4.1.2. Detailed Procedure for Synthesis of Compounds S1, 7, 8, 10, 12–17 and 9a–w
(S)-
3-
(2-
isopropyl-
5-
methyl-
5-
(3-
methylbut-
2-
en-
1-
yl)cyclopent-
1-
en-
1-
yl)phenol (
S1). To a solution of (
S)-2-isopropyl-5-methyl-5-(3-methylbut-2-en-1-yl)cyclopent-1-en-1-yl trifluoromethanesulfonate (2.1 g, 6.2 mmol) in the mixed solvents of DMF (60 mL) and EtOH (60 mL), (3-hydroxyphenyl)boronic acid (1.7 g, 12.4 mmol), palladacycle [
36] (0.18 g, 0.3 mmol), K
2CO
3 (1.7 g, 12.4 mmol) were added at room temperature under nitrogen. The resulting mixture was stirred at the same temperature for 24 h. The reaction mixture was then poured into water (60 mL) and extracted with ethyl acetate (3 × 60 mL). The organic layers were combined, washed with brine (5 × 15 mL), dried over Na
2SO
4 and concentrated in vacuum. The residue was purified by silica-gel CC to afford the desired cross-coupling product
S1 (5.50 g, 90%) as a colorless oil (
Rf = 0.48 petroleum ether/ethyl acetate = 5/1).
1H NMR (300 MHz, CDCl
3) δ 6.78–6.74 (m, 1H), 6.66 (dt,
J = 7.6, 1.2 Hz, 1H), 6.58 (dd,
J = 2.6, 1.4 Hz, 1H), 5.23–5.17 (m, 2H), 4.98 (s, 1H), 2.46–2.37 (m, 1H), 2.32 (d,
J = 14.6 Hz, 2H), 2.04–1.96 (m, 2H), 1.95–1.83 (m, 1H), 1.73 (d,
J = 1.5 Hz, 3H), 1.69–1.57 (m, 4H), 1.02 (s, 3H), 0.95 (t,
J = 7.1 Hz, 6H).
13C NMR (75 MHz, CDCl
3) δ 154.79, 144.81, 141.73, 140.41, 132.26, 128.81, 122.40, 121.9, 116.51, 113.10, 51.84, 38.16, 35.90, 28.11, 27.71, 26.40, 26.13, 21.59, 21.40, 18.01. HRESIMS
m/
z Calcd for C
20H
28O ([M + H]
+): 285.2213; Found: 285.2219.
(S)-2-(2-(3-hydroxyphenyl)-3-isopropyl-1-methylcyclopent-2-en-1-yl)acetaldehyde (7). To a solution of NaIO4 (2.75 g, 12.89 mmol), pyridine (1.03 mL, 12.89 mmol), and S1 (0.92 g, 3.22 mmol) in dioxane/H2O (30 mL/10 mL) OsO4 (0.039 M in H2O, 2.48 mL, 0.09 mmol) was added dropwise at room temperature. After stirring for 16 h, the reaction mixture was quenched with saturated aq. Na2SO3 (40 mL) and stirred for 30 min. The aqueous layer was extracted with EtOAc (3 × 20 mL). The combined organic layer was washed successively with H2O (2 × 20 mL) and brine (20 mL), dried over Na2SO4, and concentrated in vacuum. The residue was purified by silica-gel CC to provide 7 (0.68 g, 82% yield) as a colorless oil (Rf = 0.4, petroleum ether/EtOAc = 4:1).
(S)-2-(2-(3-hydroxyphenyl)-3-isopropyl-1-methylcyclopent-2-en-1-yl)acetaldehyde (8). To a solution of 7 (0.6 g, 2.32 mmol) in THF (23 mL), ethylmagnesium bromide (0.3 M in THF, 7.7 mL, 2.32 mmol) was added dropwise at −78 °C and stirred for 1 h. The reaction mixture was heated to room temperature stirred overnight, then quenched with saturated aq. NH4Cl (30 mL), and then extracted with EtOAc (3 × 20 mL). The combined organic layer was washed successively with H2O (2 × 20 mL) and brine (20 mL), dried over Na2SO4 and concentrated in vacuum. The residue was purified by silica-gel CC to give 8 (0.42 g, 70% yield) as a white solid (Rf = 0.3, petroleum ether/EtOAc = 4:1). 1H NMR (300 MHz, CDCl3) δ 7.17 (t, J = 8.0 Hz, 1H), 6.98 (d, J = 7.8 Hz, 1H), 6.78 (d, J = 8.1 Hz, 1H), 5.20 (dd, J = 10.8, 6.7 Hz, 1H), 3.21 (p, J = 6.8 Hz, 1H), 2.60–2.33 (m, 3H), 1.83–1.60 (m, 3H), 1.16 (d, J = 6.9 Hz, 3H), 1.02 (d, J = 7.8 Hz, 6H). HRESIMS m/z Calcd for C17H22O2 ([M + Na]+): 281.1512; Found: 281.1506.
(S)-1-isopropyl-3a-methyl-3,3a-dihydro-2H-cyclopenta[a]naphthalen-6-ol (10). To a solution of 7 (0.6 g, 2.32 mmol) in THF (23 mL), ethylmagnesium bromide (0.3 M in THF, 7.7 mL, 2.32 mmol) was added dropwise at −78 °C and stirred for 1 h. The reaction mixture was then heated at 85 °C for 12 h. The reaction mixture was cooled to room temperature, quenched with saturated aq. NH4Cl (30 mL), and extracted with EtOAc (3 × 20 mL). The combined organic layer was washed successively with H2O (2 × 20 mL) and brine (20 mL), dried over Na2SO4, and concentrated in vacuum. The residue was purified by silica-gel CC to give 10 (0.39 g, 70% yield) as a brown solid (Rf = 0.5, petroleum ether/EtOAc = 4:1). 1H NMR (300 MHz, CDCl3) δ 7.14–6.97 (m, 2H), 6.74–6.51 (m, 2H), 6.06 (d, J = 9.8 Hz, 1H), 3.21 (p, J = 6.7 Hz, 1H), 2.45–2.33 (m, 2H), 1.90 (t, J = 5.9 Hz, 2H), 1.20 (d, J = 6.7 Hz, 3H), 1.03 (d, J = 6.6 Hz, 3H), 0.94 (s, 3H). HRESIMS m/z Calcd for C17H20O ([M − H]−): 239.1430; Found: 239.1438.
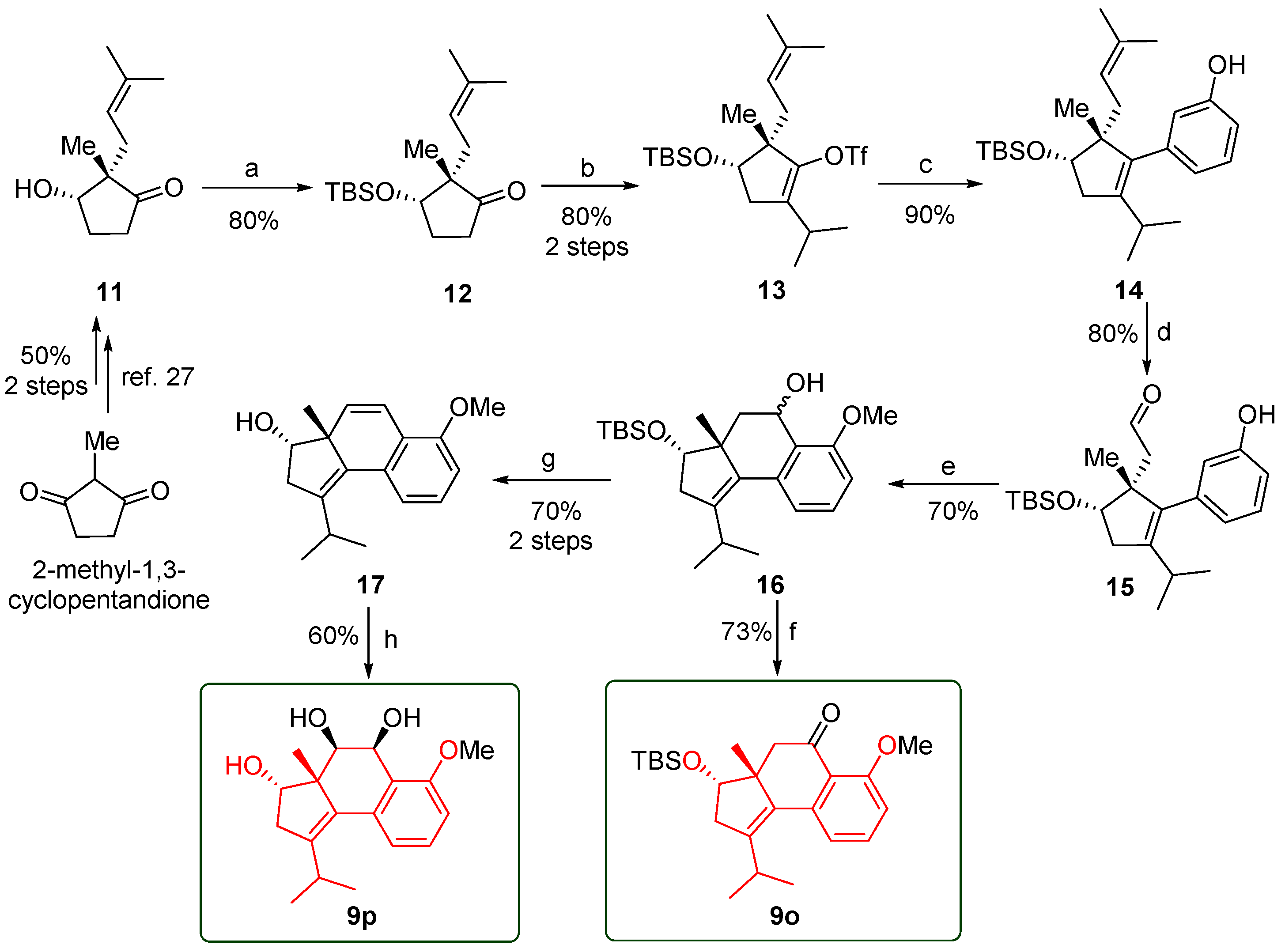
(2S,3S)-3-((tert-butyldimethylsilyl)oxy)-2-methyl-2-(3-methylbut-2-en-1-yl)cyclopentan-1-one (12). To a solution of 11 (5.70 g, 31.27 mmol) in DMF (60 mL) imidazole (2.56 g, 37.52 mmol) and TBSCl (5.66 g, 37.52 mmol) were added. After stirring overnight at room temperature, the reaction mixture was poured into aq. NaHCO3 (50 mL) and extracted with EtOAc (3 × 120 mL). The combined organic layer was washed with brine (5 × 30 mL), dried over Na2SO4 and concentrated in vacuum. The resulting residue was purified by silica-gel CC to afford 12 (7.42 g, 80%) as a colorless oil. (Rf = 0.4, petroleum ether/EtOAc = 15/1). 1H NMR (300 MHz, CDCl3) δ 5.10 (t, J = 7.7 Hz, 1H), 4.04 (t, J = 5.1 Hz, 1H), 2.47–2.36 (m, 1H), 2.29–2.00 (m, 4H), 1.96–1.87 (m, 1H), 1.70 (d, J = 1.7 Hz, 3H), 1.60 (s, 3H), 0.90 (d, J = 10.9 Hz, 12H), 0.09 (d, J = 4.8 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 220.19, 133.34, 119.71, 77.88, 53.71, 34.19, 34.15, 34.12, 28.93, 28.21, 28.19, 25.88, 25.84, 25.63, 25.60, 19.28, 19.25, 17.87, 17.82, 17.72, 17.69, 17.67, −4.39, −4.42, −5.09, −5.12. HRESIMS m/z Calcd for C17H32O2Si ([M + Na]+): 319.2056; Found: 319.2064.
(4S,5S)-4-((tert-butyldimethylsilyl)oxy)-2-isopropyl-5-methyl-5-(3-methylbut-2-en-1-yl)cyclopent-1-en-1-yl trifluoromethanesulfonate (13). To a stirred suspension of NaH (60% dispersion in m ineral oil, 5.7 g, 142.5 mmol) in THF (160 mL) a solution of 12 (8.45 g, 28.5 mmol) in THF (40 mL) was added dropwise at 0 °C, then 2-iodopropane (28.5 mL, 285.0 mmol) was added. After refluxed overnight, the reaction mixture was quenched at 0 °C by adding H2O. Then 2M HCl (80 mL) was added, and the mixture stirred at room temperature for 1.5 h. Then brine was added, and the layers were separated. The aqueous layer was extracted with EtOAc (3 × 100 mL), and the combined organic layer was washed with brine (50 mL), dried over Na2SO4 and concentrated under reduced pressure. The resulting crude residue was purified by silica-gel CC to afford intermediate (8.46 g, 88% yield) as a colorless oil. The intermediate (8.57 g, 25.31 mmol) was dissolved in dry THF (50 mL) and cooled to −78 °C. LiHMDS (1.0 M in THF, 32.9 mL, 32.90 mmol) was added and stirred for 1 h. Then PhNTf2 (11.75 g, 32.90 mmol) dissolved in dry THF (50 mL) was slowly added and the mixture was heated to room temperature stirring for 3 h. Then brine (50 mL) was added to the reaction mixture, and the layers were separated. The aqueous layer was extracted with EtOAc (3 × 50 mL), and the combined organic layer was dried over Na2SO4 and concentrated in vacuum. The residue was purified by flash chromatography to afford 13 (10.84 g, 91% yield) as a colorless oil (Rf = 0.4, petroleum ether/CH2Cl2 = 50/1). 1H NMR (300 MHz, CDCl3) δ 5.28–5.22 (m, 1H), 4.04 (t, J = 7.7 Hz, 1H), 2.83 (p, J = 6.8 Hz, 1H), 2.44 (dd, J = 15.1, 7.8 Hz, 1H), 2.24–2.05 (m, 3H), 1.68 (d, J = 1.3 Hz, 3H), 1.58 (d, J = 1.4 Hz, 3H), 1.11 (s, 3H), 1.04 (d, J = 6.8 Hz, 3H), 0.93 (d, J = 3.9 Hz, 12H), 0.10 (d, J = 2.2 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 143.10, 135.49, 133.14, 120.74, 77.20, 50.24, 34.64, 32.82, 25.99, 25.74, 25.41, 22.68, 20.28, 20.15, 17.97, 17.51, −4.44, −4.89. HRESIMS m/z Calcd for C21H38F3O4SSi ([M + H]+): 471.2207; Found: 471.2197.
3-
((4S,5R)-
4-
((tert-
butyldimethylsilyl)oxy)-
2-
isopropyl-
5-
methyl-
5-
(3-
methylbut-
2-
en-
1-
yl)cyclopent-
1-
en-
1-
yl)phenol (
14). To a solution of vinyltriflate
13 (6.60 g, 14.0 mmol) in the mixed solvents of DMF (60 mL) and EtOH (60 mL), arylboronic acid (3.86 g, 28.0 mmol), palladacycle [
36] (0.40 g, 0.06 mmol), K
2CO
3 (3.87 g, 28.0 mmol) were added at room temperature under nitrogen. When the vinyl triflate had disappeared as monitored by TLC analysis, the reaction mixture was then poured into water (60 mL) and extracted with ethyl acetate (3 × 60 mL). The organic layer was combined, washed with brine (5 × 15 mL), dried over Na
2SO
4, and concentrated in vacuum. The residue was purified by silica-gel CC to afford the desired cross-coupling product
14 (5.50 g, 80%,
Rf = 0.4, petroleum ether/EtOA c = 5/1).
1H NMR (300 MHz, CDCl
3) δ 7.17 (t,
J = 7.8 Hz, 1H), 6.75–6.69 (m, 1H), 6.64 (dt,
J = 7.6, 1.2 Hz, 1H), 6.55 (dd,
J = 2.7, 1.4 Hz, 1H), 5.33–5.23 (m, 1H), 4.80 (dd,
J = 6.8, 2.8 Hz, 1H), 4.02 (t,
J = 7.6 Hz, 1H), 2.49–2.38 (m, 2H), 2.33–2.18 (m, 2H), 1.94 (dd,
J = 15.0, 5.8 Hz, 1H), 1.68–1.56 (m, 6H), 0.91 (d,
J = 8.6 Hz, 18H), 0.09 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 154.87, 142.02, 130.45, 128.86, 123.00, 122.13, 116.36, 113.32, 80.67, 53.55, 37.32, 33.21, 27.50, 26.10, 25.95, 25.40, 21.46, 21.31, 18.21, 18.16, −4.27, −4.74. HRESIMS
m/
z Calcd for C
26H
42O
2Si ([M + Na]
+): 437.2846; Found: 437.2840.
2-((1R,5S)-5-((tert-butyldimethylsilyl)oxy)-2-(3-hydroxyphenyl)-3-isopropyl-1-methylcyclopent-2-en-1-yl)acetaldehyde (15). To a solution of 14 (6.34 g, 15.5 mmol) in the mixed solvent of dioxane (125 mL) and H2O (25 mL), NaIO4 (16.58 g, 77.5 mmol), pyridine (3.74 g, 46.5 mmol), and 15.5 mL solution of OsO4 in H2O (0.04 M) were added. The mixture was stirred at 80 °C until compound 14 had disappeared as monitored by TLC. H2O (100 mL) and EtOAc (200 mL) were added to the mixture. The organic phase was separated, and the aqueous layer was extracted with EtOAc (3 × 80 mL). The combined organic layer was dried over Na2SO4 and concentrated in vacuum. The resulting residue was purified by flash column chromatography to provide crude 15 (4.35 g, 80%) as a brown liquid (Rf = 0.5, petroleum ether/EtOAc = 4:1). 1H NMR (300 MHz, CDCl3) δ 7.54 (s, 1H), 7.17 (t, J = 7.8 Hz, 1H), 6.90–6.70 (m, 2H), 4.97 (s, 1H), 4.87 (d, J = 9.5 Hz, 1H), 4.22 (t, J = 8.4 Hz, 1H), 2.60–2.35 (m, 2H), 2.28 (dd, J = 15.4, 8.5 Hz, 1H), 2.10 (d, J = 14.6 Hz, 1H), 1.69 (d, J = 7.9 Hz, 1H), 1.04 (s, 3H), 0.96 (s, 9H), 0.91 (d, J = 7.7 Hz, 6H), 0.14 (d, J = 3.8 Hz, 6H).
(3S,3aR)-3-((tert-butyldimethylsilyl)oxy)-1-isopropyl-6-methoxy-3a-methyl-3,3a,4,5-tetrahydro-2H-cyclopenta[a]naphthalen-5-ol (16). To a solution of aldehyde 15 (4.8 g) in THF (120 mL), ethylmagnesium bromide (0.5 M in THF, 27 mmol) was added dropwise at −78 °C and stirred for 1 h. After heating at 50 °C for 12 h, the reaction mixture was cooled to room temperature, quenched with saturated aq. NH4Cl (30 mL), and then extracted with EtOAc (3 × 80 mL). The combined organic layer was washed successively with H2O (150 mL) and brine (150 mL), dried over Na2SO4, and concentrated in vacuum. The resulting residue was purified by silica-gel CC to provide 16 (3.5 g, 70% yield) as a brown solid (Rf = 0.5, petroleum ether/EtOAc = 5:1). HRESIMS m/z Calcd for C24H38O3Si ([M + Na]+): 425.2482; Found: 425.2470.
(3S,3aR)-1-isopropyl-6-methoxy-3a-methyl-3,3a-dihydro-2H-cyclopenta[a]naphthalen-3-ol (17). To a solution of compound 16 (201 mg, 0.5 mmol) in THF (5 mL), TBAF (0.5 mL, 1M in THF) was added dropwise at 0 °C and stirred for 2 h. The reaction mixture was heated to room temperature, quenched with ammonium chloride aqueous (10 mL), and then extracted with EtOAc (3 × 10 mL). The combined organic layer was washed successively with brine (20 mL), dried over Na2SO4, and concentrated in vacuum. The resulting residue was purified by silica-gel CC to provide an intermediate (110 mg, 76% yield) as a white solid (Rf = 0.3, petroleum ether/EtOAc = 1:1). To a solution of intermediate (77 mg, 0.25 mmol) in THF (3 mL), p-toluenesulfonic acid (2.2 mg, 0.01 mmol) was added at room temperature. The reaction mixture was stirred at 75 °C for 8 h, quenched with aq. NH4Cl (5 mL), and then extracted with EtOAc (3 × 5 mL). The combined organic layer was washed successively with brine (10 mL), dried over Na2SO4, and concentrated in vacuum. The resulting residue was purified by silica-gel CC to provide 17 (62 mg, 92% yield) as a white solid (Rf = 0.7, petroleum ether/EtOAc = 1:1). 1H NMR (300 MHz, CDCl3) δ 7.19 (t, J = 8.0 Hz, 1H), 7.05 (d, J = 7.8 Hz, 1H), 6.89 (d, J = 10.0 Hz, 1H), 6.78 (d, J = 8.2 Hz, 1H), 6.15 (d, J = 9.9 Hz, 1H), 4.14 (d, J = 4.5 Hz, 1H), 3.85 (s, 3H), 3.30 (h, J = 6.9 Hz, 1H), 2.81 (dd, J = 17.0, 4.4 Hz, 1H), 2.45 (d, J = 17.0 Hz, 1H), 1.20 (d, J = 6.9 Hz, 3H), 1.09 (d, J = 6.9 Hz, 3H), 1.02 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 155.22, 145.29, 133.38, 132.56, 131.19, 127.52, 120.63, 119.63, 109.50, 79.42, 55.61, 55.31, 38.84, 26.92, 22.12, 21.62, 21.27. HRESIMS m/z Calcd for C18H22O2 ([M + Na]+): 293.1512; Found: 293.1509.
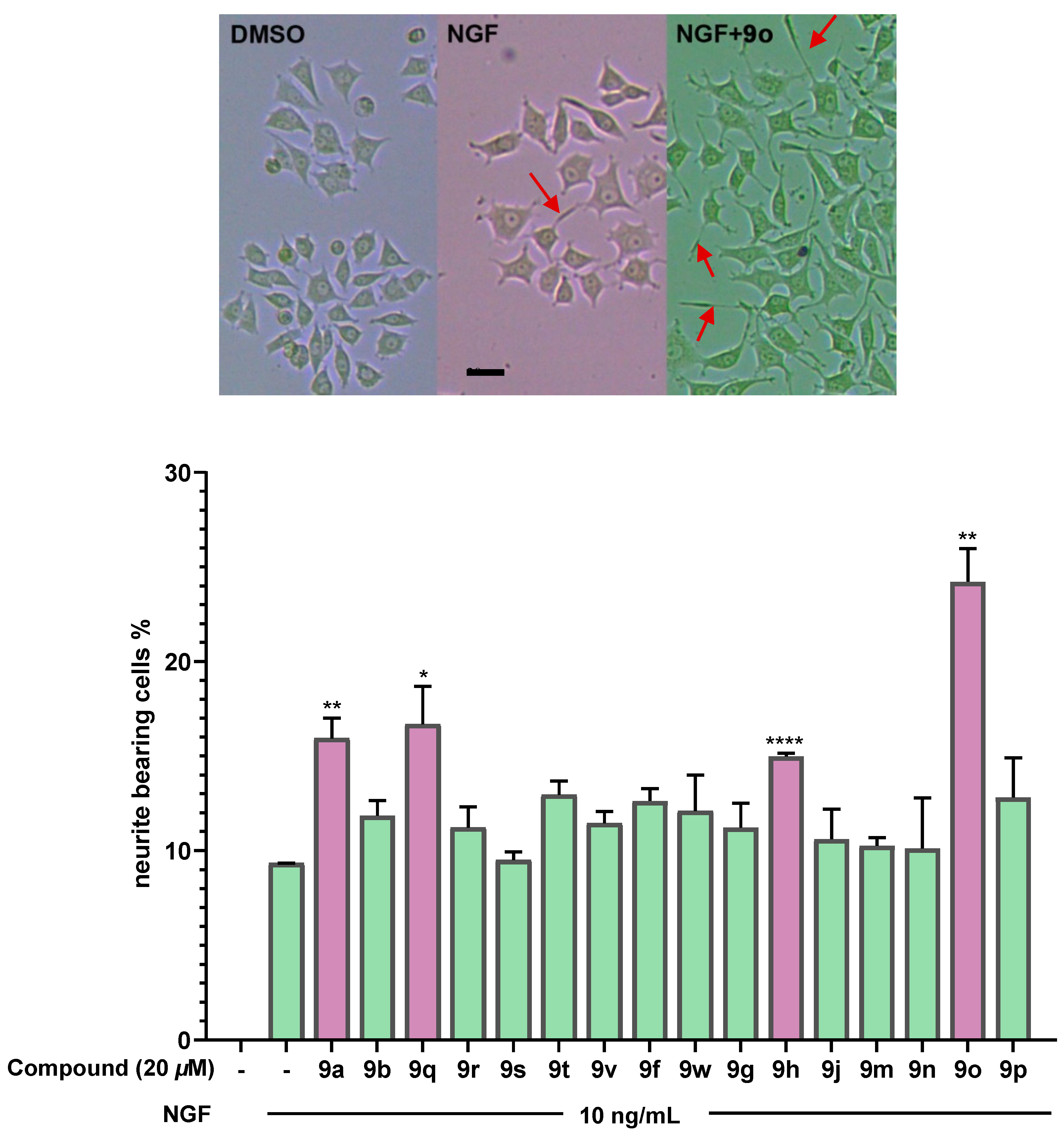
(3aR,4R,5S)-1-isopropyl-3a-methyl-3,3a,4,5-tetrahydro-2H-cyclopenta[a]naphthalene-4,5,6-triol (9a). To a solution of 4-methylmorpholine N-oxide (0.78 g, 5.80 mmol) and 10 (0.35 g, 1.45 mmol) in THF/H2O (12 mL/3 mL), osmium tetroxide (0.039 M in H2O, 1.86 mL, 0.072 mmol) was added dropwise at room temperature. The reaction mixture was stirred at room temperature for 12 h. The reaction mixture was then quenched with saturated solution of sodium sulfite (20 mL) and stirred for 30 min. The mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic layer was washed successively with H2O (2 × 10 mL) and brine (10 mL), dried over Na2SO4 and concentrated under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide 9a (0.24 g, 60% yield) as a white solid (Rf = 0.3, petroleum ether/ethyl acetate = 4:1). 1H NMR (300 MHz, Chloroform-d) δ 8.65 (s, 1H), 7.19 (t, J = 7.9 Hz, 1H), 6.97 (d, J = 7.7 Hz, 1H), 6.87–6.77 (m, 1H), 5.03 (dd, J = 11.0, 4.0 Hz, 1H), 3.77–3.58 (m, 2H), 3.27 (p, J = 6.9 Hz, 1H), 2.63–2.50 (m, 2H), 2.19 (dt, J = 13.4, 9.0 Hz, 1H), 1.61–1.70 (m, 1H), 1.44 (d, J = 11.0 Hz, 1H), 1.20 (d, J = 6.9 Hz, 3H), 1.10–1.00 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 158.57, 149.47, 131.85, 129.09, 128.94, 119.86, 119.06, 115.83, 77.45, 77.02, 76.60, 75.52, 70.18, 54.68, 31.15, 29.83, 27.51, 22.29, 22.04, 21.19. HMRS Calcd for C17H22O3 ([M + Na]+): 297.1461; Found: 297.1461.
(S)-6-hydroxy-1-isopropyl-3a-methyl-2,3,3a,4-tetrahydro-5H-cyclopenta[a]naphthalen-5-one (9b). The compound 8 (26 mg, 0.1 mmol) in dichloromethane (0.5 mL) was added to a stirred solution of PCC (86 mg, 0.4 mmol), NaOAc (33 mg, 0.4 mmol), and celite (20 mg) in dichloromethane (1 mL) at 0 °C. Then the solution was stirred at room temperature for 2 h. The reaction mixture was filtered through celite and washed repeatedly with Et2O. The solvent was removed under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide the ketone 9b (18 mg, 70% yield) as a white solid (Rf = 0.6, petroleum ether/ethyl acetate = 4:1). 1H NMR (300 MHz, CDCl3) δ 12.71 (s, 1H), 7.45 (t, J = 8.0 Hz, 1H), 6.96 (d, J = 7.6 Hz, 1H), 6.85 (d, J = 8.4 Hz, 1H), 3.28–3.14 (m, 1H), 2.74 (s, 2H), 2.60 (dd, J = 8.8, 6.5 Hz, 2H), 1.94–1.78 (m, 2H), 1.22 (d, J = 6.9 Hz, 3H), 1.13–1.02 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 205.59, 162.91, 146.94, 138.43, 136.21, 133.21, 118.25, 116.15, 77.45, 77.02, 76.60, 54.45, 49.99, 36.70, 29.57, 27.40, 24.56, 21.42, 21.18. HRMS Calcd for C17H20O2 ([M + H]+): 257.1536; Found: 257.1532.
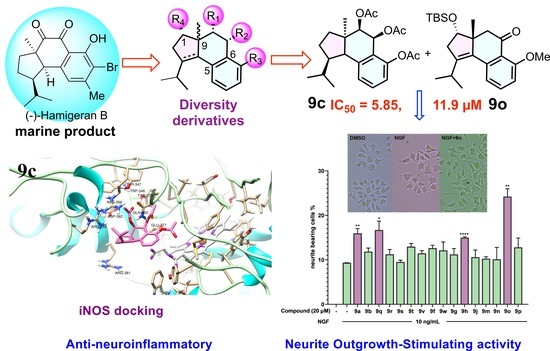
(1R,3aR,4R,5S,9bR)-1-isopropyl-3a-methyl-2,3,3a,4,5,9b-hexahydro-1H-cyclopenta[a]naphthalene-4,5,6-triyl triacetate (9c). To a solution of compound 9h (100 mg, 0.25 mmol) in EtOH (5 mL), Pd(OH)2/C (25 mg, 10%) was added in a high-pressure autoclave. The reaction mixture was hydrogenated for 24 h under 32 atm H2 at room temperature. Pd(OH)2/C was removed by filtration and the filtration was concentrated under reduced pressure without further purification to provide 9c (100 mg, quant) as a white solid (Rf = 0.6, petroleum ether/ethyl acetate = 2:1). 1H NMR (500 MHz, CDCl3) δ 7.30 (t, J = 8.6 Hz, 1H), 7.12 (d, J = 7.9 Hz, 1H), 6.97 (d, J = 8.0 Hz, 1H), 6.34 (d, J = 3.7 Hz, 1H), 4.90 (d, J = 3.6 Hz, 1H), 3.19 (d, J = 7.1 Hz, 1H), 2.45–2.38 (m, 1H), 2.23 (t, J = 1.7 Hz, 3H), 2.04–1.89 (m, 7H), 1.96–1.88 (m, 1H), 1.66–1.57 (m, 2H), 1.46–1.35 (m, 2H), 1.19 (d, J = 1.4 Hz, 3H), 1.14 (dd, J = 6.2, 1.4 Hz, 3H), 0.71 (dd, J = 6.5, 1.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 170.65, 170.19, 169.44, 149.40, 139.26, 128.85, 127.78, 125.76, 120.02, 77.48, 77.06, 76.63, 75.02, 63.38, 53.61, 52.98, 42.59, 34.38, 30.95, 28.10, 26.10, 24.07, 21.44, 20.94, 20.87, 20.84. HRMS Calcd for C23H30O6 ([M + Na]+): 425.1935; Found: 425.1926.
(1R,3aR,9bR)-6-hydroxy-1-isopropyl-3a-methyl-2,3,3a,9b-tetrahydro-1H-cyclopenta[a]naphthalene-4,5-dione (9d). To a solution of lithium hydroxide (105 mg, 2.5 mmol) in H2O (2.5 mL), a solution of compound 9c (100 mg, 0.25 mmol) in THF (2.5 mL) was added at 0 °C. The reaction mixture was stirred at room temperature for 12 h under oxygen atmosphere. To the mixture, 2N HCl (5 mL) was added and extracted with ethyl acetate (3 × 10 mL). The combined organic layer was washed successively with H2O (2 × 10 mL) and brine (10 mL), dried over Na2SO4, and concentrated under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide 9d (48 mg, 70% yield) as a yellow solid (Rf = 0.5, petroleum ether/ethyl acetate = 4:1). 1H NMR (300 MHz, CDCl3) δ 11.91 (s, 1H), 7.56 (t, J = 8.0 Hz, 1H), 6.92 (dd, J = 13.0, 7.9 Hz, 2H), 3.49 (d, J = 9.0 Hz, 1H), 2.64 (dd, J = 9.1, 3.9 Hz, 1H), 2.38–2.24 (m, 1H), 1.92–1.52 (m, 3H), 1.33 (s, 3H), 1.22 (dt, J = 13.4, 6.5 Hz, 1H), 0.56 (d, J = 6.5 Hz, 3H), 0.42 (d, J = 6.5 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 199.69, 185.32, 164.45, 144.48, 138.52, 122.15, 118.58, 116.01, 77.45, 77.02, 76.60, 56.98, 56.58, 51.48, 33.82, 28.21, 26.98, 24.31, 23.15, 19.87. HRMS Calcd for C17H20O3 ([M + Na]+): 295.1305; Found: 295.1303.
(3aR,3bR,6R,6aR,10bS)-6-isopropyl-2,2,3b-trimethyl-3a,3b,5,6,6a,10b-hexahydro-4H-cyclopenta [3,4]naphtho[1,2-d][1,3]dioxol-10-ol (9e). To a solution of compound 9j (31 mg, 0.1 mmol) in CH2Cl2 (1 mL), Pd(OH)2/C (25 mg, 10%) was added in a high-pressure autoclave. The reaction mixture was hydrogenated for 24 h under 30 atm H2 at room temperature. Pd(OH)2/C was removed by filtration and the filtration was concentrated under reduced pressure without further purification to provide 9e (31 mg, quant) as a white solid (Rf = 0.7, petroleum ether/ethyl acetate = 4:1). 1H NMR (500 MHz, CDCl3) δ 7.42 (s, 1H), 7.08 (t, J = 7.9 Hz, 1H), 6.80 (dd, J = 7.8, 1.0 Hz, 1H), 6.72 (dt, J = 8.0, 0.9 Hz, 1H), 5.18 (d, J = 4.7 Hz, 1H), 4.14 (d, J = 4.8 Hz, 1H), 3.09 (d, J = 8.3 Hz, 1H), 2.24–2.14 (m, 2H), 2.05–1.98 (m, 1H), 1.94–1.87 (m, 1H), 1.72–1.64 (m, 1H), 1.49–1.43 (m, 4H), 1.25 (s, 3H), 1.12 (s, 3H), 0.66 (d, J = 6.7 Hz, 3H), 0.51 (d, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 156.77, 137.22, 128.07, 123.15, 120.23, 113.39, 109.96, 79.80, 73.47, 51.96, 51.72, 41.45, 35.52, 28.90, 28.01, 27.37, 26.91, 26.44, 24.21, 21.38. HRMS Calcd for C20H28O3 ([M + Na]+): 339.1931; Found: 339.1927.
(1R,3aR,4R,9bR)-
4,6-
dihydroxy-
1-
isopropyl-
3a-
methyl-
1,2,3,3a,4,9b-
hexahydro-
5H-
cyclopenta[a]naphthalen-
5-
one (
9f). To a solution of compound
9e (32 mg, 0.1 mmol) in CH
2Cl
2 (1 mL), trifluoroacetic acid (35 μL, 0.4 mmol) was added at 0 °C. Then the reaction mixture was stirred at that temperature for 2 h and concentrated in vacuum. The crude reaction mixture was purified by silica-gel CC to provide
9f (23 mg, 84% yield) as a white solid (
Rf = 0.6, petroleum ether/ethyl acetate = 5:1) [
37].
1H NMR (300 MHz, CDCl
3) δ 7.09 (t,
J = 7.8 Hz, 1H), 6.84 (d,
J = 7.7 Hz, 1H), 6.69 (d,
J = 7.9 Hz, 1H), 5.55 (s, 1H), 3.74 (d,
J = 21.4 Hz, 1H), 3.47–3.32 (m, 2H), 2.58–2.51 (m, 1H), 2.31–2.21 (m, 1H), 1.84–1.68 (m, 1H), 1.61–1.46 (m, 1H), 1.45–1.24 (m, 2H), 1.21 (s, 3H), 0.54 (dd,
J = 17.7, 6.6 Hz, 6H).
13C NMR (75 MHz, CDCl
3) δ 214.89, 152.69, 137.31, 126.78, 123.18, 120.46, 112.49, 77.45, 77.03, 76.61, 57.10, 54.33, 50.74, 36.64, 34.86, 28.23, 27.14, 24.83, 23.30, 19.58. HRMS Calcd for C
20H
28O
3 ([M − H]
−): 273.1485; Found: 273.1478.
(3bR,6R,6aR)-
6-
isopropyl-
2,2,3b-
trimethyl-
2,3b,4,5,6,6a-
hexahydrocyclopenta[3,4]naphtho[1,2-
d]imidazol-
10-
ol (
9g). To a stirred solution of compound
9d (27 mg, 0.1 mmol) and ammonium acetate (54 mg, 0.7 mmol) in AcOH (0.5 mL), acetone (8 μL, 0.11 mmol) was added. Then the reaction mixture was heated to 120 °C for 7 h. To the mixture was added ethyl acetate (5 mL) and washed with brine (10 mL) and sodium bicarbonate solution (10 mL), dried over Na
2SO
4 and concentrated under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide
9g (22 mg, 71% yield) as a yellow solid (
Rf = 0.5, petroleum ether/ethyl acetate = 4:1) [
38].
1H NMR (300 MHz, CDCl
3) δ 10.88 (s, 1H), 7.35 (t,
J = 7.9 Hz, 1H), 6.90 (dd,
J = 17.9, 7.9 Hz, 2H), 3.32 (d,
J = 9.7 Hz, 1H), 2.61–2.49 (m, 1H), 2.32–2.19 (m, 1H), 1.84 (q,
J = 7.2 Hz, 2H), 1.55 (m, 7H), 1.29 (s, 3H), 1.06 (q,
J = 6.7 Hz, 1H), 0.42 (dd,
J = 8.4, 6.6 Hz, 6H).
13C NMR (75 MHz, CDCl
3) δ 172.37, 160.54, 159.18, 141.56, 132.99, 122.18, 114.19, 113.24, 104.61, 77.44, 77.02, 76.60, 55.68, 51.96, 45.23, 37.53, 28.02, 27.82, 24.39, 24.02, 23.48, 19.66. HRMS Calcd for C
20H
26O
3 ([M + H]
+): 311.2118; Found: 311.2115.
(3aR,4R,5S)-1-isopropyl-3a-methyl-3,3a,4,5-tetrahydro-2H-cyclopenta[a]naphthalene-4,5,6-triyl triacetate (9h). To a solution of 9a (0.1 g, 0.37 mmol) in pyridine (5 mL), acetic anhydride (0.5 mL, 5.5 mmol) was added at room temperature. The reaction mixture was heated at 100 °C for 3 h. The reaction mixture was cooled to room temperature. To the mixture, 2N HCl (5 mL) was added and stirred for 10 min. The mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic layer was washed successively with H2O (2 × 10 mL) and brine (10 mL), dried over Na2SO4 and concentrated under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide 9h (0.13 g, 86% yield) as a white solid (Rf = 0.5, petroleum ether/ethyl acetate = 2:1). 1H NMR (300 MHz, CDCl3) δ 7.37 (t, J = 7.9 Hz, 1H), 7.31–7.26 (m, 1H), 6.96 (dd, J = 8.0, 1.3 Hz, 1H), 5.18 (d, J = 4.6 Hz, 1H), 3.20 (p, J = 6.8 Hz, 1H), 2.62–2.45 (m, 2H), 2.26 (s, 3H), 2.02 (d, J = 11.4 Hz, 6H), 1.97–1.88 (m, 1H), 1.68–1.60 (m, 1H), 1.20 (d, J = 6.9 Hz, 3H), 1.09 (s, 3H), 1.02 (d, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 170.93, 170.03, 169.26, 149.71, 146.22, 135.98, 131.65, 129.23, 125.08, 124.35, 121.15, 77.47, 77.04, 76.62, 75.32, 66.63, 52.04, 32.92, 29.68, 27.29, 23.46, 21.43, 21.25, 20.98, 20.91, 20.71. HRMS Calcd for C23H28O6 ([M + Na]+): 423.1778; Found: 423.1771.
(3aR,3bR,10bS)-
10-
hydroxy-
6-
isopropyl-
3b-
methyl-
3a,3b,5,10b-
tetrahydro-
4H-
cyclopenta[3,4]naphtho[1,2-
d][1,3]dioxol-
2-
one (
9i). To a stirred solution of compound
9a (27 mg, 0.1 mmol) and pyridine (80 μL, 1 mmol) in CH
2Cl
2 (1.5 mL), a solution of triphosgene (45 mg, 0.15 mmol) in CH
2Cl
2 (0.5 mL) was added dropwise at −78 °C. Then the reaction mixture was allowed to warm to room temperature slowly. The reaction mixture was quenched using saturated aqueous NH
4Cl followed by extraction of the aqueous phase with CH
2Cl
2. The organic extracts were washed with 1 M HCl, saturated NaHCO
3, saturated NaCl, H
2O, dried with MgSO
4, and filtered. The crude reaction mixture was purified by silica-gel CC to provide
9i (25 mg, 82% yield) as a white solid (
Rf = 0.2, petroleum ether/ethyl acetate = 2:1) [
39].
1H NMR (300 MHz, CDCl
3) δ 7.24 (t,
J = 8.0 Hz, 1H), 6.91 (d,
J = 7.7 Hz, 1H), 6.73 (d,
J = 8.1 Hz, 1H), 6.06 (d,
J = 7.7 Hz, 1H), 5.63 (s, 1H), 4.87 (d,
J = 7.7 Hz, 1H), 3.21–3.00 (m, 1H), 2.58 (td,
J = 9.9, 5.7 Hz, 2H), 2.39–2.24 (m, 1H), 1.79–1.69 (m, 1H), 1.19 (d,
J = 6.8 Hz, 3H), 1.00 (d,
J = 7.5 Hz, 6H).
13C NMR (75 MHz, CDCl
3) δ 155.27, 148.98, 135.51, 130.76, 128.47, 119.97, 116.86, 114.50, 83.47, 77.45, 77.02, 76.60, 72.74, 51.98, 31.59, 29.96, 27.45, 23.09, 21.41, 21.34. HRMS Calcd for C
18H
20O
4 ([M + Na]
+): 323.1254; Found: 323.1250.
(3aR,3bR,10bS)-
6-
isopropyl-
2,2,3b-
trimethyl-
3a,3b,5,10b-
tetrahydro-
4H-
cyclopenta[3,4]naphtho[1,2-
d][1,3]dioxol-
10-
ol (
9j). To a stirred solution of compound
9a (55 mg, 0.2 mmol) and pyridinium p-toluenesulfonate acid (5 mg, 2% mmol) in CH
2Cl
2 (2 mL), 2,2-dimethoxypropane (52 mg, 0.5 mmol) was added at room temperature. Then the reaction mixture was stirred for 6 h at 40 °C. The solvent was removed under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide
9j (51 mg, 82% yield) as a white solid (
Rf = 0.5, petroleum ether/ethyl acetate = 4:1) [
40].
1H NMR (300 MHz, CDCl
3) δ 7.19 (t,
J = 7.9 Hz, 1H), 6.98 (d,
J = 7.7 Hz, 1H), 6.81 (d,
J = 8.0 Hz, 1H), 6.54 (s, 1H), 5.29 (d,
J = 5.1 Hz, 1H), 4.25 (d,
J = 5.1 Hz, 1H), 3.27–3.13 (m, 1H), 2.54 (t,
J = 7.6 Hz, 2H), 2.31–2.17 (m, 1H), 1.69–1.56 (m, 1H), 1.44 (s, 3H), 1.19 (d,
J = 6.8 Hz, 3H), 1.13 (s, 3H), 1.05–0.96 (m, 6H).
13C NMR (75 MHz, CDCl
3) δ 156.16, 145.93, 133.34, 130.50, 128.71, 120.96, 119.54, 114.59, 110.18, 82.36, 73.51, 52.16, 31.80, 29.58, 27.83, 27.37, 26.98, 23.53, 21.62, 21.30. HRMS Calcd for C
20H
26O
3 ([M + Na]
+): 337.1774; Found: 337.1771.
(3aR,4R,5S)-
4,6-
bis((tert-
butyldimethylsilyl)oxy)-
1-
isopropyl-
3a-
methyl-
3,3a,4,5-
tetrahydro-
2H-
cyclopenta[a]naphthalen-
5-
ol (
9k). To a stirred solution of compound
9a (27 mg, 0.1 mmol) and 2,6-lutidine (70 μL, 0.7 mmol) in CH
2Cl
2 (1 mL), TBSOtf (114 μL, 0.5 mmol) was added at −40 °C. Then the reaction mixture was stirred for 5 h at −40°C. The mixture was dilute with CH
2Cl
2 (10 mL) and washed with 1N HCl aqueous (4 mL) and brine (10 mL), dried over Na
2SO
4 and concentrated under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide
9k (33 mg, 66% yield) as a white solid (
Rf = 0.5, petroleum ether/ethyl acetate = 40:1) [
41].
1H NMR (300 MHz, CDCl
3) δ 7.18 (t,
J = 7.9 Hz, 1H), 7.08 (d,
J = 7.8 Hz, 1H), 6.76 (d,
J = 7.9 Hz, 1H), 4.72 (d,
J = 2.0 Hz, 1H), 3.87 (s, 1H), 3.34 (d,
J = 2.0 Hz, 1H), 3.22 (q,
J = 6.8 Hz, 1H), 2.48 (dd,
J = 8.8, 5.9 Hz, 2H), 2.15 (dt,
J = 12.9, 7.7 Hz, 1H), 1.59–1.45 (m, 1H), 1.21 (d,
J = 6.9 Hz, 3H), 1.08 (d,
J = 7.8 Hz, 12H), 1.00 (d,
J = 6.7 Hz, 3H), 0.84 (s, 9H), 0.39 (s, 3H), 0.25 (s, 3H), 0.15 (d,
J = 2.8 Hz, 6H).
13C NMR (75 MHz, CDCl
3) δ 154.47, 145.48, 134.50, 131.88, 127.52, 120.66, 116.07, 78.18, 71.17, 51.05, 32.14, 29.99, 27.38, 25.80, 25.72, 23.79, 21.66, 21.51, 18.02, 17.92, −3.35, −4.47, −4.74, −4.81. HRMS Calcd for C
29H
50O
3Si
2 ([M + Na]
+): 525.3191; Found: 525.3185.
(3aR,3bR,10bS)-6-isopropyl-2,2,3b-trimethyl-3a,3b,5,10b-tetrahydro-4H-cyclopenta[3,4]naphtho[1,2-d][1,3]dioxol-10-yl (tert-butoxycarbonyl)alaninate (9l). To a stirred solution of compound 9j (31 mg, 0.1 mmol) in CHCl3/THF (0.5/1.5 mL), Boc-l-Ala (68 mg, 0.36 mmol), DMAP (4 mg, 0.03 mmol), and EDCI (63 mg, 0.33 mmol) were added. The reaction was stirred for 18 h at room temperature. The solvent was removed in vacuum, and the crude reaction mixture was purified by silica-gel CC to provide 9l (39 mg, 82% yield) as a white solid (Rf = 0.7, petroleum ether/ethyl acetate = 1:1). 1H NMR (300 MHz, CDCl3) δ 7.33 (t, J = 7.8 Hz, 1H), 7.23 (d, J = 7.7 Hz, 1H), 6.98 (d, J = 7.9 Hz, 1H), 5.36 (d, J = 8.1 Hz, 1H), 5.20 (d, J = 5.8 Hz, 1H), 4.73–4.61 (m, 1H), 4.24 (d, J = 5.9 Hz, 1H), 3.14 (p, J = 6.9 Hz, 1H), 2.53 (dt, J = 9.4, 5.1 Hz, 2H), 2.31–2.22 (m, 1H), 1.61–1.56 (m, 4H), 1.47 (d, J = 6.5 Hz, 9H), 1.36 (s, 3H), 1.20 (d, J = 6.9 Hz, 3H), 1.05 (s, 3H), 1.01–0.93 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 155.29, 150.21, 146.67, 135.17, 130.46, 128.50, 127.07, 125.04, 120.85, 109.12, 82.52, 79.73, 72.03, 52.64, 49.56, 31.93, 29.99, 28.38, 27.42, 27.39, 26.39, 23.81, 21.48, 18.42. HRMS Calcd for C28H39NO6 ([M + Na]+): 508.2670; Found: 508.2661.
(3aR,4R,5S)-1-isopropyl-3a-methyl-3,3a,4,5-tetrahydro-2H-cyclopenta[a]naphthalene-4,5,6-triyl tris(2-((tert-butoxycarbonyl)amino)propanoate) (
9m). To a stirred solution of compound
9a (27 mg, 0.1 mmol) in CHCl
3/THF (0.5/1.5 mL), Boc-l-Ala (68 mg, 0.36 mmol), DMAP (4 mg, 0.03 mmol), and EDCI (63 mg, 0.33 mmol) were added. The reaction was stirred for 18 h at room temperature. The solvent was removed in vacuum, and the crude reaction mixture was purified by silica-gel CC to provide
9m (25 mg, 83% yield) as a white solid (R
f = 0.7, petroleum ether/ethyl acetate = 1:1) [
42].
1H NMR (300 MHz, CDCl
3) δ 7.40 (t, J = 7.9 Hz, 1H), 7.06 (d, J = 8.1 Hz, 1H), 6.29 (d, J = 4.0 Hz, 1H), 5.68 (s, 1H), 5.17 (s, 2H), 4.45–4.18 (m, 3H), 3.23–3.07 (m, 1H), 2.50 (dt, J = 9.6, 5.2 Hz, 2H), 2.00 (d, J = 11.0 Hz, 1H), 1.64 (s, 3H), 1.55 (d, J = 7.2 Hz, 3H), 1.51–1.37 (m, 30H), 1.33 (d, J = 7.3 Hz, 3H), 1.18 (d, J = 6.8 Hz, 3H), 1.10–0.99 (m, 6H).
13C NMR (75 MHz, CDCl
3) δ 172.83, 172.30, 171.83, 155.25, 154.90, 149.44, 145.53, 133.32, 130.01, 125.08, 124.17, 120.95, 79.73, 77.50, 77.28, 77.08, 76.65, 67.05, 50.23, 49.18, 33.41, 28.75, 28.35, 28.30, 26.88, 24.63, 21.26, 18.88, 18.14, 17.52. HRMS Calcd for C
41H
61N
3O
12 ([M + Na]
+): 810.4147; Found: 810.4133.
(3aR,4R,5S)-
1-
isopropyl-
3a-
methyl-
3,3a,4,5-
tetrahydro-
2H-
cyclopenta[a]naphthalene-
4,5,6-
triyltris(4-
nitrobenzoate) (
9n). To a stirred solution of compound
9a (55 mg, 0.2 mmol) and DMAP (54 mg, 0.8 mmol) in CH
2Cl
2 (1.5 mL), a solution of 4-nitrobenzoyl chloride (100 mg, 0.11 mmol) in CH
2Cl
2 (0.5 mL) was added. Then the reaction mixture was stirred for 3 h at room temperature. The mixture was dilute with CH
2Cl
2 (10 mL) and washed with brine (10 mL), dried over Na
2SO
4 and concentrated under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide
9n (104 mg, 72% yield) as a white solid (
Rf = 0.5, petroleum ether/ethyl acetate = 2:1) [
43].
1H NMR (300 MHz, CDCl
3) δ 8.24–7.94 (m, 10H), 7.73–7.65 (m, 2H), 7.55 (d,
J = 3.9 Hz, 2H), 7.15 (dd,
J = 5.5, 3.8 Hz, 1H), 6.83 (d,
J = 4.4 Hz, 1H), 5.60 (d,
J = 4.4 Hz, 1H), 3.41 (p,
J = 6.7 Hz, 1H), 2.74–2.53 (m, 2H), 2.06 (dt,
J = 13.5, 9.0 Hz, 1H), 1.80 (dd,
J = 12.7, 8.7, 3.4 Hz, 1H), 1.35–1.25 (m, 6H), 1.19 (d,
J = 6.6 Hz, 3H).
13C NMR (75 MHz, CDCl
3) δ 164.26, 163.32, 162.95, 150.75, 150.69, 150.42, 149.55, 147.01, 135.98, 134.85, 134.39, 133.89, 131.29, 131.12, 130.69, 130.35, 130.11, 125.92, 123.60, 123.39, 123.27, 123.18, 121.57, 77.48, 77.05, 76.63, 76.49, 68.51, 52.21, 32.70, 29.74, 27.39, 23.71, 21.54, 21.49. HRMS Calcd for C
38H
31N
3O
12 ([M + Na]
+): 744.1800; Found: 744.1782.
(3S,3aR)-3-((tert-butyldimethylsilyl)oxy)-1-isopropyl-6-methoxy-3a-methyl-2,3,3a,4-tetrahydro-5H-cyclopenta[a]naphthalen-5-one (9o). The compound 16 (4.02 g, 10 mmol) in dichloromethane (10 mL) was added to a stirred solution of PCC (4.7 g, 22 mmol), NaOAc (1.5 g, 11 mmol) and celite (4 g) in dichloromethane (40 mL) at 0 °C. Then the solution was stirred at room temperature for 2 h. The reaction mixture was filtered through celite and washed repeatedly with Et2O. The solvent was removed under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide the ketone 9o (2.9 g, 73% yield) as a colorless liquid (Rf = 0.3, petroleum ether/ethyl acetate = 5:1). 1H NMR (300 MHz, CDCl3) δ 7.43 (t, J = 8.1 Hz, 1H), 7.03 (d, J = 7.7 Hz, 1H), 6.87 (d, J = 8.4 Hz, 1H), 4.01 (dd, J = 6.5, 3.2 Hz, 1H), 3.91 (s, 3H), 3.25–3.00 (m, 2H), 2.85 (dd, J = 17.1, 6.4 Hz, 1H), 2.47–2.27 (m, 2H), 1.17 (d, J = 6.9 Hz, 3H), 1.07–0.98 (m, 6H), 0.89 (s, 9H), 0.07 (d, J = 2.0 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 199.08, 160.23, 143.06, 139.83, 133.73, 132.77, 120.49, 119.85, 110.52, 77.59, 77.51, 77.09, 76.66, 56.01, 53.96, 49.01, 40.07, 27.19, 25.82, 23.40, 21.22, 21.17, 18.09, −4.60, −4.89. HRMS Calcd for C24H36O3Si ([M + Na]+): 423.2326; Found: 423.2319.
(3S,3aR,4S,5R)-1-isopropyl-6-methoxy-3a-methyl-3,3a,4,5-tetrahydro-2H-cyclopenta[a]naphthalene-3,4,5-triol (9p). To a solution of 4-methylmorpholine N-oxide (68 mg, 0.5 mmol) and compound 17 (27 mg, 0.1 mmol) in THF/H2O (0.8 mL/0.2 mL), osmium tetroxide (0.039 M in H2O, 0.25 mL, 0.01 mmol) was added dropwise at room temperature. After stirring for 36 h, the reaction mixture was quenched with saturated solution of sodium sulfite (10 mL) and stirred for 30 min. The mixture was extracted with ethyl acetate (3 × 50 mL). The combined organic layer was washed successively with H2O (2 × 50 mL) and brine (10 mL), dried over Na2SO4, and concentrated under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide 9p (18 mg, 60% yield) as a dark green solid (Rf = 0.3, petroleum ether/ethyl acetate = 1:1). 1H NMR (500 MHz, CDCl3) δ 7.25 (td, J = 7.9, 2.3 Hz, 1H), 6.97 (dd, J = 7.7, 2.3 Hz, 1H), 6.79 (dd, J = 8.4, 2.3 Hz, 1H), 5.16 (t, J = 3.0 Hz, 1H), 4.07 (dd, J = 5.5, 2.8 Hz, 1H), 3.93 (t, J = 3.0 Hz, 1H), 3.85 (d, J = 2.3 Hz, 3H), 3.12 (pd, J = 6.9, 2.2 Hz, 1H), 2.93–2.88 (m, 1H), 2.36 (dt, J = 17.3, 2.3 Hz, 1H), 1.13 (dd, J = 6.9, 2.4 Hz, 3H), 1.01–0.93 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 157.44, 142.94, 134.76, 132.16, 128.70, 124.53, 120.24, 109.00, 78.64, 77.51, 77.08, 76.66, 76.20, 66.54, 55.53, 54.29, 40.59, 26.84, 23.14, 21.55, 21.14. HMRS Calcd for C18H24O4 ([M + Na]+): 327.1567; Found: 327.1563.
(3S,3aR)-
3-
((tert-
butyldimethylsilyl)oxy)-
6-
hydroxy-
1-
isopropyl-
3a-
methyl-
2,3,3a,4-
tetrahydro-
5H-
cyclopenta[a]naphthalen-
5-
one (
9q). To a solution of compound
9o (40 mg, 0.1 mmol) in THF (1.5 mL), BBr
3 (48 μL, 0.5 mmol) was added dropwise at −78 °C and stirred for 2 h. The reaction mixture was heated to room temperature slowly, quenched with ice water (5 mL), and then extracted with ethyl acetate (3 × 5 mL). The combined organic layer was washed successively with brine (10 mL), dried over Na
2SO
4 and concentrated under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide
9q (29 mg, 76% yield) as a white solid (
Rf = 0.2, petroleum ether/ethyl acetate = 2:1) [
44].
1H NMR (300 MHz, CDCl
3) δ 12.76 (s, 1H), 7.45 (t,
J = 8.0 Hz, 1H), 7.00 (d,
J = 7.7 Hz, 1H), 6.86 (dd,
J = 8.4, 1.0 Hz, 1H), 4.08 (dd,
J = 6.3, 2.1 Hz, 1H), 3.33–3.21 (m, 2H), 3.02 (dd,
J = 17.8, 6.2 Hz, 1H), 2.58–2.45 (m, 2H), 1.20 (d,
J = 7.0 Hz, 3H), 1.13–1.06 (m, 6H), 0.89 (s, 9H), 0.07 (d,
J = 2.0 Hz, 6H).
13C NMR (75 MHz, CDCl
3) δ 205.99, 162.90, 144.34, 137.68, 136.29, 131.04, 118.33, 116.48, 114.92, 77.45, 77.03, 76.66, 76.61, 53.92, 45.88, 40.19, 27.32, 23.41, 21.39, 20.91. HRMS Calcd for C
23H
34O
3Si ([M + Na]
+): 409.2169; Found: 409.1616.
(3S,3aR)-3-hydroxy-1-isopropyl-6-methoxy-3a-methyl-2,3,3a,4-tetrahydro-5H-cyclopenta[a]naphthalen-5-one (9r). To a solution of compound 9o (40 mg, 0.1 mmol) in THF (1 mL), TBAF (1M in THF, 0.15 mmol) was added dropwise at 0 °C and stirred for 1 h. The reaction mixture was cooled to room temperature, quenched with saturated solution of ammonium chloride (5 mL), and then extracted with ethyl acetate (3 × 5 mL). The combined organic layer was washed successively with H2O (10 mL) and brine (10 mL), dried over Na2SO4, and concentrated under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide 9r (22 mg, 76% yield) as a white solid (Rf = 0.1, petroleum ether/ethyl acetate = 2:1). 1H NMR (300 MHz, CDCl3) δ 7.45 (t, J = 8.1 Hz, 1H), 7.06 (d, J = 7.8 Hz, 1H), 6.89 (d, J = 8.4 Hz, 1H), 4.08 (dd, J = 6.6, 2.8 Hz, 1H), 3.91 (s, 3H), 3.24–2.91 (m, 3H), 2.52–2.41 (m, 2H), 2.16 (s, 1H), 1.18 (d, J = 6.9 Hz, 3H), 1.10–1.01 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 198.54, 160.21, 143.18, 139.50, 133.92, 132.53, 120.38, 119.88, 110.67, 77.49, 77.12, 77.07, 76.65, 56.00, 53.58, 48.11, 39.78, 27.22, 23.52, 21.32, 21.10. HRMS Calcd for C18H22O3 ([M + Na]+): 309.1461; Found: 309.1458.
(3S,3aR)-3,6-dihydroxy-1-isopropyl-3a-methyl-2,3,3a,4-tetrahydro-5H-cyclopenta[a]naphthalen-5-one (9s). To a solution of compound 9r (43 mg, 0.15 mmol) in THF (1.5 mL), BBr3 (58 μL, 0.6 mmol) was added dropwise at −78 °C and stirred for 2 h. The reaction mixture was heated to room temperature slowly, quenched with ice water (5 mL), and then extracted with ethyl acetate (3 × 5 mL). The combined organic layer was washed successively with brine (10 mL), dried over Na2SO4, and concentrated under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide 9s (31 mg, 76% yield) as a white solid (Rf = 0.6, petroleum ether/ethyl acetate = 1:1). 1H NMR (300 MHz, CDCl3) δ 12.75 (s, 1H), 7.43 (t, J = 8.0 Hz, 1H), 6.98 (d, J = 7.5 Hz, 1H), 6.84 (dd, J = 8.4, 1.0 Hz, 1H), 4.05 (s, 1H), 3.30–3.20 (m, 2H), 3.05–2.96 (m, 1H), 2.56–2.45 (m, 2H), 1.19 (d, J = 6.9 Hz, 3H), 1.09 (dd, J = 3.7, 2.9 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 205.98, 162.90, 144.34, 137.68, 136.30, 131.04, 118.33, 116.49, 114.92, 77.46, 77.03, 76.66, 76.61, 53.92, 45.88, 40.19, 27.32, 23.41, 21.39, 20.91. HRMS Calcd for C17H20O3 ([M + Na]+): 273.1485; Found: 273.1486.
(S)-1-isopropyl-6-methoxy-3a-methyl-3a,4-dihydro-5H-cyclopenta[a]naphthalen-5-one (9t). To a stirred solution of compound 9r (31 mg, 0.11 mmol) in THF (1 mL) containing triphenylphophine (58 mg, 0.22 mmol) and p-nitrobenzoic acid (37mg, 0.22 mmol), DEAD (3 μL, 0.22 mmol) was added dropwise over 10 min period at 0 °C under argon, and the reaction mixture was stirred at 50 °C for 12 h. The solvent was removed under reduced pressure to leave an oil, which was purified by column chromatography to provide 9t (21 mg, 72% yield) as a white solid (Rf = 0.4, petroleum ether/ethyl acetate = 2:1). 1H NMR (300 MHz, CDCl3) δ 7.52 (t, J = 8.1 Hz, 1H), 7.06 (dd, J = 7.8, 1.0 Hz, 1H), 6.89 (dd, J = 8.5, 1.0 Hz, 1H), 6.60 (d, J = 5.3 Hz, 1H), 6.53 (d, J = 5.3 Hz, 1H), 3.96 (s, 3H), 3.24 (p, J = 6.8 Hz, 1H), 2.92 (d, J = 15.3 Hz, 1H), 2.15 (d, J = 15.2 Hz, 1H), 1.33 (d, J = 6.9 Hz, 3H), 1.17–1.08 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 197.41, 160.69, 147.11, 146.54, 139.83, 139.39, 134.27, 130.63, 119.20, 118.86, 109.95, 77.47, 77.04, 76.62, 56.52, 56.01, 50.95, 26.28, 22.54, 22.09, 19.09. HRMS Calcd for C18H20O2 ([M + H]+): 269.1536; Found: 269.1539.
(3S,3aR,4S,5R)-1-isopropyl-6-methoxy-3a-methyl-3,3a,4,5-tetrahydro-2H-cyclopenta[a]naphthalene-3,4,5-triyl triacetate (9u). To a solution of 9p (30 g, 0.1 mmol) in pyridine (1.5 mL), acetic anhydride (0.13 mL, 1.5 mmol) was added at room temperature. The reaction mixture was heated at 100 °C for 3 h. The reaction mixture was cooled to room temperature. To the mixture 2N HCl (3 mL) was added and stirred for 10 min. The mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic layer was washed successively with H2O (5 × 10 mL) and brine (50 mL), dried over Na2SO4, and concentrated under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide 9u (37 mg, 86% yield) as a white solid (Rf = 0.6, petroleum ether/ethyl acetate = 1:1). 1H NMR (300 MHz, CDCl3) δ 7.31 (t, J = 8.1 Hz, 1H), 6.94 (d, J = 7.7 Hz, 1H), 6.80 (d, J = 8.3 Hz, 1H), 6.40 (d, J = 5.2 Hz, 1H), 5.44 (d, J = 5.1 Hz, 1H), 5.14 (dd, J = 9.0, 6.3 Hz, 1H), 3.78 (s, 3H), 3.24 (p, J = 6.9 Hz, 1H), 3.06 (dd, J = 17.3, 9.0 Hz, 1H), 2.39 (dd, J = 17.3, 6.3 Hz, 1H), 2.05–1.97 (m, 9H), 1.22–1.14 (m, 6H), 1.02 (d, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 171.13, 170.25, 169.81, 158.63, 142.83, 134.85, 130.28, 129.30, 119.72, 119.47, 109.52, 79.33, 77.49, 77.06, 76.64, 71.34, 66.77, 55.48, 55.41, 38.51, 27.22, 22.13, 21.43, 21.24, 21.09, 20.88, 20.57. HRMS Calcd for C24H30O7 ([M + Na]+): 453.1884; Found: 453.1875.
(3S,3aR,4S,5R)-
5,6-
dihydroxy-
1-
isopropyl-
3a-
methyl-
3,3a,4,5-
tetrahydro-
2H-
cyclopenta[a]naphthalene-
3,4-
diyl diacetate (
9v). To a solution of compound
9u (43 mg, 0.1 mmol) in THF (1.5 mL), BBr
3 (48 μL, 0.5 mmol) was added dropwise at −78 °C and stirred for 2 h. The reaction mixture was warm to room temperature slowly, quenched with ice water (5 mL), and then extracted with ethyl acetate (3 × 5 mL). The combined organic layer was washed successively with brine (10 mL), dried over Na
2SO
4, and concentrated under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide
9v (28 mg, 75% yield) as a white solid (
Rf = 0.2, petroleum ether/ethyl acetate = 2:1) [
42].
1H NMR (300 MHz, CDCl
3) δ 7.22 (t,
J = 7.9 Hz, 1H), 6.92–6.76 (m, 2H), 5.40–5.24 (m, 2H), 5.13 (dd,
J = 9.0, 6.0 Hz, 1H), 3.24–2.98 (m, 2H), 2.40 (dd,
J = 17.4, 6.0 Hz, 1H), 2.06 (d,
J = 2.8 Hz, 6H), 1.23–1.15 (m, 6H), 1.00 (d,
J = 6.8 Hz, 3H).
13C NMR (75 MHz, CDCl
3) δ 171.99, 171.23, 157.12, 142.39, 133.15, 130.22, 129.24, 119.31, 118.35, 115.59, 79.70, 77.45, 77.03, 76.60, 73.69, 69.27, 55.49, 38.59, 27.11, 22.89, 21.45, 21.31, 21.21, 20.93. HRMS Calcd for C
21H
26O
6 ([M + Na]
+): 397.1622; Found: 397.1617.
(3aS,3bR,4S,10bR)-6-isopropyl-10-methoxy-2,2,3b-trimethyl-3a,3b,5,10b-tetrahydro-4H-cyclopenta[3,4]naphtho[1,2-d][1,3]dioxol-4-ol (9w). To a stirred solution of compound 9p (46 mg, 0.15 mmol) and Pyridinium p-Toluenesulfonate acid (5 mg, 0.02 mmol) in CH2Cl2 (2 mL), 2,2-dimethoxypropane (40 mg, 0.38 mmol) was added at room temperature. Then the reaction mixture was stirred for 6 h at 40 °C. The solvent was removed under reduced pressure. The crude reaction mixture was purified by silica-gel CC to provide 9w (51 mg, 67% yield) as a white solid (Rf = 0.3, petroleum ether/ethyl acetate = 3:1). 1H NMR (300 MHz, CDCl3) δ 7.27 (t, J = 8.0 Hz, 1H), 6.82 (t, J = 7.9 Hz, 2H), 5.59 (d, J = 6.8 Hz, 1H), 4.51 (d, J = 6.8 Hz, 1H), 4.24–4.14 (m, 1H), 3.88 (d, J = 1.4 Hz, 3H), 3.13–2.90 (m, 3H), 2.35 (dd, J = 16.9, 6.5 Hz, 1H), 1.44 (s, 3H), 1.24 (s, 3H), 1.16 (d, J = 6.9 Hz, 3H), 0.96 (d, J = 6.8 Hz, 3H), 0.91 (d, J = 1.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 158.42, 143.06, 135.70, 130.93, 128.97, 122.69, 119.73, 109.23, 109.01, 80.42, 79.79, 77.50, 77.08, 76.65, 71.51, 55.88, 55.22, 41.71, 27.08, 27.00, 25.50, 22.88, 21.84, 21.15. HRMS Calcd for C21H28O4 ([M + Na]+): 367.1880; Found: 367.1879.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










