Chemistry of Renieramycins. Part 19: Semi-Syntheses of 22-O-Amino Ester and Hydroquinone 5-O-Amino Ester Derivatives of Renieramycin M and Their Cytotoxicity against Non-Small-Cell Lung Cancer Cell Lines

 ,
,  ,
,
Abstract
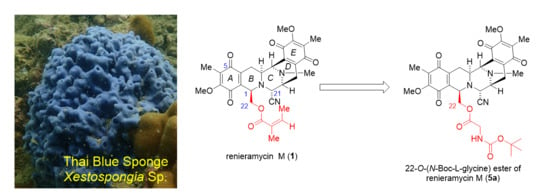
:
1. Introduction
2. Results and Discussion
2.1. Extraction and Isolation of 1 from the Thai Blue Sponge Xestospongia sp.
2.2. Syntheses of 22-O-amino Ester and Hydroquinone 5-O-amino Ester Derivatives of 1
2.3. Cytotoxicity of the Semi-Synthetic Amino Ester Derivatives against Non-Small Cell Lung Cancer Cell Lines
3. Materials and Methods
3.1. General for Synthetic Procedure
3.2. Isolation of Renieramycin M (1)
3.2.1. Sample Collection
3.2.2. Extraction and Isolation
3.3. Semi-Synthesis Procedures
3.3.1. Preparation of Jorunnamycin A (7)
3.3.2. General Procedure for Preparation of 22-O-amino Ester Derivatives of Renieramycin M (5a–5e)
3.3.3. General Procedure for Preparation of Hydroquinone 5-O-amino Ester Derivatives of Renieramycin M (6a–6e)
3.3.4. Cytotoxic Evaluations against Non-Small-Cell Lung Cancer Cell Lines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hu, Y.; Chen, J.; Hu, G.; Yu, J.; Zhu, X.; Lin, Y.; Chen, S.; Yuan, J. Statistical research on the bioactivity of new marine natural products discovered during the 28 years from 1985 to 2012. Mar. Drugs 2015, 13, 202–221. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.M.; Glaser, K.B.; Cuevas, C.; Jacobs, R.S.; Kem, W.; Little, R.D.; McIntosh, J.M.; Newman, D.J.; Potts, B.C.; Shuster, D.E. The odyssey of marine pharmaceuticals: A current pipeline perspective. Trends Pharmacol. Sci. 2010, 31, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-K. Handbook of Anticancer Drugs from Marine Origin; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Newman, D.; Cragg, G. Current status of marine-derived compounds as warheads in anti-tumor drug candidates. Mar. Drugs 2017, 15, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalifa, S.A.M.; Elias, N.; Farag, M.A.; Chen, L.; Saeed, A.; Hegazy, M.-E.F.; Moustafa, M.S.; Abd El-Wahed, A.; Al-Mousawi, S.M.; Musharraf, S.G.; et al. Marine natural products: A source of novel anticancer drugs. Mar. Drugs 2019, 17, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez, P.C.; Wilke, D.V.; Branco, P.C.; Bauermeister, A.; Rezende-Teixeira, P.; Gaudêncio, S.P.; Costa-Lotufo, L.V. Enriching cancer pharmacology with drugs of marine origin. Br. J. Pharmacol. 2020, 177, 3–27. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J.; Cragg, G.M. Marine-sourced anti-cancer and cancer pain control agents in clinical and late preclinical development. Mar. Drugs 2014, 12, 255–278. [Google Scholar] [CrossRef] [Green Version]
- Mayer, A.M.; Nguyen, M.; Kalwajtys, P.; Kerns, H.; Newman, D.J.; Glaser, K.B. The marine pharmacology and pharmaceuticals pipeline in 2016. FASEB J. 2017, 31, 818.1. [Google Scholar]
- Scott, J.D.; Williams, R.M. Chemistry and biology of the tetrahydroisoquinoline antitumor antibiotics. Chem. Rev. 2002, 102, 1669–1730. [Google Scholar] [CrossRef]
- Avendaño, C.; de la Cuesta, E. Recent synthetic approaches to 6,15-iminoisoquino[3,2-b]3-benzazocine compounds. Eur. J. Chem. 2010, 16, 9722–9734. [Google Scholar] [CrossRef]
- Lane, J.W.; Estevez, A.; Mortara, K.; Callan, O.; Spencer, J.R.; Williams, R.M. Antitumor activity of tetrahydroisoquinoline analogues 3-epi-jorumycin and 3-epi-renieramycin G. Bioorg. Med. Chem. Lett. 2006, 16, 3180–3183. [Google Scholar] [CrossRef] [Green Version]
- Le, V.H.; Inai, M.; Williams, R.M.; Kan, T. Ecteinascidins. A review of the chemistry, biology and clinical utility of potent tetrahydroisoquinoline antitumor antibiotics. Nat. Prod. Rep. 2015, 32, 328–347. [Google Scholar] [CrossRef] [Green Version]
- Zewail-Foote, M.; Hurley, L.H. Ecteinascidin 743: A minor groove alkylator that bends DNA toward the major groove. J. Med. Chem. 1999, 42, 2493–2497. [Google Scholar]
- Suwanborirux, K.; Amnuoypol, S.; Plubrukarn, A.; Pummangura, S.; Kubo, A.; Tanaka, C.; Saito, N. Chemistry of renieramycins. Part 3. Isolation and tructure of stabilized renieramycin type derivatives possessing antitumor activity from Thai sponge Xestospongia species, pretreated with potassium cyanide. J. Nat. Prod. 2003, 66, 1441–1446. [Google Scholar] [CrossRef]
- Amnuoypol, S.; Suwanborirux, K.; Pummangura, S.; Kubo, A.; Tanaka, C.; Saito, N. Chemistry of renieramycins. Part 5. Structure elucidation of renieramycin-type derivatives O, Q, R, and S from Thai marine sponge Xestospongia species pretreated with potassium cyanide. J. Nat. Prod. 2004, 67, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Charupant, K.; Daikuhara, N.; Saito, E.; Amnuoypol, S.; Suwanborirux, K.; Owa, T.; Saito, N. Chemistry of renieramycins. Part 8: Synthesis and cytotoxicity evaluation of renieramycin M-jorunnamycin A analogues. Bioorg. Med. Chem. 2009, 17, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Sirimangkalakitti, N.; Chamni, S.; Charupant, K.; Chanvorachote, P.; Mori, N.; Saito, N.; Suwanborirux, K. Chemistry of renieramycins. Part 15. Synthesis of 22-O-ester derivatives of jorunnamycin A and their cytotoxicity against non-small-cell lung cancer Cells. J. Nat. Prod. 2016, 79, 2089–2093. [Google Scholar] [CrossRef] [PubMed]
- Chamni, S.; Sirimangkalakitti, N.; Chanvorachote, P.; Saito, N.; Suwanborirux, K. Chemistry of renieramycins. Part 17. A new generation of renieramycins: Hydroquinone 5-O-monoester analogues of renieramycin M as potential cytotoxic agents against non-small-cell lung cancer cells. J. Nat. Prod. 2017, 80, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- WHO. The Top 10 Causes of Death Fact Sheet N°310; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- Halim, H.; Chunhacha, P.; Suwanborirux, K.; Chanvorachote, P. Anticancer and antimetastatic activities of renieramycin M, a marine tetrahydroisoquinoline alkaloid, in human non-small cell lung cancer cells. Anticancer Res. 2011, 31, 193–201. [Google Scholar]
- Cheun-Arom, T.; Chanvorachote, P.; Sirimangkalakitti, N.; Chuanasa, T.; Saito, N.; Abe, I.; Suwanborirux, K. Replacement of a quinone by a 5-O-acetylhydroquinone abolishes the accidental necrosis inducing effect while preserving the apoptosis-inducing effect of renieramycin M on lung cancer cells. J. Nat. Prod. 2013, 76, 1468–1474. [Google Scholar] [CrossRef]
- Sirimangkalakitti, N.; Chamni, S.; Suwanborirux, K.; Chanvorachote, P. Renieramycin M sensitizes anoikis-resistant H460 lung cancer cells to anoikis. Anticancer Res. 2016, 36, 1665–1671. [Google Scholar] [PubMed]
- Pinkhien, T.; Maiuthed, A.; Chamni, S.; Suwanborirux, K.; Saito, N.; Chanvorachote, P. Bishydroquinone renieramycin M induces apoptosis of human lung cancer cells through a mitochondria-dependent pathway. Anticancer Res. 2016, 36, 6327–6333. [Google Scholar] [CrossRef] [PubMed]
- Sirimangkalakitti, N.; Chamni, S.; Suwanborirux, K.; Chanvorachote, P. Renieramycin M attenuates cancer stem cell-like phenotypes in H460 lung cancer cells. Anticancer Res. 2017, 37, 615–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiuthed, A.; Pinkhien, T.; Chamni, S.; Suwanborirux, K.; Saito, N.; Petpiroon, N.; Chanvorachote, P. Apoptosis-inducing effect of hydroquinone 5-O-cinnamoyl ester analog of renieramycin M on non-small cell lung cancer cells. Anticancer Res. 2017, 37, 6259–6267. [Google Scholar]
- Saito, N.; Tanaka, C.; Koizumi, Y.; Suwanborirux, K.; Amnuoypol, S.; Pummangura, S.; Kubo, A. Chemistry of renieramycins. Part 6: Transformation of renieramycin M into jorumycin and renieramycin J including oxidative degradation products, mimosamycin, renierone, and renierol acetate. Tetrahedron 2004, 60, 3873–3881. [Google Scholar] [CrossRef]
- Neises, B.; Steglich, W. Simple Method for the Esterification of Carboxylic Acids. Angew. Chem. Int. Ed. Engl. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M.J. Organic carbamates in drug design and medicinal chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar]
- Jornada, D.H.; dos Santos Fernandes, G.F.; Chiba, D.E.; De Melo, T.R.F.; Dos Santos, J.L.; Chung, M.C. The prodrug approach: A successful tool for improving drug solubility. Molecules 2016, 21, 42. [Google Scholar] [CrossRef] [Green Version]
- Polakis, P. Antibody Drug Conjugates for Cancer Therapy. Pharmacol. Rev. 2016, 68, 3–19. [Google Scholar] [CrossRef]
- Taylor, J. Toxicological Profile for Cyanide; U.S. Dept. of Health and Human Services, Public Health Service, Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2006.
- Riss, T.L.; Moravec, R.A.; Niles, A.L.; Benink, H.A.; Worzella, T.J.; Minor, L. Assay Guidance Manual; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Compound | O–R | H292 | H460 |
|---|---|---|---|---|
| IC50 ± S.D. (nM) | IC50 ± S.D. (nM) | |||
| 1 | 1 | Angeloyl | 24.56 ± 1.12 | 6.50 ± 0.39 |
| 2 | 7 | H | 217.43 ± 21.67 | 164.30 ± 11.07 |
| 3 | 5a | N-Boc-l-Gly | 3.56 ± 0.62 | 9.94 ± 0.82 |
| 4 | 5b | N-Boc-l-Ala | 10.32 ± 1.95 | 19.75 ± 2.38 |
| 5 | 5c | N-Boc-l-Phe | 110.50 ± 2.53 | 51.43 ± 5.67 |
| 6 | 5d | N-Boc-l-Val | 32.39 ± 3.33 | 20.95 ± 3.53 |
| 7 | 5e | N-Boc-l-Pro | 66.55 ± 9.47 | 45.91 ± 6.94 |
| 8 | 6a | N-Boc-l-Gly | 10.24 ± 0.94 | 6.14 ± 0.68 |
| 9 | 6b | N-Boc-l-Ala | 24.03 ± 2.61 | 15.94 ± 1.32 |
| 10 | 6c | N-Boc-l-Phe | 57.35 ± 5.36 | 42.72 ± 4.91 |
| 11 | 6d | N-Boc-l-Val | 127.20 ± 2.65 | 200.53 ± 29.27 |
| 12 | 6e | N-Boc-l-Pro | 85.00 ± 8.62 | 43.28 ± 0.68 |
| 13 | cisplatin | - | 12.13 × 103 ± 1.12 × 103 | 8.15 × 103 ± 0.64 × 103 |
| 14 | doxorubicin | - | 350.70 ± 32.94 | 33.70 ± 4.56 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamni, S.; Sirimangkalakitti, N.; Chanvorachote, P.; Suwanborirux, K.; Saito, N. Chemistry of Renieramycins. Part 19: Semi-Syntheses of 22-O-Amino Ester and Hydroquinone 5-O-Amino Ester Derivatives of Renieramycin M and Their Cytotoxicity against Non-Small-Cell Lung Cancer Cell Lines. Mar. Drugs 2020, 18, 418. https://0-doi-org.brum.beds.ac.uk/10.3390/md18080418
Chamni S, Sirimangkalakitti N, Chanvorachote P, Suwanborirux K, Saito N. Chemistry of Renieramycins. Part 19: Semi-Syntheses of 22-O-Amino Ester and Hydroquinone 5-O-Amino Ester Derivatives of Renieramycin M and Their Cytotoxicity against Non-Small-Cell Lung Cancer Cell Lines. Marine Drugs. 2020; 18(8):418. https://0-doi-org.brum.beds.ac.uk/10.3390/md18080418
Chicago/Turabian StyleChamni, Supakarn, Natchanun Sirimangkalakitti, Pithi Chanvorachote, Khanit Suwanborirux, and Naoki Saito. 2020. "Chemistry of Renieramycins. Part 19: Semi-Syntheses of 22-O-Amino Ester and Hydroquinone 5-O-Amino Ester Derivatives of Renieramycin M and Their Cytotoxicity against Non-Small-Cell Lung Cancer Cell Lines" Marine Drugs 18, no. 8: 418. https://0-doi-org.brum.beds.ac.uk/10.3390/md18080418