NS3-4A protease to cleave peroxisomal and mitochondrial MAVS was analyzed, as well as whether this would have any effect on the cellular antiviral response, by creating a Myc-tagged mutant of MAVS. | The HCV NS3-4A protease cleaved the mitochondrial MAVS, inhibiting the downstream response by blocking the RIG-I-like (RLR) signaling from peroxisomes, thus inhibiting the antiviral gene expression. | [82] |
| Dengue virus (Flaviviridae) | This paper investigated the role of human dendritic cells in the dengue virus infection. | The dendritic cells infected by the dengue virus showed hypertrophy and proliferation of the ER, as well as swollen mitochondria. | [88] |
| Dengue virus (Flaviviridae) | Exanimating bone marrow-derived dendritic cells prepared from Tlr9-knockout mice, a previously unrecognized phenomenon in which DENV infection activates TLR9 signaling (important sensors that recognize pathogen-associated molecular patterns) was unraveled by inducing mt-DNA release. | Dengue virus infection induced an mt-DNA release into the cytosol, activating the TLR9 signal pathway and leading to the production of IFNs. This caused ROS generation and inflammasome activation. | [84] |
| West Nile (Flaviviridae) | In this in vitro study, cell death in the brain-derived tumor cell line, T98G, was induced to illuminate the molecular mechanism of WNV-induced neural cell death. | WNV replication decreased cell viability and induced apoptosis. The intrinsic apoptosis pathway established the signaling of pro-apoptotic proteins such as Bax, which trigger the mitochondrion OM, followed by cytochrome C release from the mitochondria into the cytoplasm. | [89] |
| Dengue (Flaviviridae) | To confirm the interaction between GR978, a cellular chaperone, and VDAC and DENV E protein, HEK293T/17 cells infected with DENV were used to determine the percentage of infection by flow cytometry. | An interaction between DENV proteins, GRP78 and VDAC, located in the mitochondrial OMM, resulted in the movement of VDAC towards the ER, causing the formation of pores in the mitochondrial membrane and the subsequent release of cytochrome C in the cytosol. | [90] |
| Japanese Encephalitis (Flaviviridae) | To address the role of fatty-acid β-oxidation in JEV infection, the oxygen consumption rate in JEV-infected cells cultured with or without LCFA palmitate was measured. | JEV nonstructural protein 5 (NS5) interacted with the mitochondrial trifunctional protein, an enzyme complex involved in LCFA B-oxidation, enhancing cytokine production and contributing to JEV pathogenesis. | [91] |
| Dengue (Flaviviridae) | In this study, the associations between dengue virus-induced cell death and mitochondrial function in HepG2, a human hepatoma cell line, were evaluated. | HepG2 cells infected with the dengue virus suffer continuous metabolic stress, promoting mitochondrial bioenergetics changes, such as a cellular respiration increase, causing mitochondrial swelling and other morphological changes and inducing mitochondrial dysfunction and alterations in the cellular ATP balance. | [92] |
| Respiratory syncytial (Paramyxoviridae) | Employing bioenergetic measurements, mitochondrial redox-sensitive dye, and high-resolution quantitative imaging, the importance of mitochondrial complex I in RSV infection was studied. | RSV attacked the mitochondrial Complex I subunit knock-out (KO) cells, leading to a mitochondrial respiration decrease and an increased ROS, which favors RSV infection. | [93] |
| Hepatitis B (Hepadnaviridae) | In this review, the interaction between mitochondria and oncoviruses, particularly in hepatitis B and C and human papilloma and HIV viruses, were discussed. | HBx, a protein of the HBV, targets and integrates into the mitochondrial OMM, inducing ROS overproduction and mt-DNA oxidative damage. | [80] |
| Hepatitis C (Flaviviridae) | The HCV core protein disturbs both the ER and the mitochondrial OMM, inducing a specific inhibition of Complex I, which adapts the mitochondria for hypoxia and enhances the ROS production at the same time. Meanwhile, the virus core protein facilitates ER Ca2+ release and increases the mitochondrial Ca2+ uptake, thus increasing the ROS production. | ||
| Papillomavirus (Papillomaviridae) | HPV 18 E2 interacted directly with the mitochondrial respiratory chain and increased the mitochondrial ROS release. | ||
| Human Immunodeficiency (Retroviridae) | HIV targets Mfn2 and reduces its protein level, damaging the integrity of the outer mitochondrial membrane and inducing a progressive mitochondrial deformation. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdés-Aguayo, J.J.; Garza-Veloz, I.; Badillo-Almaráz, J.I.; Bernal-Silva, S.; Martínez-Vázquez, M.C.; Juárez-Alcalá, V.; Vargas-Rodríguez, J.R.; Gaeta-Velasco, M.L.; González-Fuentes, C.; Ávila-Carrasco, L.; et al. Mitochondria and Mitochondrial DNA: Key Elements in the Pathogenesis and Exacerbation of the Inflammatory State Caused by COVID-19. Medicina 2021, 57, 928. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina57090928
Valdés-Aguayo JJ, Garza-Veloz I, Badillo-Almaráz JI, Bernal-Silva S, Martínez-Vázquez MC, Juárez-Alcalá V, Vargas-Rodríguez JR, Gaeta-Velasco ML, González-Fuentes C, Ávila-Carrasco L, et al. Mitochondria and Mitochondrial DNA: Key Elements in the Pathogenesis and Exacerbation of the Inflammatory State Caused by COVID-19. Medicina. 2021; 57(9):928. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina57090928
Chicago/Turabian StyleValdés-Aguayo, José J., Idalia Garza-Veloz, José I. Badillo-Almaráz, Sofia Bernal-Silva, Maria C. Martínez-Vázquez, Vladimir Juárez-Alcalá, José R. Vargas-Rodríguez, María L. Gaeta-Velasco, Carolina González-Fuentes, Lorena Ávila-Carrasco, and et al. 2021. "Mitochondria and Mitochondrial DNA: Key Elements in the Pathogenesis and Exacerbation of the Inflammatory State Caused by COVID-19" Medicina 57, no. 9: 928. https://0-doi-org.brum.beds.ac.uk/10.3390/medicina57090928