Analysis of the Transcriptome of the Red Seaweed Grateloupia imbricata with Emphasis on Reproductive Potential


Abstract
:1. Introduction
2. Results and Discussion
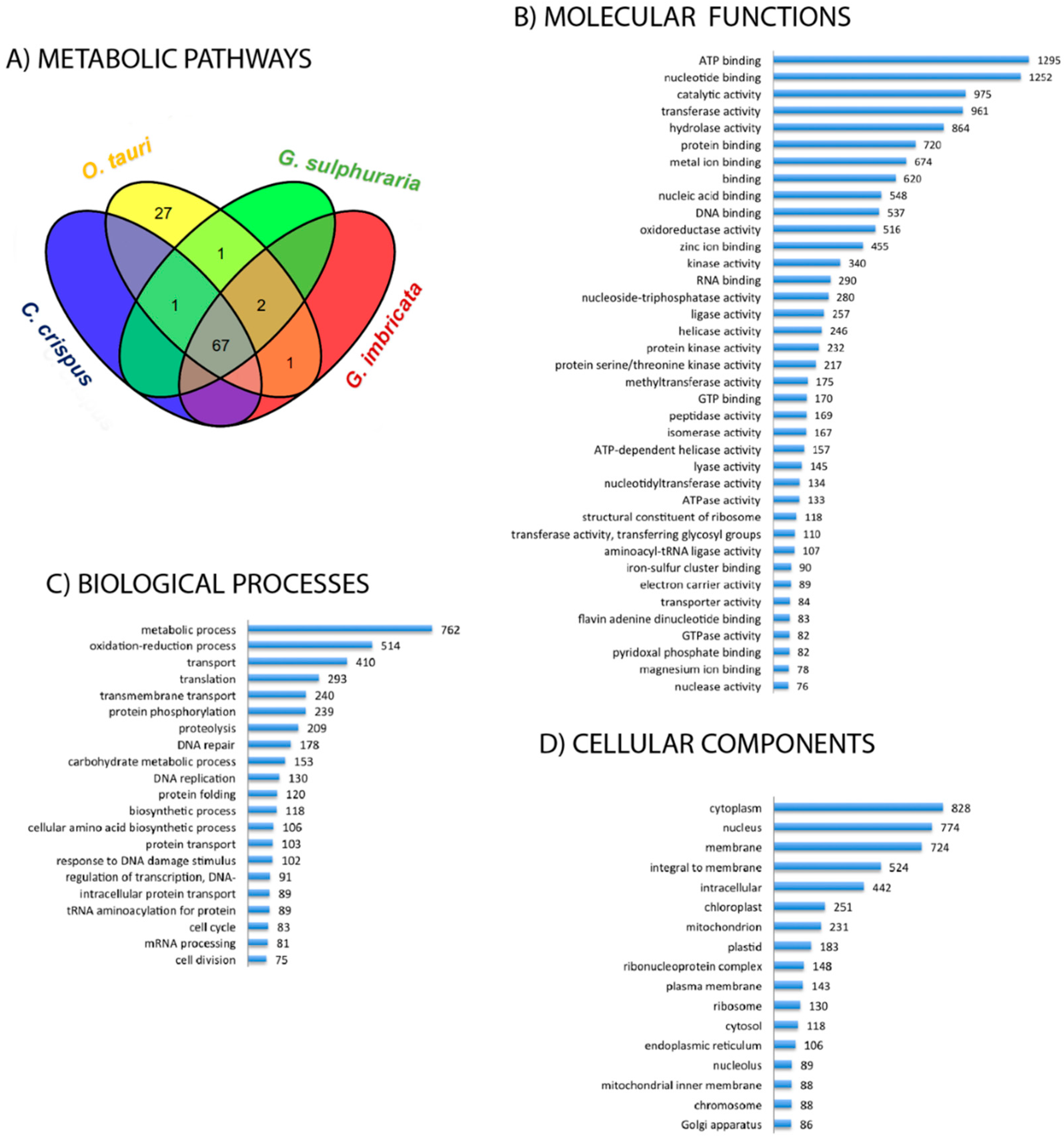
2.1. De novo Reconstruction and Annotation of the Red Seaweed Grateloupia imbricata Transcriptome
2.2. Functional Profile of the Grateloupia imbricata Transcriptome
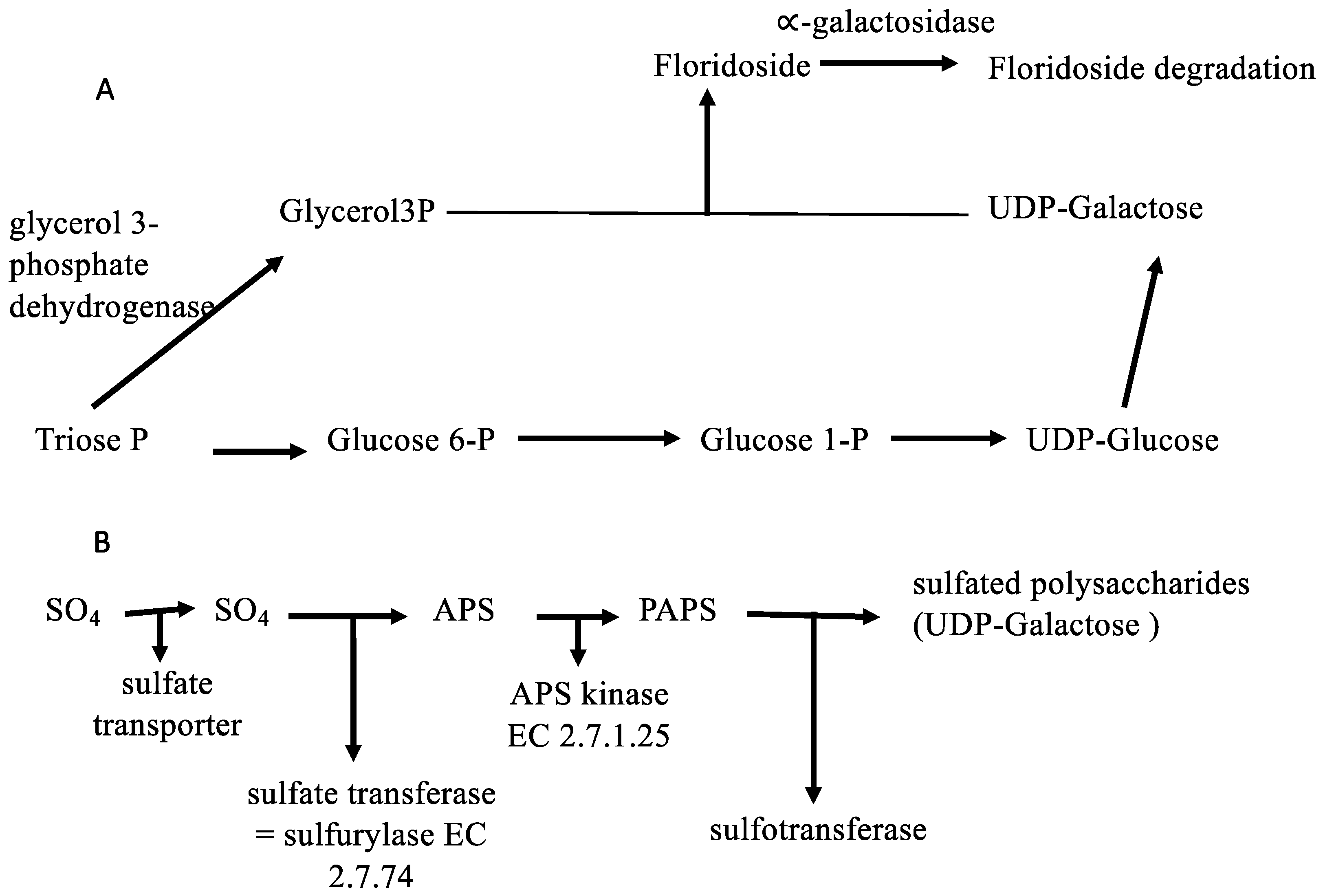
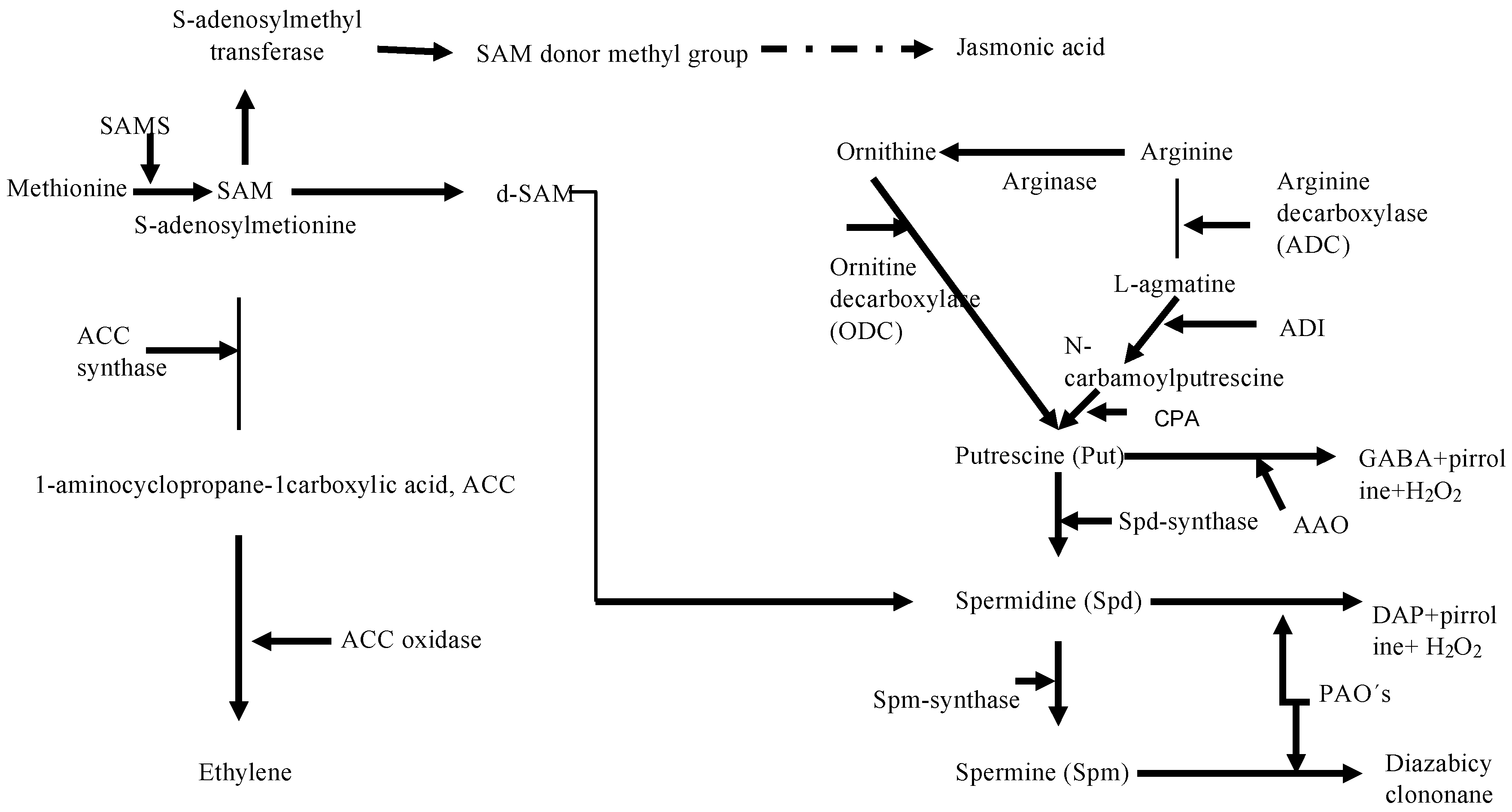
2.3. Metabolic Perspectives on the Growth, Development, and Reproduction of Grateloupia
3. Materials and Methods
3.1. Sampling
3.2. RNA Extraction and Poly(A)-RNA Enrichment
3.3. RNA Library Construction and Transcriptome Sequencing
3.4. Preprocessing, De Novo Assembly, and Annotations
3.5. Data Mining and Comparative Analyses
3.6. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chan, C.X.; Blouin, N.A.; Zhuang, Y.; Zaüner, S.; Prochnik, S.E.; Lindquist, E.; Lin, S.; Benning, C.; Lohr, M.; Yarish, C.; et al. Porphyra (Bangiophyceae) transcriptomes provide insights into red algal development and metabolism. J. Phycol. 2012, 48, 1328–1342. [Google Scholar] [CrossRef] [PubMed]
- Collén, J.; Porcel, B.; Carré, W.; Ball, S.G.; Chaparro, C.; Tonon, T.; Barbeyron, T.; Michel, G.; Noel, B.; Valentin, K.; et al. Genome structure and metabolic features in the red seaweed Chondrus crispus shed light on evolution of the Archeaplastida. Proc. Natl. Acad. Sci. USA 2013, 26, 5247–5252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Jimenez, P.; Robaina, R.R.; Luque, A.; Tsekos, I. Glycerol-activated cellular division and biosynthetic activity during growth and morphogenesis of carpospore seedlings of Grateloupia doryphora. Phycologia 1996, 35, 261–269. [Google Scholar] [CrossRef]
- Garcia-Jimenez, P.; Rodrigo, M.; Robaina, R.R. Plant growth regulators and glycerol induced cell proliferation and morphogenesis in carpospore-seedlings cultivated in vitro. J. Appl. Phycol. 1998, 10, 95–100. [Google Scholar] [CrossRef]
- Garcia-Jimenez, P.; Brito-Romano, O.; Robaina, R.R. Occurrence of jasmonates during cystocarp development in the red alga Grateloupia imbricata. J. Phycol. 2016, 52, 1085–1093. [Google Scholar] [CrossRef]
- Sacramento, A.T.; Garcia-Jimenez, P.; Robaina, R.R. The polyamine spermine induces cystocarp development in the seaweed Grateloupia (Rhodophyta). Plant Growth Reg. 2007, 53, 147–154. [Google Scholar] [CrossRef]
- Sacramento, A.T.; Garcia-Jimenez, P.; Alcázar, R.; Tiburcio, A.; Robaina, R.R. Influence of polyamines on the sporulation of Grateloupia (Halymeniaceae, Rhodophyta). J. Phycol. 2004, 50, 887–894. [Google Scholar] [CrossRef]
- Garcia-Jimenez, P.; Robaina, R.R. Effects of ethylene on tetrasporogenesis in Pterocladiella capillacea (Rhodophyta). J. Phycol. 2012, 48, 710–715. [Google Scholar] [CrossRef]
- Garcia-Jimenez, P.; Robaina, R.R. Volatiles in the aquatic marine ecosystem: Ethylene and related plant hormones and sporulation in red seaweeds. In System Biology of Marine Ecosystems; Kumar, M., Ralph, P., Eds.; Springer: Sydney, Australia, 2017; pp. 99–116. [Google Scholar]
- Garcia-Jimenez, P.; García-Maroto, F.; Garrido-Cárdenas, J.A.; Ferrandiz, C.; Robaina, R.R. Differential expression of the ornithine decarboxylase gene during carposporogenesis in the thallus of the red seaweed Grateloupia imbricata (Halymeniaceae). J. Plant Physiol. 2009, 166, 1745–1754. [Google Scholar] [CrossRef]
- Garcia-Jimenez, P.; Montero-Fernández, M.; Robaina, R.R. Molecular mechanisms underlying Grateloupia imbricata (Rhodophyta) carposporogenesis induced by methyl jasmonate. J. Phycol. 2017, 53, 1340–1344. [Google Scholar] [CrossRef]
- Montero-Fernandez, M.; Robaina, R.R.; Garcia-Jimenez, P. In silico characterization of DNA motifs associated with the differential expression of the ornithine decarboxylase gene during in vitro cystocarp development in the red seaweed Grateloupia imbricata. J. Plant Physiol. 2016, 195, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Blanc, G.; Etten, J.L.V. The Chlorella variabilis NC64A genome reveals adaptation to photosymbiosis, coevolution with viruses, and cryptic sex. Plant Cell. 2010, 22, 2943–2955. [Google Scholar] [CrossRef] [PubMed]
- Shuangxiu, W.; Jing, S.; Shan, C.; Liang, W.; Xumin, W.; Cui, L.; Xingang, L.; Jinlong, Y.; Tao, L.; Jun, Y. Transcriptome sequencing of essential marine brown and red algal species in China and its significance in algal biology and phylogeny. Acta Oceanol. Sin. 2014, 33, 1–12. [Google Scholar] [CrossRef]
- Lee, W.K.; Lim, Y.Y.; Thean-Chor Leow, A.; Namasivayam, P.; Ong Abdullah, J.; Ho, C.L. Biosynthesis of agar in red seaweeds: A review. Carbohydr. Polym. 2017, 164, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Shabtai, Y.; Arad, S. Floridoside as a carbon precursor for the synthesis of cell-wall polysaccharide in the red microalga Porphyridium sp. (Rhodophyta). J. Phycol. 2002, 38, 931–938. [Google Scholar] [CrossRef]
- Robaina, R.R.; Garcia-Jimenez, P.; García-Reina, G.; Luque, A. Morphogenetic effect of glycerol on tissue cultures of the red seaweed Grateloupia doryphora. J. Appl. Phycol. 1990, 2, 137–143. [Google Scholar] [CrossRef]
- Robaina, R.R.; Garcia-Jimenez, P.; Brito, I.; Luque, A. Light control of the respiration of exogenous glycerol in the red macroalga Grateloupia doryphora. Eur. J. Phycol. 1995, 30, 81–86. [Google Scholar] [CrossRef]
- Kremer, B.P.; Kirst, G.O. Biosynthesis of 2-O-d-glycerol-a-d-galactopyranoside (Floridoside) in marine Rhodophyceae. Plant Sci. Lett. 1981, 23, 349–357. [Google Scholar] [CrossRef]
- Weïwer, M.; Sherwood, T.; Linhardt, R.J. Synthesis of floridoside. J. Carbohydr. Chem. 2008, 27, 420–427. [Google Scholar] [CrossRef]
- Kopriva, S. Regulation of sulfate assimilation in Arabidopsis and beyond. Ann. Bot. 2006, 97, 479–495. [Google Scholar] [CrossRef] [PubMed]
- Oesterhelt, C.; Schnarrenberger, C.; Gross, W. Characterization of a sugar/polyol uptake system in the red alga Galdieria sulphuraria. Eur. J. Phycol. 1999, 34, 271–277. [Google Scholar] [CrossRef]
- Guzman-Urióstegui, A.; Garcia-Jimenez, P.; Marián, F.; Robledo, D.; Robaina, R.R. Polyamines influence maturation in reproductive structures of Gracilaria cornea (Gracilariales, Rhodophyta). J. Phycol. 2002, 38, 1169–1175. [Google Scholar] [CrossRef]
- De Oliveira, L.S.; Gregoracci, G.B.; Silva, G.G.; Salgado, L.T.; Filho, G.A.; Alves-Ferreira, M.A.; Pereira, R.C.; Thompson, F. Transcriptomic analysis of the red seaweed Laurencia dendroidea (Florideophyceae, Rhodophyta) and its microbiome. BMC Genomics 2012, 13, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Jimenez, P.; Robaina, R.R. On reproduction in red algae: Further research needed at the molecular level. Front. Plant Sci. 2015, 6, 1–6. [Google Scholar] [CrossRef]
- Roje, S. S-Adenosyl-L-methionine: Beyond the universal methyl group donor. Phytochemistry 2006, 67, 1686–1698. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Asamizu, E.; Shibata, D.; Nakamura, Y.; Kaneko, T.; Awai, K.; Amagai, M.; Kuwata, C.; Tsugane, T.; Masuda, T.; et al. Monitoring of methyl jasmonate-responsive genes in Arabidopsis by cDNA macroarray: Self-activation of jasmonic acid biosynthesis and crosstalk with other phytohormone signaling pathways. DNA Res. 2001, 8, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Devoto, A.; Turner, J.G. Regulation of jasmonate-mediated plant responses in Arabidopsis. Ann. Bot. 2003, 92, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Ecker, J.R. The ethylene signaling pathway: New insights. Curr. Opin. Plant Biol. 2004, 7, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Harter, K. Mitogen-activated protein kinase cascades and ethylene: Signaling, biosynthesis, or both? Plant Physiol. 2009, 149, 1207–1210. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Jimenez, P.; Marian, F.D.; Rodrigo, M.; Robaina, R.R. Sterilization and sporulation method for axenic culture of Gelidium canariensis. J. Biotech. 1999, 70, 227–229. [Google Scholar] [CrossRef]
- Dring, M.J. Photo control of development in algae. Ann. Rev. Plant Physiol. Plant Mol. Biol. 1988, 39, 157–174. [Google Scholar] [CrossRef]
- Rodrigo-Sanz, M. Crecimiento, Desarrollo y Diferenciación de Macroalgas Marinas; Servicio de Publicaciones de la Universidad: Las Palmas de Gran Canaria, Spain, 1998; 320p. [Google Scholar]
- Schaller, A. Bioactive peptides as signal molecules in plant defense, growth and development. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, Neederlands, 2001; pp. 367–411. [Google Scholar]
- Van Alstyne, K.L.; Houser, L.T. Dimethylsulfide release during macroinvertebrate grazing and its role as an activated chemical defense. Mar. Ecol. Prog. Ser. 2003, 250, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Jimenez, P.; Brito-Romano, O.; Robaina, R.R. Production of volatiles by the red seaweed Gelidium arbuscula (Rhodophyta): Emission of ethylene and dimethyl sulfide. J. Phycol. 2013, 49, 661–669. [Google Scholar] [CrossRef]
- Kocsis, M.; Nolte, K.D.; Rhodes, D.; Shen, T.; Gage, D.A.; Hanson, A.D. Dimethylsulfoniopropionate biosynthesis in Spartina alterniflora. Evidence that S-methylmethionine and dimethylsulfoniopropylamine are intermediates. Plant Physiol. 1998, 117, 273–281. [Google Scholar] [CrossRef]
- Giordano, M.; Norici, A.; Hell, R. Sulfur and phytoplankton: Acquisition, metabolism and impact on the environment. New Phytol. 2005, 166, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futami, R.; Muñoz-Pomer, L.; Viu, J.M.; Dominguez-Escriba, L.; Covelli, L.; Bernet, G.P.; Sempere, J.M.; Moya, A.; Llorens, C. GPRO: The professional tool for management, functional analysis and annotation of omic sequences and databases. SOFTWARE ARTICLE. Biotechvana Bioinform. 2011, 1, 1–5. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium. The Gene Ontology project in 2008. Nucleic Acids Res. 2008, 36, 440–444. [Google Scholar]
- Kotera, M.; Hirakawa, M.; Tokimatsu, T.; Goto, S.; Kanehisa, M. The KEGG Databases and Tools Facilitating Omics Analysis: Latest Developments Involving Human Diseases and Pharmaceuticals. Methods Mol. Biol. 2012, 802, 19–23. [Google Scholar] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; et al. The COG database: An updated version includes eukaryotes. BMC Bioinform. 2003, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Amode, M.R.; Barrell, D.; Beal, K.; Billis, K.; Brent, S.; Flicek, P. Ensembl 2015. Nucleic Acids Res. 2015, 43, D662–D669. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
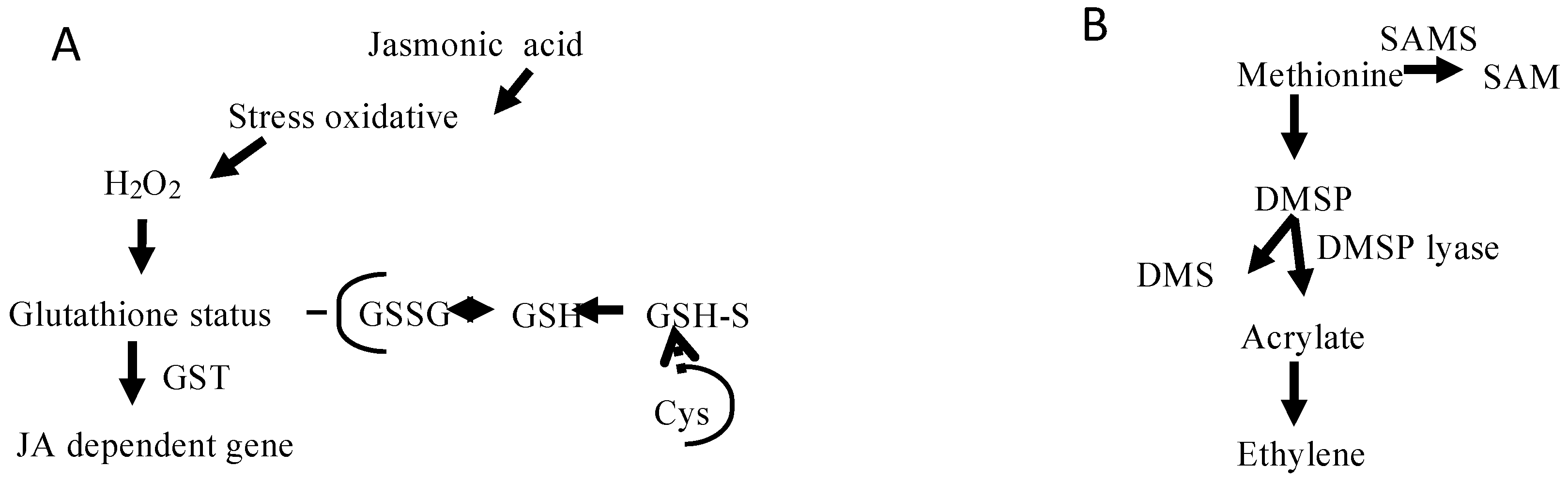{kind=link}
| Contigs/Transcripts | 19,284 | |
| Total transcriptome size | 11,681,373 | |
| Longest transcript (nt) | 7289 | |
| Shortest transcript (nt) | 200 | |
| Number of transcripts < 1K | 16,664 (86.3%) | |
| Number of transcripts > 1K | 2640 (13.7%) | |
| Mean transcript size | 606 | |
| Median transcript size | 400 | |
| N50 (nt) | 734 | |
| L50 (nt) | 4176 | |
| %A | 25.72 | |
| %C | 24.99 | |
| %G | 24.45 | |
| %T | 24.84 | |
| %Ns | 0 | |
| Average coverage | 42.28 | |
| Databases/Systems | NR annotations | Sequences |
| NR/NT Gene Identifiers (GIs) | 8326 | 10,866 |
| Gene Ontology (GO) terms | 3323 | 5686 |
| Enzyme Codes (ECs) | 637 | 2087 |
| Metabolic Path Maps | 88 | 2031 |
| Orthologs (KOG) Clusters | 2304 | 5394 |
| BUSCO Parameters | ||
| Complete | Total | 25.5% |
| single-copy | 24.8% | |
| duplicated | 0.7% | |
| Fragmented | 51.5% | |
| Missing | 23.0% | |
| Metrics were Inferred using the script assemblathon_stats.pl available (version 3.0. Korf Lab. Genome Center, UC Davis. USA) | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia-Jimenez, P.; Llorens, C.; Roig, F.J.; Robaina, R.R. Analysis of the Transcriptome of the Red Seaweed Grateloupia imbricata with Emphasis on Reproductive Potential. Mar. Drugs 2018, 16, 490. https://0-doi-org.brum.beds.ac.uk/10.3390/md16120490
Garcia-Jimenez P, Llorens C, Roig FJ, Robaina RR. Analysis of the Transcriptome of the Red Seaweed Grateloupia imbricata with Emphasis on Reproductive Potential. Marine Drugs. 2018; 16(12):490. https://0-doi-org.brum.beds.ac.uk/10.3390/md16120490
Chicago/Turabian StyleGarcia-Jimenez, Pilar, Carlos Llorens, Francisco J. Roig, and Rafael R. Robaina. 2018. "Analysis of the Transcriptome of the Red Seaweed Grateloupia imbricata with Emphasis on Reproductive Potential" Marine Drugs 16, no. 12: 490. https://0-doi-org.brum.beds.ac.uk/10.3390/md16120490