
Divergent Strategy in Marine Tetracyclic Meroterpenoids Synthesis


1
Department of Chemical Engineering, Escuela Politécnica Superior, University of Sevilla, 41011 Sevilla, Spain
2
Organic Chemistry, ceiA3, University of Almería, 04120 Almeria, Spain
*
Author to whom correspondence should be addressed.
Mar. Drugs 2021, 19(5), 273; https://0-doi-org.brum.beds.ac.uk/10.3390/md19050273
Submission received: 29 April 2021
/
Revised: 11 May 2021
/
Accepted: 12 May 2021
/
Published: 13 May 2021
(This article belongs to the Special Issue Synthesis of Marine Natural Products and Molecules Inspired by Marine Substances II)
Abstract
:The divergent total synthesis strategy can be successfully applied to the preparation of families of natural products using a common late-stage pluripotent intermediate. This approach is a powerful tool in organic synthesis as it offers opportunities for the efficient preparation of structurally related compounds. This article reviews the synthesis of the marine natural product aureol, as well as its use as a common intermediate in the divergent synthesis of other marine natural and non-natural tetracyclic meroterpenoids.

1. Introduction
The original definition of divergent total synthesis (Figure 1) was reported by Boger et al. [1] and was defined as the synthesis in which “at least two members of the class of compounds” can be separately prepared from a common, advanced synthetic intermediate. Therefore, the most important challenge in a divergent synthesis is the choice of a common intermediate which could be transformed into a target array of natural products and non-natural derivatives. This strategy is a powerful tool that has attracted the attention of numerous research groups as it improves the efficiency of chemical processes [2,3], and attains special relevance when structure-activity studies are the ultimate goals. Later, other terms such as “diverted total synthesis” [4] and “collective total synthesis” [5] were introduced, thus extending the amplitude of divergent synthesis. In this way, “diverted total synthesis” can be applied to the preparation of a natural product-like compound library by appropriate transformations of a common intermediate, avoiding the limitations inherent to partial syntheses from the natural product caused by the presence of multiple similar functional groups. In addition, the term “collective total synthesis” is used when the common intermediate is endowed with functional characteristics suitable for the preparation of structurally diverse natural products belonging to different families.
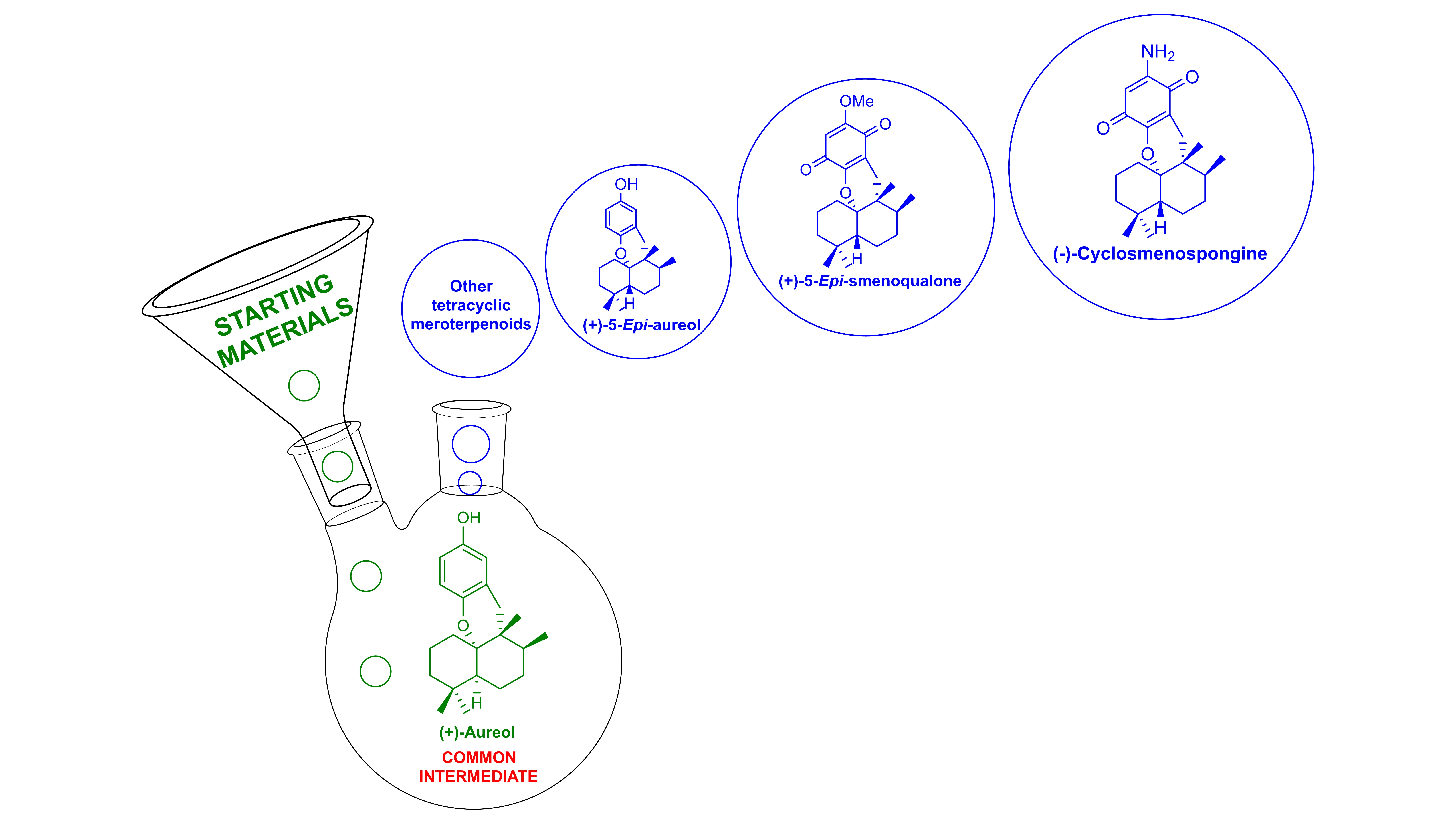
On the other hand, tetracyclic meroterpenoids [6,7] are a unique class of marine natural compounds with significant biological activities. Representative examples of marine natural and non-natural tetracyclic meroterpenoids, including (+)-aureol (1) [8,9,10], (+)-strongylin A (2) [8,11,12], (+)-smenoqualone (3) [8,13], (−)-cyclosmenospongine (4) [8,14], (+)-5-epi-aureol (5) [8,15,16,17], and (+)-5-epi-smenoqualone (6) [8] have been considered of interest by the chemical community due to their interesting biological properties and unique molecular structures (Scheme 1). In fact, structure–activity relationship (SAR) studies show that variations on the nature and substituents on the aromatic ring have a strong impact on the observed activity [8,18]. These natural products (Scheme 1) contain a compact tetracyclic system with a substituted benzopyran moiety, four consecutive asymmetric carbon atoms, and a well-defined trans- or cis-relationship between the two cyclohexane rings of the decalin system. Although several synthetic methods have been described, a divergent approach to this class of compounds has not been previously reported as such.
This article focuses on the synthetic efforts towards aureol (1), a marine natural meroterpenoid present in the Caribbean sponges Smenospongia aurea [9] and Verongula gigantea [10] which has shown an important biological profile [8,19,20]. Aureol (1) can be an excellent advanced and common synthetic intermediate for the divergent synthesis of other natural and non-natural tetracyclic terpenoids. In this article, we present a unified and versatile approach for the diversification of this class of compounds with the aim to contribute to the development of new desirable drugs for the pharmaceutical industry and the medicinal chemistry. The divergent synthesis of either natural or fully synthetic derivatives could be achieved through aureol (1) as a common intermediate, by adequate sequential functionalization of the aromatic ring, or by epimerization of the decalin core of aureol (1) to 5-epi-aureol (5) followed by functionalization of the aromatic ring (Scheme 2).
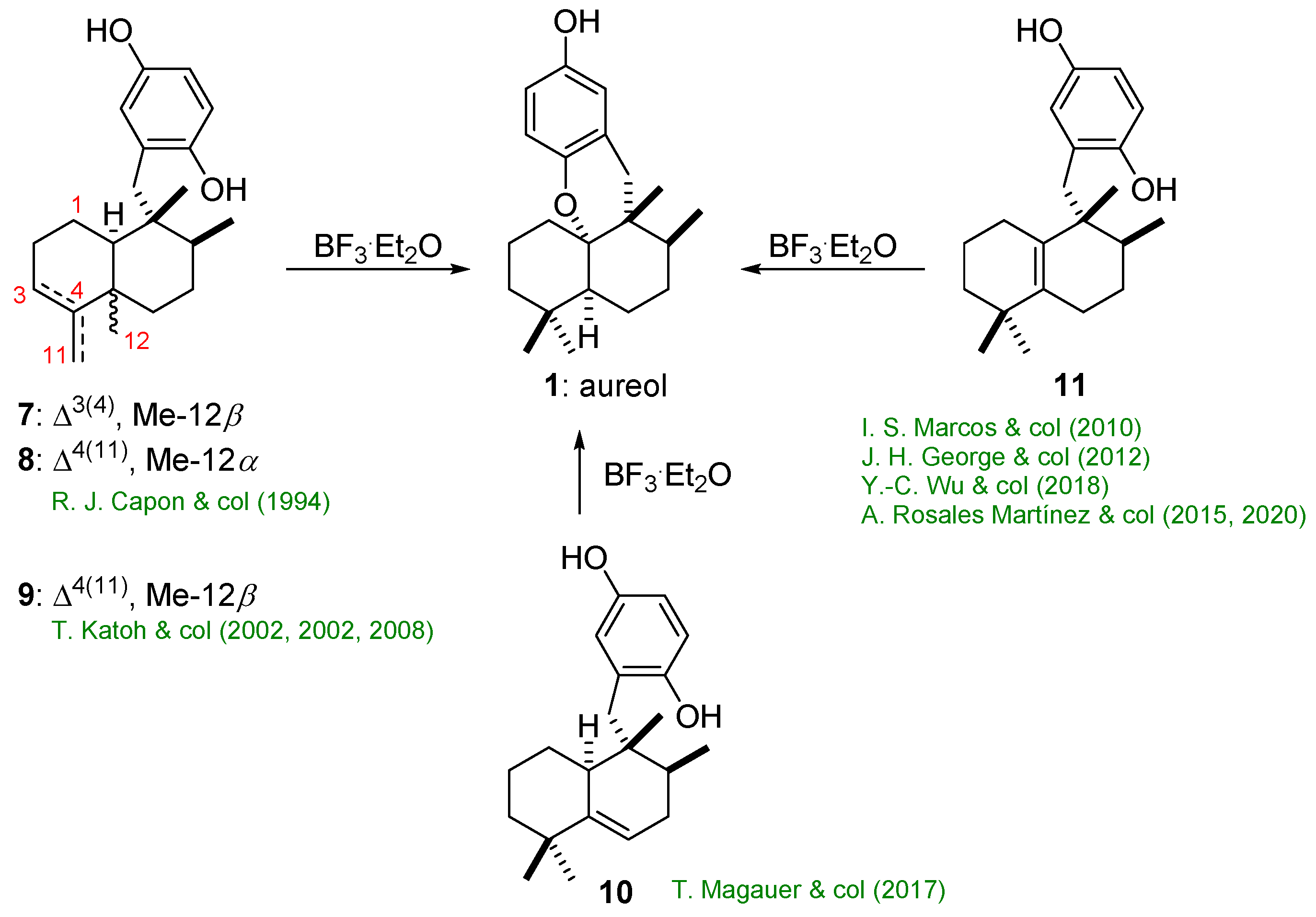
Scheme 3 summarizes the last step of previous syntheses towards aureol (1). All of them have as key step a cationic cyclization of an olefinic intermediate (7–11).
The different synthetic sequences for aureol (1) are listed below, classified according to the olefinic key intermediate shown in Scheme 3.
2. Synthesis of Aureol
2.1. Synthesis of Aureol from Key Intermediates 7–9
2.1.1. Capon’s Synthesis of (+)-Aureol
The first work on the synthesis of the marine product (+)-aureol (1) was published by the group of R. J. Capon [15] using natural sesquiterpene hydroquinones ((+)-avarol (7) and (+)-arenarol (8)) as starting materials (Scheme 4). In these processes (+)-aureol (1) could be formed via a concerted 1,2-migration of Me-12 and H-10. However, formation of (+)-epi-aureol (5) is better understood considering that, after methyl migration, there is a loss of the C-10 proton to give a Δ5,10 olefin intermediate, which would later suffer trans addition of the OH group. This lack of stereocontrol of the process was later confirmed by Lakshmi et al. [21], as they could determine the structure of 5 by X-ray analysis (Scheme 4b).
2.1.2. Katoh’s Synthesis of (+)-Aureol
Katoh and colleagues [22,23,24] reported the first enantioselective total synthesis of (+)-aureol (1) in 2002 [22], a process they later improved in 2003 [23]. The retrosynthetic plan of the improved synthesis is shown in Scheme 5a. This approach obtains aureol (1) in one step by acid-induced rearrangement/cyclization of (−)-neoavarol (9). In turn, 9 can be prepared by reduction of the quinone moiety present in (−)-neovarone (13), a compound which can be readily obtained by strategic salcomine oxidation of 14. This can be assembled by stereocontrolled reductive alkylation of (+)-5-methyl-Wieland-Miescher ketone (15) with 2-methoxybenzyl bromide (16) (Scheme 5a).
As shown in Scheme 5b, Katoh’s synthesis of (+)-aureol (1) used enantiopure (+)-5-methyl-Wieland-Miescher ketone (15) as the starting material. A C-C bond-forming reaction between 15 and a lithiated arene unit, prepared from 2-methoxybenzyl bromide (16), gave the coupling product 17 as a single diastereomer in 74%. The Wittig methylenation of 17 produced the exo-double bond present in decaline 18 in 86% yield. Removal of the acetal protective group by acid treatment (97% yield), followed by hydrogenation of the exocyclic double bond present in the resulting ketone 19, led to the product 20 (80% yield) together with its C8 epimer (13% yield). Subsequent Wittig methylenation of 20 quantitatively gave 21, which was submitted to a deprotection of the O-methyl group in order to form the phenol 14 (92% yield). O2/salcomine oxidation of 14 gave the quinone 13 (91% yield). Finally, NaBH4 reduction of quinone 13 gave (−)-neoavarol (9) (86% yield). Once 9 was synthesized, the crucial step was the BF3.Et2O-induced rearrangement of 9, which led to the desired (+)-aureol (1) (93% yield). This rearrangement occurred via stereospecific 1,2-hydride and methyl shifts, as shown in Scheme 5b. In this reaction the three intermediate carbocations I, II, and III are involved. This synthesis was completed in nine steps (33% overall yield) from the starting material 15.
2.2. Synthesis of Aureol from Key Intermediate 10
Magauer’s Synthesis of (+)-Aureol
The synthesis of (+)-aureol (1) reported by Magauer and colleagues [8] used a highly robust and modular synthetic platform, developed for the preparation of natural and fully synthetic analogues of tetracyclic meroterpenoids. The retrosynthetic plan for (+)-aureol (1) (Scheme 6a) is based on the stereospecific acid-promoted cyclization of the Δ5(6) olefin intermediate 10. Its methoxymethyl ether derivative 22 could be prepared from 23 following the Barton–McCombie deoxygenation protocol. This diastereoisomeric mixture of benzyl alcohols results from the addition of 2-lithiohydroquinone dimethyl ether to aldehyde 24, which can be obtained by Fukuyama’s reduction [25] of thioester 25.
The keystone in the asymmetric process is the formation of the three chiral centers present in 25 through the asymmetric Diels–Alder reaction [26,27] between 26 and enantiopure 27 followed by lithium ethanethiolate removal of the chiral auxiliar (Scheme 6a).
Scheme 6b details the synthesis of (+)-aureol (1) from diene 26. The first key step was the asymmetric construction of the 5,6-dehydrodecaline component 28 employing an exo-selective Diels–Alder cycloaddition between diene 26 and tiglic acid-derived dienophile 27 to afford 28 (61% yield). The oxazolidinone chiral auxiliar was replaced by nucleophilic 1,2-addition of lithium ethanethiolate to the carbonyl group of the Diels–Alder product 28, a process which afforded thioester 25 in 98% yield. A smooth Fukuyama reduction [25] of 25 gave aldehyde 24 (85% yield), which was coupled with the lithiated arene unit to afford a mixture of diastereomeric benzylic alcohols 23 (86% yield). The free hydroxy group of 23 was removed using the two-step Barton–McCombie deoxygenation protocol, which afforded 22 in 84% yield (two steps). The subsequent deprotection of 22 with HCl/MeOH gave hydroquinone sesquiterpene 10, which was directly subjected to cyclization conditions to give (+)-aureol (1) (83% yield). In this reaction, the proton formed by coordination of BF3.OEt2 to one of the OH-groups in 10 possibly triggers the cationic rearrangement. When the cyclization is carried out under kinetic conditions (at temperatures below −10 °C), a cis-decaline framework is formed exclusively. On the other hand, under thermodynamic control, only the trans-decaline is obtained. This total synthesis was achieved in eight steps (30% overall yield) from the starting material 26.
2.3. Synthesis of Aureol from Key Intermediate 11
2.3.1. Marcos’s Synthesis of (−)-Aureol
Marcos and colleagues [28] reported the total synthesis of the (−) enantiomer of aureol (ent-1) from the methyl ester of natural ent-halimic acid (Scheme 7a). Their approach was based on: (a) the acid-induced cyclization of sesquiterpene hydroquinone ent-11, (b) the Barton decarboxylation reaction/p-benzoquinone addition sequence and the subsequent reduction with Raney® nickel (ent-11 from 29), and (c) the side-chain degradation of ent-halimic acid methyl ester 30 and the subsequent reduction of C-18 methyl ester.
As shown in Scheme 7b the synthesis of (−)-aureol (ent-1) used ent-halimic acid methyl ester 30 as the starting material. The degradation of the side chain of 30 was achieved [29,30] by oxidation with OsO4 followed by Pb(OAc)4, which gave ketone 31 (94% yield, two steps). The synthesis of the endo-olefin 33 required the Wittig methylenation of 31 (87% yield) and subsequent acid isomerization of 32 (99% yield). In order to remove the C-18 methyl ester, a three steps sequence from 33 to 36 was used, a process which gave a very good global yield. The synthesis of product 39 was achieved in four steps: a) the chemoselective epoxidation of the side-chain double bond in 36 (98% yield), b) the oxidative cleavage with H5IO6 in H2O/THF to afford 37 (94% yield); c) reduction with LiAlH4 (99% yield) to give 38 (99% yield), and d) the acetylation of the hydroxy group in 38 to afford 39 (99% yield). The isomerization of the olefin double bond present in acetate 39 with HI (97% yield) followed by the saponification of the acetoxy group (98% yield) gave the rearranged product ent-40. Finally, the oxidation of ent-40 to acid 29 via aldehyde 41 was achieved with pyridinium dichromate (PDC) in a moderate yield. Once intermediate 29 was available, the key precursor ent-11 of (−)-aureol (ent-1) could be readily prepared by Barton decarboxylation reaction in the presence of p-benzoquinone, a methodology reported by Theodorakis and colleagues [31,32] for the synthesis of ilimaquinone. In this way, when 29 was treated with 2-mercaptopyridine N-oxide in the presence of N,N′-dicyclohexylcarbodiimide (DCC) a photo labile thio-hydroxamic ester (42) was obtained. Then, 43 was prepared by light-induced decarboxylation (halogen lamp 500W) of 42 in the presence of benzoquinone in a 65% yield from 29. The subsequent reduction of 43 with Raney® nickel gave ent-11 in a 99% yield. With the key precursor ent-11 in their hands, the treatment of this compound with BF3.Et2O at low temperature exclusively afforded (−)-aureol (ent-1) with complete stereoselectivity (60% yield). This total synthesis was achieved in 19 steps (10.3% overall yield) from ent-halimic acid methyl ester 30 as the chiral pool starting material.
2.3.2. George’s Synthesis of (+)-Aureol
George’s group [33] published in 2012 the second total synthesis of (+)-aureol (1). Their biosynthetically inspired retrosynthesis of (+)-aureol (1) (Scheme 8a) rests upon the biomimetic acid-mediated cyclization of the key tetrasubstituted olefin intermediate 11, which could be prepared through a process involving the addition of an aryllithium derivative to aldehyde 44. This aldehyde could be formed using a one-carbon dehomologation sequence from 45. Another key step in the process is the biomimetic sequence of 1,2-hydride and 1,2-methyl shifts, which converts alcohol 46 into 45. Finally, the reduction and selective protection of the commercially available enantiopure starting material (+)-sclareolide (47) would form the intermediate 46.
As shown in Scheme 8b, George’s synthesis of (+)-aureol (1) used 11 as the key intermediate, which was prepared from natural (+)-sclareolide (47). Its reduction with LiAlH4 gave a diol, which was selectively protected at the primary hydroxy group with Ac2O in pyridine to afford the acetate 46 in an 84% yield (two steps). Monoacetate 46 was stereoselectively converted to the single stereoisomer olefin 45 (70% yield) in a rearrangement induced by BF3.Et2O, which occurred via stereospecific sequential 1,2-hydride and 1,2-methyl shifts. Saponification of the acetate in 45 gave alcohol 40 (83% yield), which was readily converted into aldehyde 44 through a one-carbon dehomologation sequence using the Grieco–Sharples elimination protocol [34,35] (67% yield in two steps) followed by oxidative cleavage of the resulting terminal alkene 48 (45% yield in two steps). With aldehyde 44 in their hands, the coupling between 44 and an aryllithium species gave the mixture of diastereomeric benzylic alcohols 49. In order to remove the OH group, this mixture of alcohols (49) was treated with lithium in liquid ammonia followed by NH4Cl aqueous solution to afford deoxygenated compound 50 in a 78% yield (two steps). Removal of the TBS protecting groups in 50 with tetrabutylammonium fluoride provided the key intermediate 11 in an 86% yield. To complete the synthesis of (+)-aureol (1), the intermediate 11 was treated with BF3.Et2O to afford (+)-aureol (1) in a 66% yield. This total synthesis was achieved in 12 steps (6% overall yield) from (+)-sclareolide (47).
2.3.3. Wu’s Synthesis of (+)-Aureol
In 2018, Wu and colleagues [36] published the formal synthesis of (+)-aureol (1). Their retrosynthesis of (+)-aureol is outlined in Scheme 9a. This retrosynthetic analysis is based on: (a) the biomimetic acid-mediated cyclization of the hydroquinone 11 to generate (+)-aureol (1), (b) the removal of the two O-Me protecting groups of 51 to afford the key intermediate 11, (c) the cross-coupling reaction between alkyl iodide 52 and Grignard reagent 53 to give the intermediate 51, (d) the rearrangement reaction of 54 to afford 52, and (e) the reduction of (+)-sclareolide (47) and subsequent C-C bond cleavage to give 54.
As shown in Scheme 9b, the synthesis of intermediate 11 was carried out starting from commercially available (+)-sclareolide (47). Reduction of 47 using diisobutylaluminium hydride (DIBAL-H) generated sclareal 55 in a 98% yield. The treatment of 55 under the C-C bond cleavage conditions described by Suárez and colleagues [37] gave drimanal iodoformate (54) in a 78% yield. The crucial step was the BF3.Et2O-mediated rearrangement of 54, which occurred via stereospecific sequential 1,2-hydride and 1,2-methyl shifts to generate the desired alkyl iodide 52 (63%), together with a minor amount of by-product 56 (20%). In this reaction, the intermediate carbocations V-VII could be involved. With alkyl iodide 52 in their hands, the cross-coupling reaction between Grignard reagent 53 and alkyl iodide 52 generated the key intermediate 51 in a 56% yield. As olefin 51 was an advanced intermediate in the Rosales’s synthesis [38,39] of (±)-aureol (1), their strategy constituted a formal synthesis of (+)-aureol (1). This formal synthesis was completed in four steps (27% overall yield) from starting material (+)-sclareolide (47).
2.3.4. Rosales Martínez’s Synthesis of (±)-Aureol
As a part of our efforts directed towards the synthesis of marine terpenoids [40], we embarked on a project aimed at the divergent synthesis of tetracyclic meroterpenoids. Our endeavors started with the racemic preparation of (±)-aureol (1) in 2015 [38], a process we latter improved in 2020 [39]. This effort continues with the divergent synthesis of other tetracyclic meroterpenoids using aureol (1) as a common synthetic intermediate. The retrosynthetic plan for each synthesis is shown in Scheme 10. Our strategy is based on the preparation of (±)-aureol (1) through the biomimetic acid cyclization of hydroquinone 11, an intermediate that could be generated from 57 through a sequence of 1,2-hydride and 1,2-methyl shifts and the subsequent deprotection of both O-Me groups. 57 is an intermediate common to both synthetic approaches. In one of them, 57 is prepared through Cp2TiCl-catalyzed reductive epoxide cyclization cascade of epoxyfarnesol derivative 58 and the subsequent deoxygenation of the OH-group. In the other, a cross-coupling reaction between albicanal (59) and 2-lithiohydroquinone is used.
Initially [38], we pursued the synthesis of the key intermediate 57 using epoxyfarnesol 60 as the starting material (Scheme 11). The one-pot mesylation of product 60 with MsCl, and the subsequent addition of LiBr quantitatively gave a yield of bromide 61. The cross-coupling reaction between 61 and 2,5-dimethoxyphenylmagnesium bromide afforded the epoxyfarnesol derivative 58 (97% yield). A very elegant Cp2TiCl-catalyzed [40] radical cascade cyclization of 58 gave 62 in a moderate 48% yield. The subsequent deoxygenation of alcohol 62 was carried out using the Barton–McCombie procedure, which afforded 57 in an 86% overall yield (two steps). Later [39], the key intermediate 57 was also prepared through a C-C bond-forming reaction between 2-lithiohydroquinone dimethyl ether and (±)-albicanal (59) as starting material, which was previously obtained by oxidation of (±)-albicanol (63) with the Dess–Martin reagent (99.7% yield). In this way, the coupling of 59 with 2-lithiohydroquinone dimethyl ether gave a mixture of diastereomeric benzylic alcohols which, without separation, was treated with lithium in liquid NH3/THF followed by aqueous NH4Cl to give the deoxygenated product 57 in a 90% yield (two steps). With 57 in our hands, tetrasubstituted olefin 51 was synthesized by biomimetic-type rearrangement of 57 mediated by BF3.Et2O.
Under these conditions, 51 was obtained in a 63% yield, together with the by-product 64 in a 30% yield. In this reaction, the intermediate carbocations IX-XI could be involved [39]. The cationic rearrangement might be initiated by a proton from HF, which could be formed through hydrolysis of BF3, since is known that BF3.Et2O is very moisture sensitive. The demethylation of 51 gave 11 in an 82% yield over the two steps. Finally, the treatment of the hydroquinone 11 with BF3.Et2O afforded aureol (1) (62%). This cyclization was originally explored by Marcos et al. [28] This synthesis of racemic (±)-aureol (1) was completed in eight steps (14% overall yield) from the starting material epoxyfarnesol (60) or in seven steps (28% yield overall yield) from the starting material (±)-albicanal (59).
3. Aureol as Pluripotent Late-Stage Intermediate for the Synthesis of Tetracyclic Meroterpenoids
The possibility of using aureol (1) as a late-intermediate for the divergent synthesis of other tetracyclic terpenoids stems from the fact that these compounds have differences mostly on the aromatic moiety. Furthermore, the easy epimerization of aureol (1) into 5-epi-aureol (5) previously described by Magauer and colleagues [8] opens the door to the preparation of trans-decaline. From both cis- or trans-decaline frameworks, it should be quite straightforward the access to a library of natural or non-natural tetracyclic meroterpenoid analogues, just by simple variation of the arene moiety. The examples represented in Scheme 12 illustrate how other compounds can be obtained from aureol (1). The non-natural 5-epi-aureol (5) was synthesized by thermal isomerization of (+)-aureol (1) using hydroiodic acid in benzene at 90 °C (87% yield) [8]. From 5-epi-aureol (5), the compounds (−)-cyclosmenospongine (4) and 5-epi-smenoqualone (6) were prepared by sequential functionalization of their aromatic core. In this way, selective bromination of 5 with Br2, and the subsequent methylation gave the compound 65 in an excellent yield. The non-natural 5-epi-smenoqualone (6) was prepared from 65 via a boronation-oxidation sequence in a 58% yield (two steps). Eventually, non-natural 6 was converted to (−)-cyclomenospongine (4) via aminolysis (60% yield). In addition, the application of this sequential functionalization of the aromatic core to (+)-aureol (1) could be used to prepare natural (+)-smenoqualone (4).
4. Conclusions
The divergent synthesis is a valuable tool in the design of efficient routes for the synthesis of natural products using a common intermediate. Although several unified strategies have been reported for some families of natural products, it is desirable to extrapolate this methodology to the synthesis of tetracyclic meroterpenoids. In this context, this article reviews the synthesis of the marine natural product aureol (1), with special emphasis on their strategies and methodologies. In addition, this natural tetracyclic meroterpenoid can be used as pluripotent late-stage intermediate for the synthesis of other natural and non-natural tetracyclic meroterpenoids. In this article, we proposed a methodology based on a diversification strategy that we believe will be useful in future research for the preparation of other tetracyclic meroterpenoids as substances that could be used as new drugs or in structure–activity relationship studies.
Author Contributions
A.R.M.: design and coordination of the project, writing—original draft, writing—review and editing. I.R.-G.: writing—review and editing. J.L.L.-M.: writing—review. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Vicerrectorado de Investigación (Project 2020/00001014) of University of Sevilla (Spain) and by the Vicerrectorado de Investigación e Innovación of University of Almería (Project PPUENTE2020/010).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
A. Rosales Martínez acknowledges University of the Sevilla for his position as professor and for financial support (Project 2020/00001014).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Boger, D.L.; Brotherton, C.E. Total synthesis of azafluoranthene alkaloids: Rufescine and imeluteine. J. Org. Chem. 1984, 49, 4050. [Google Scholar] [CrossRef]
- Li, L.; Chen, Z.; Zhang, X.; Jia, Y. Divergent Strategy in Natural Product Total Synthesis. Chem. Rev. 2018, 118, 3752–3832. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, J. Divergent strategy in natural product total synthesis. Tetrahedron Lett. 2014, 55, 6156–6162. [Google Scholar] [CrossRef] [Green Version]
- Njardarson, J.T.; Gaul, C.; Shan, D.; Huang, X.-Y.; Danishefsky, S.J. Discovery of Potent Cell Migration Inhibitors through Total Synthesis: Lessons from Structure-Activity Studies of (+)-Migrastatin. J. Am. Chem. Soc. 2004, 126, 1038–1040. [Google Scholar] [CrossRef]
- Jones, S.B.; Simmons, B.; Mastracchio, A.; MacMillan, D.W.C. Collective synthesis of natural products by means of organocascade catalysis. Nature 2011, 475, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Katoh, T. Total synthesis of decahydrobenzo[d]xanthene sesquiterpenoids aureol, strongylin A, and stachyflin. Development of a new strategy for the construction of a common tetracyclic core structure. Heterocycles 2013, 87, 2199–2224. [Google Scholar] [CrossRef]
- Zong, Y.; Wang, W.; Xu, T. Total synthesis of bioactive marine meroterpenoids: The cases of liphagal and frondosin B. Mar. Drugs 2018, 16, 115. [Google Scholar] [CrossRef] [Green Version]
- Wildermuth, R.; Speck, K.; Haut, F.-L.; Mayer, P.; Karge, B.; Broenstrup, M.; Magauer, T. A modular synthesis of tetracyclic meroterpenoid antibiotics. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djura, P.; Stierle, D.B.; Sullivan, B.; Faulkner, D.J.; Arnold, E.V.; Clardy, J. Some metabolites of the marine sponges Smenospongia aurea and Smenospongia (. ident. Polyfibrospongia) echina. J. Org. Chem. 1980, 45, 1435. [Google Scholar] [CrossRef]
- Ciminiello, P.; Dell’Aversano, C.; Fattorusso, E.; Magno, S.; Pansini, M. Chemistry of Verongida sponges. 10. Secondary metabolite composition of the Caribbean sponge Verongula gigantea. J. Nat. Prod. 2000, 63, 263–266. [Google Scholar] [CrossRef]
- Wright, A.E.; Rueth, S.A.; Cross, S.S. An antiviral sesquiterpene hydroquinone from the marine sponge Strongylophora hartmani. J. Nat. Prod. 1991, 54, 1108. [Google Scholar] [CrossRef]
- Coval, S.J.; Conover, M.A.; Mierzwa, R.; King, A.; Puar, M.S.; Phife, D.W.; Pai, J.-K.; Burrier, R.E.; Ahn, H.-S.; Boykow, G.C.; et al. Wiedendiol-A and -B, cholesteryl ester transfer protein inhibitors from the marine sponge Xestospongia wiedenmayeri. Bioorg. Med. Chem. Lett. 1995, 5, 605–610. [Google Scholar] [CrossRef]
- Bourguet-Kondracki, M.L.; Martin, M.T.; Guyot, M. Smenoqualone, a novel sesquiterpenoid from the marine sponge Smenospongia sp. Tetrahedron Lett. 1992, 33, 8079. [Google Scholar] [CrossRef]
- Utkina, N.K.; Denisenko, V.A.; Scholokova, O.V.; Makarchenko, A.E. Determination of the Absolute Stereochemistry of Cyclosmenospongine. J. Nat. Prod. 2003, 66, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Urban, S.; Capon, R.J. Marine sesquiterpene quinones and hydroquinones: Acid-catalyzed rearrangements and stereochemical investigations. Aust. J. Chem. 1994, 47, 1023–1029. [Google Scholar] [CrossRef]
- Speck, K.; Wildermuth, R.; Magauer, T. Convergent Assembly of the Tetracyclic Meroterpenoid (-)-Cyclosmenospongine by a Non-Biomimetic Polyene Cyclization. Angew. Chem. Int. Ed. 2016, 55, 14131–14135. [Google Scholar] [CrossRef] [PubMed]
- Speck, K.; Magauer, T. Evolution of a Polyene Cyclization Cascade for the Total Synthesis of (-)-Cyclosmenospongine. Chem. Eur. J. 2017, 23, 1157–1165. [Google Scholar] [CrossRef]
- Prokof’eva, N.G.; Utkina, N.K.; Chaikina, E.L.; Makarchenko, A.E. Biological activities of marine sesquiterpenoid quinones: Structure-activity relationships in cytotoxic and hemolytic assays. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2004, 139B, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.E.; Cross, S.S.; Burres, N.S.; Koehn, F. Antiviral and Antitumor Terpene Hydroquinones from Marine Sponge and Methods of Use. U.S. Patent WO9,112,250, 22 August 1991. [Google Scholar]
- Longley, R.E.; McConnell, O.J.; Essich, E.; Harmody, D. Evaluation of marine sponge metabolites for cytotoxicity and signal transduction activity. J. Nat. Prod. 1993, 56, 915. [Google Scholar] [CrossRef]
- Lakshmi, V.; Gunasekera, S.P.; Schmitz, F.J.; Ji, X.; Van der Helm, D. Acid-catalyzed rearrangement of arenerol. J. Org. Chem. 1990, 55, 4709. [Google Scholar] [CrossRef]
- Nakamura, M.; Suzuki, A.; Nakatani, M.; Fuchikami, T.; Inoue, M.; Katoh, T. An efficient synthesis of (+)-aureol via boron trifluoride etherate-promoted rearrangement of (+)-arenarol. Tetrahedron Lett. 2002, 43, 6929–6932. [Google Scholar] [CrossRef]
- Suzuki, A.; Nakatani, M.; Nakamura, M.; Kawaguchi, K.; Inoue, M.; Katoh, T. Highly improved synthesis of (+)-aureol via (-)-neoavarone and (-)-neoavarol, by employing salcomine oxidation and acid-induced rearrangement/cyclization strategy. Synlett 2003, 329–332. [Google Scholar] [CrossRef]
- Sakurai, J.; Oguchi, T.; Watanabe, K.; Abe, H.; Kanno, S.-I.; Ishikawa, M.; Katoh, T. Highly efficient total synthesis of the marine natural products (+)-avarone, (+)-avarol, (-)-neoavarone, (-)-neoavarol and (+)-aureol. Chem. Eur. J. 2008, 14, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Fukuyama, T.; Lin, S.C.; Li, L. Facile reduction of ethyl thiol esters to aldehydes: Application to a total synthesis of (+)-neothramycin A methyl ether. J. Am. Chem. Soc. 1990, 112, 7050. [Google Scholar] [CrossRef]
- Yoon, T.; Danishefsky, S.J.; de Gala, S. A Concise Total Synthesis of (±)-Mamanuthaquinone by Using an exo-Diels–Alder Reaction. Angew. Chem. Int. Ed. Engl. 1994, 33, 853–855. [Google Scholar] [CrossRef]
- Buter, J.; Heijnen, D.; Wan, I.C.; Bickelhaupt, F.M.; Young, D.C.; Otten, E.; Moody, D.B.; Minnaard, A.J. Stereoselective Synthesis of 1-Tuberculosinyl Adenosine; a Virulence Factor of Mycobacterium tuberculosis. J. Org. Chem. 2016, 81, 6686–6696. [Google Scholar] [CrossRef]
- Marcos, I.S.; Conde, A.; Moro, R.F.; Basabe, P.; Díez, D.; Urones, J.G. Synthesis of quinone/hydroquinone sesquiterpenes. Tetrahedron 2010, 66, 8280–8290. [Google Scholar] [CrossRef]
- Marcos, I.S.; Hernández, F.A.; Sexmero, M.J.; Díez, D.; Basabe, P.; Pedrero, A.B.; García, N.; Sanz, F.; Urones, J.G. Synthesis and absolute configuration of (-)-chettaphanin II. Tetrahedron Lett. 2002, 43, 1243–1245. [Google Scholar] [CrossRef]
- Marcos, I.S.; Hernández, F.A.; Sexmero, M.J.; Díez, D.; Basabe, P.; Pedrero, A.B.; García, N.; Urones, J.G. Synthesis and absolute configuration of (-)-chettaphanin I and (-)-chettaphanin II. Tetrahedron 2003, 59, 685–694. [Google Scholar] [CrossRef]
- Ling, T.; Poupon, E.; Rueden, E.J.; Kim, S.H.; Theodorakis, E.A. Unified Synthesis of Quinone Sesquiterpenes Based on a Radical Decarboxylation and Quinone Addition Reaction. J. Am. Chem. Soc. 2002, 124, 12261–12267. [Google Scholar] [CrossRef]
- Ling, T.; Xiang, A.X.; Theodorakis, E.A. Enantioselective total synthesis of avarol and avarone. Angew. Chem. Int. Ed. 1999, 38, 3089–3091. [Google Scholar] [CrossRef]
- Kuan, K.K.W.; Pepper, H.P.; Bloch, W.M.; George, J.H. Total Synthesis of (+)-Aureol. Org. Lett. 2012, 14, 4710–4713. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, K.B.; Young, M.W. Olefin synthesis. Rate enhancement of the elimination of alkyl aryl selenoxides by electron-withdrawing substituents. J. Org. Chem. 1975, 40, 947. [Google Scholar] [CrossRef]
- Majetich, G.; Grieco, P.A.; Nishizawa, M. Total synthesis of β-elemenone. J. Org. Chem. 1977, 42, 2327. [Google Scholar] [CrossRef]
- Wang, J.-L.; Li, H.-J.; Wang, M.; Wang, J.-H.; Wu, Y.-C. A six-step synthetic approach to marine natural product (+)-aureol. Tetrahedron Lett. 2018, 59, 945–948. [Google Scholar] [CrossRef]
- Concepción, J.I.; Francisco, C.G.; Freire, R.; Hernández, R.; Salazar, J.A.; Suárez, E. Iodosobenzene diacetate, an efficient reagent for the oxidative decarboxylation of carboxylic acids. J. Org. Chem. 1986, 51, 402. [Google Scholar] [CrossRef]
- Rosales, A.; Muñoz-Bascón, J.; Roldán-Molina, E.; Rivas-Bascón, N.; Padial, N.M.; Rodríguez-Maecker, R.; Rodríguez-García, I.; Oltra, J.E. Synthesis of (±)-Aureol by Bioinspired Rearrangements. J. Org. Chem. 2015, 80, 1866–1870. [Google Scholar] [CrossRef]
- Rosales Martínez, A.; Enríquez, L.; Jaraíz, M.; Pozo Morales, L.; Rodríguez-García, I.; Díaz Ojeda, E. A Concise Route for the Synthesis of Tetracyclic Meroterpenoids: (±)-Aureol Preparation and Mechanistic Interpretation. Mar. Drugs 2020, 18, 441. [Google Scholar] [CrossRef]
- Rosales Martínez, A.; Pozo Morales, L.; Díaz Ojeda, E.; Castro Rodríguez, M.; Rodríguez-García, I. The Proven Versatility of Cp2TiCl. J. Org. Chem. 2021, 86, 1311–1329. [Google Scholar] [CrossRef]
Figure 1.
Divergent total synthesis.

Scheme 1.
Representative examples of marine natural and non-natural tetracyclic meroterpenoids and their reported biological activity.
Scheme 1.
Representative examples of marine natural and non-natural tetracyclic meroterpenoids and their reported biological activity.

Scheme 2.
Conceptual model of the divergent synthesis of tetracyclic meroterpenoids using aureol (1) as a pluripotential late-stage intermediate.
Scheme 2.
Conceptual model of the divergent synthesis of tetracyclic meroterpenoids using aureol (1) as a pluripotential late-stage intermediate.

Scheme 3.
Key olefinic intermediates of previous syntheses of aureol (1).

Scheme 4.
(a): Acid-induced rearrangement of (+)-avarol (7). (b): Acid-induced rearrangement of (+)-arenarol (8).
Scheme 4.
(a): Acid-induced rearrangement of (+)-avarol (7). (b): Acid-induced rearrangement of (+)-arenarol (8).

Scheme 5.
Strategy for the synthesis of (+)-aureol (1) according to Katoh and colleagues [22,23,24]. (a): Retrosynthetic plan. (b): Synthesis of (+)-aureol (1). HMPA = hexamethylphosphoramide; salcomine = N,N′-bis(salicylidene)ethylenediamino cobalt(II); DMF = N,N′-dimethylformamide.

Scheme 6.
Strategy for the synthesis of (+)-aureol (1) according to Magauer et al. [8]. (a): Retrosynthetic plan. (b): Synthesis of (+)-aureol (1). TMEDA = N,N,N′,N′-tetramethylethane-1,2-diamine; NaHDMS = sodium bis(trimethylsilyl)amide; AIBN = 2,2′-azobis(2-methylpropionitrile).
Scheme 6.
Strategy for the synthesis of (+)-aureol (1) according to Magauer et al. [8]. (a): Retrosynthetic plan. (b): Synthesis of (+)-aureol (1). TMEDA = N,N,N′,N′-tetramethylethane-1,2-diamine; NaHDMS = sodium bis(trimethylsilyl)amide; AIBN = 2,2′-azobis(2-methylpropionitrile).

Scheme 7.
Strategy for the synthesis of (−)-aureol (ent-1) according to Marcos and colleagues [26]. (a): Retrosynthetic plan. (b): Synthesis of (−)-aureol (ent-1). NMO = N-methylmorpholine-N-oxide; TPAP = tetra-n-propylammonium perruthenate; mCPBA = m-chloroperoxybenzoic acid; PDC = pyridinium dichromate; DCC = N,N′-dicyclohexylcarbodiimide.
Scheme 7.
Strategy for the synthesis of (−)-aureol (ent-1) according to Marcos and colleagues [26]. (a): Retrosynthetic plan. (b): Synthesis of (−)-aureol (ent-1). NMO = N-methylmorpholine-N-oxide; TPAP = tetra-n-propylammonium perruthenate; mCPBA = m-chloroperoxybenzoic acid; PDC = pyridinium dichromate; DCC = N,N′-dicyclohexylcarbodiimide.

Scheme 8.
Strategy for the synthesis of (+)-aureol (1) according to George and colleagues [33]. (a) Retrosynthetic plan. (b) Synthesis of (+)-aureol (1). DMAP = 4-(dimethylamino)pyridine; NMO= N-methylmorpholine-N-oxide; TBAF = tetrabutylammonium fluoride.
Scheme 8.
Strategy for the synthesis of (+)-aureol (1) according to George and colleagues [33]. (a) Retrosynthetic plan. (b) Synthesis of (+)-aureol (1). DMAP = 4-(dimethylamino)pyridine; NMO= N-methylmorpholine-N-oxide; TBAF = tetrabutylammonium fluoride.

Scheme 9.
Strategy for the synthesis of (+)-aureol (1) according to Wu and colleagues [36]. (a): Retrosynthetic plan. (b): Synthesis of (+)-aureol (1). DIBAL-H = diisobutylaluminium hydride; PIDA = (diacetoxyiodo)benzene.
Scheme 9.
Strategy for the synthesis of (+)-aureol (1) according to Wu and colleagues [36]. (a): Retrosynthetic plan. (b): Synthesis of (+)-aureol (1). DIBAL-H = diisobutylaluminium hydride; PIDA = (diacetoxyiodo)benzene.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Scheme 11.
Synthesis of (±)-aureol (1) according to Rosales Martínez et al. [38,39]. DMAP = 4-(dimethylamino)pyridine; AIBN = 2,2′-azobis(2-methylpropionitrile).

Scheme 12.
Synthesis of (-)-cyclomenospongine (4) and 5-epi-smenoquealone (6) from (+)-aureol (1). DMF = N,N′-dimethylformamide.
Scheme 12.
Synthesis of (-)-cyclomenospongine (4) and 5-epi-smenoquealone (6) from (+)-aureol (1). DMF = N,N′-dimethylformamide.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rosales Martínez, A.; Rodríguez-García, I.; López-Martínez, J.L. Divergent Strategy in Marine Tetracyclic Meroterpenoids Synthesis. Mar. Drugs 2021, 19, 273. https://0-doi-org.brum.beds.ac.uk/10.3390/md19050273
AMA Style
Rosales Martínez A, Rodríguez-García I, López-Martínez JL. Divergent Strategy in Marine Tetracyclic Meroterpenoids Synthesis. Marine Drugs. 2021; 19(5):273. https://0-doi-org.brum.beds.ac.uk/10.3390/md19050273
Chicago/Turabian StyleRosales Martínez, Antonio, Ignacio Rodríguez-García, and Josefa L. López-Martínez. 2021. "Divergent Strategy in Marine Tetracyclic Meroterpenoids Synthesis" Marine Drugs 19, no. 5: 273. https://0-doi-org.brum.beds.ac.uk/10.3390/md19050273
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.