
Attenuating Effects of Dieckol on High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease by Decreasing the NLRP3 Inflammasome and Pyroptosis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
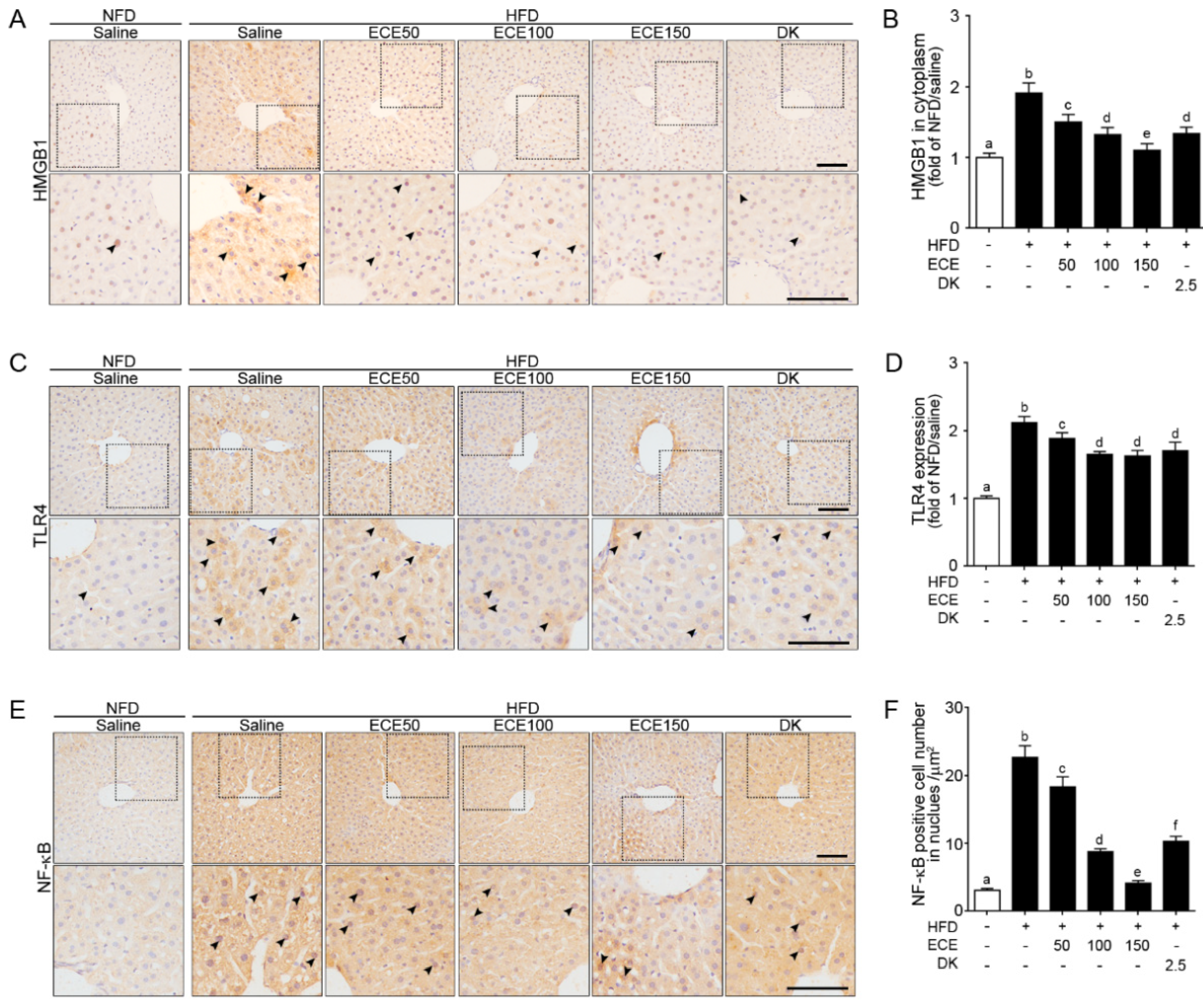
2.1. ECE and DK Decreased HMGB1, TLR4, and NF-κB Expression in the Liver of HFD Mice
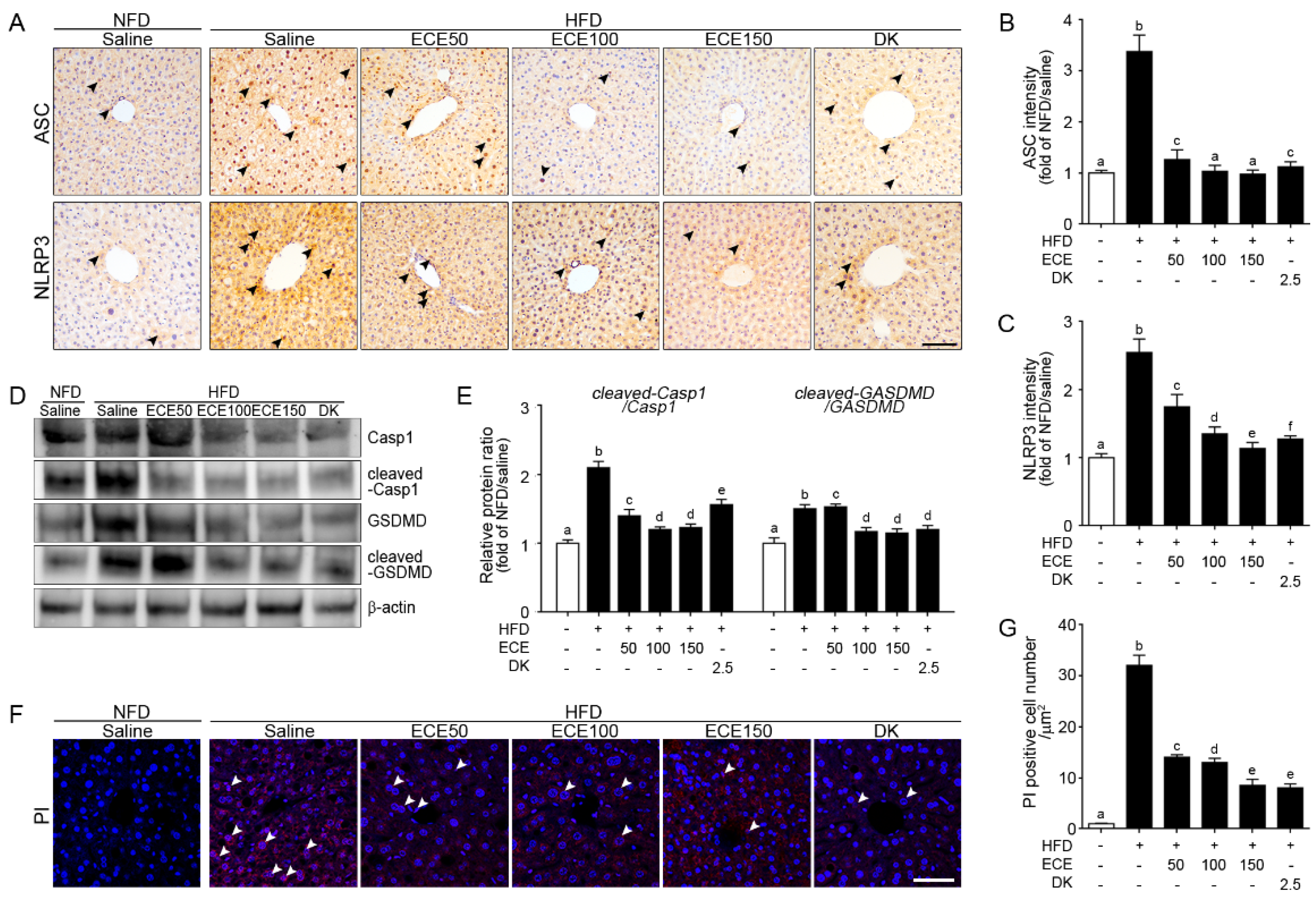
2.2. ECE and DK Attenuated the NLRP3 Inflammasome and Pyroptosis in the Liver of HFD Mice
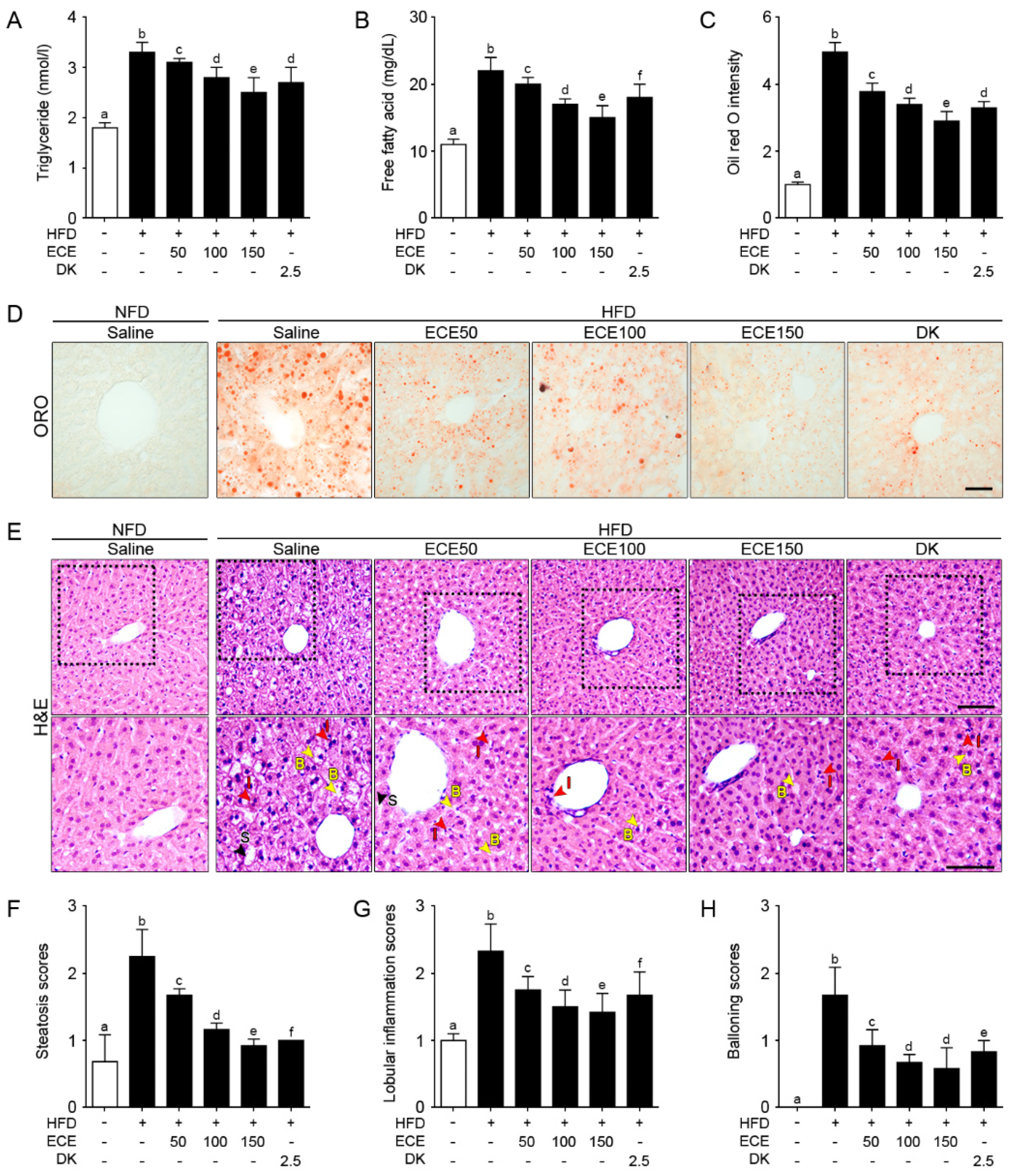
2.3. ECE and DK Decreased NAFLD Activity in HFD Mice
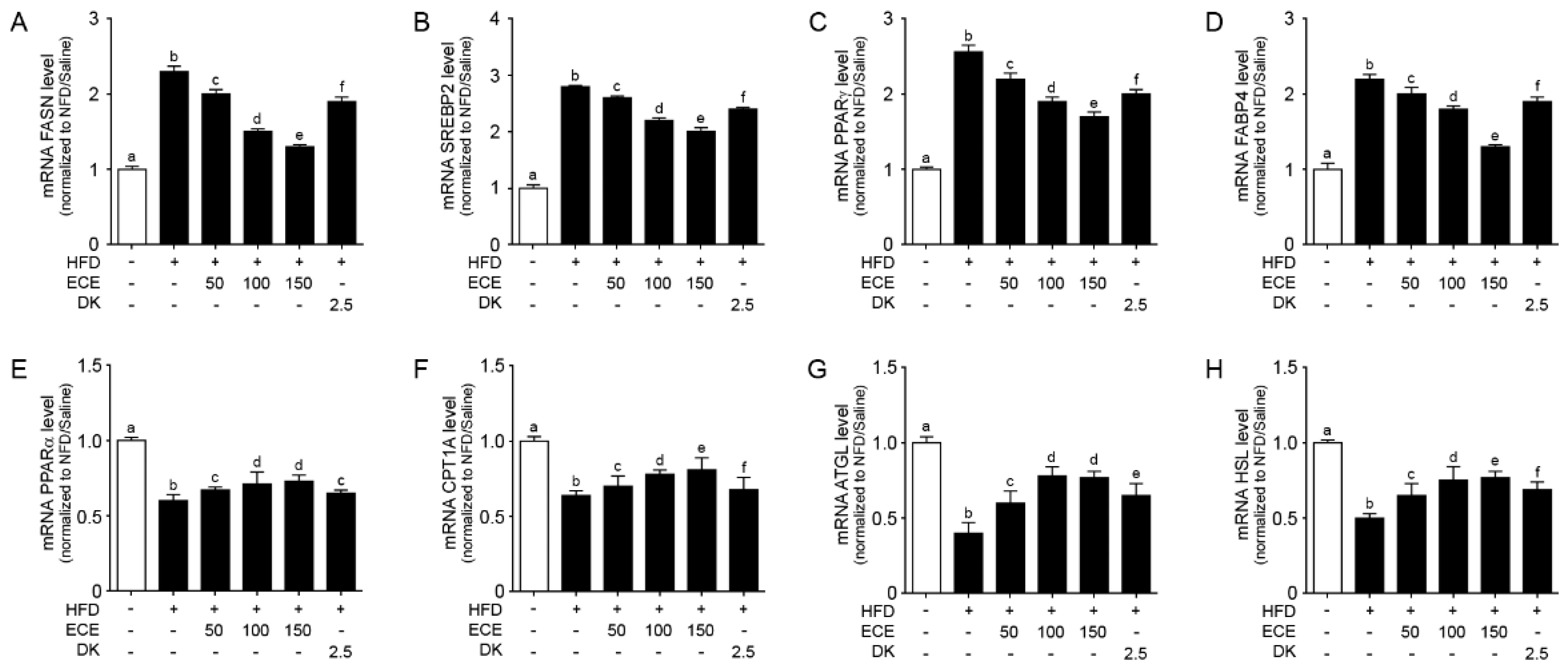
2.4. ECE and DK Reduced Lipogenesis and Increased Lipolysis in the Liver of HFD Mice
3. Discussion
4. Materials and Methods
4.1. ECE and DK Preparation
4.2. HFD-Induced NAFLD Mice Model
4.3. Isolation of RNA and Quantitative Real-Time-Polymerase Chain Reaction (qRT-PCR)
4.4. 3,3′-Diaminobenzidine (DAB) Staining Immunohistochemistry
4.5. Protein Extraction and Immunoblotting
4.6. Propidium Iodide (PI) Staining
4.7. Oil Red O (ORO) Staining for Hepatic Lipid Accumulation Measurement
4.8. Measurement of Triglycerides and Free Fatty Acids
4.9. Hematoxylin and Eosin (H&E) Staining for Histological Measurement
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Fazel, Y.; Koenig, A.B.; Sayiner, M.; Goodman, Z.D.; Younossi, Z.M. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism 2016, 65, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Machado, M.; Marqu’es-Vidal, P.; Machado, M. Hepatic histology in obese patients undergoing bariatric surgery. J. Hepatol. 2006, 45, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Mells, J.E.; Fu, P.P.; Kumar, P.; Smith, T.; Karpen, S.J.; Anania, F.A. Saturated fat and cholesterol are critical to inducing murine metabolic syndrome with robust nonalcoholic steatohepatitis. J. Nutr. Biochem. 2015, 26, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Alkhouri, N.; Dixon, L.J.; Feldstein, A.E. Lipotoxicity in nonalcoholic fatty liver disease: Not all lipids are created equal. Expert Rev. Gastroenterol. Hepatol. 2009, 3, 445–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.; Liu, J.; Jackson, M.I.; Zhao, F.Q.; Yan, L.; Combs, G.F., Jr. Fatty liver accompanies an increase in lactobacillus species in the hind gut of C57BL/6 mice fed a high-fat diet. J. Nutr. 2013, 143, 627–631. [Google Scholar] [CrossRef] [Green Version]
- Goossens, N.; Jornayvaz, F.R. Translational aspects of diet and non-alcoholic fatty liver disease. Nutrients 2017, 9, 1077. [Google Scholar] [CrossRef] [Green Version]
- Lau, J.K.; Zhang, X.; Yu, J. Animal models of non-alcoholic fatty liver disease: Current perspectives and recent advances. J. Pathol. 2017, 241, 36–44. [Google Scholar] [CrossRef]
- Shi, C.; Yang, H.; Zhang, Z. Involvement of Nucleotide-Binding Oligomerization Domain-Like Receptor Family Pyrin Domain Containing 3 Inflammasome in the Pathogenesis of Liver Diseases. Front. Cell Dev. Biol. 2020, 10, 139. [Google Scholar] [CrossRef] [Green Version]
- Bergsbaken, T.; Fink, S.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, L.T.; Van Rooyen, D.M.; Koina, M.E.; McCuskey, R.S.; Teoh, N.C.; Farrell, G.C. Hepatocyte free cholesterol lipotoxicity results from JNK1-mediated mitochondrial injury and is HMGB1 and TLR4-dependent. J. Hepatol. 2014, 61, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Guzmán-Ruiz, R.; Ortega, F.; Rodríguez, A.; Vázquez-Martínez, R.; Díaz-Ruiz, A.; García-Navarro, S.; Giralt, M.; Garcia-Rios, A.; Cobo-Padilla, D.; Tinahones, F.J.; et al. Alarmin high-mobility group B1 (HMGB1) is regulated in human adipocytes in insulin resistance and influences insulin secretion in β-cells. Int. J. Obes. 2014, 38, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Yu, S.; Chen, X.; Ye, R.; Zhu, W.; Liu, X. NLRP3 Is involved in ischemia/reperfusion injury. CNS Neurol. Disord. Drug Targets 2016, 15, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Hayward, J.A.; Man, S.M. Molecular mechanisms of inflammasome signaling. J. Leukoc. Biol. 2018, 103, 233–257. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Pitzer, A.L.; Chen, Y.; Wang, L.; Li, P.L. Coronary endothelial dysfunction induced by nucleotide oligomerization domain-like receptor protein with pyrin domain containing 3 inflammasome activation during hypercholesterolemia: Beyond inflammation. Antioxid. Redox Signal. 2015, 22, 1084–1096. [Google Scholar] [CrossRef] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [Green Version]
- An, N.; Gao, Y.; Si, Z.; Zhang, H.; Wang, L.; Tian, C.; Yuan, M.; Yang, X.; Li, X.; Shang, H.; et al. Regulatory mechanisms of the nlrp3 inflammasome, a novel immune-inflammatory marker in cardiovascular diseases. Front. Immunol. 2019, 10, 1592. [Google Scholar] [CrossRef]
- Yang, Y.I.; Woo, J.H.; Seo, Y.J.; Lee, K.T.; Lim, Y.; Choi, J.H. Protective effect of brown alga phlorotannins against hyper-inflammatory responses in lipopolysaccharide-induced sepsis models. J. Agric. Food Chem. 2016, 64, 570–578. [Google Scholar] [CrossRef]
- Yang, Y.I.; Shin, H.C.; Kim, S.H.; Park, W.Y.; Lee, K.T.; Choi, J.H. 6,6′-Bieckol, isolated from marine alga Ecklonia cava, suppressed LPS-induced nitric oxide and PGE2 production and inflammatory cytokine expression in macrophages: The inhibition of NFκB. Int. Immunopharmacol. 2012, 12, 510–517. [Google Scholar] [CrossRef]
- Lee, M.S.; Shin, T.; Utsuki, T.; Choi, J.S.; Byun, D.S.; Kim, H.R. Isolation and identification of phlorotannins from Ecklonia stolonifera with antioxidant and hepatoprotective properties in tacrine-treated HepG2 cells. J. Agric. Food Chem. 2012, 60, 5340–5349. [Google Scholar] [CrossRef]
- Choi, J.; Oh, S.; Son, M.; Byun, K. Pyrogallol-phloroglucinol-6,6-bieckol alleviates obesity and systemic inflammation in a mouse model by reducing expression of RAGE and RAGE Ligands. Mar. Drugs 2019, 17, 612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.; Oh, S.; Lee, H.S.; Chung, D.-M.; Jang, J.T.; Jeon, Y.; Choi, C.H.; Park, K.Y.; Son, K.H.; Byun, K. Ecklonia cava extract attenuates endothelial cell dysfunction by modulation of inflammation and brown adipocyte function in perivascular fat tissue. Nutrients 2019, 11, 2795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.; Oh, S.; Choi, J.; Jang, J.T.; Choi, C.H.; Park, K.Y.; Son, K.H.; Byun, K. The phlorotannin-rich fraction of Ecklonia cava extract attenuated the expressions of the markers related with Inflammation and leptin resistance in adipose tissue. Int. J. Endocrinol. 2020, 2020, 9142134. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, D.; Liu, G.M.; Chen, Q.; Lu, Z. Ameliorative effect of dieckol-enriched extraction from Laminaria japonica on hepatic steatosis induced by a high-fat diet via β-oxidation pathway in ICR mice. J. Funct. Food 2019, 58, 44–55. [Google Scholar] [CrossRef]
- Vande Walle, L.; Lamkanfi, M. Pyroptosis. Curr. Biol. 2016, 26, R568–R572. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.W.; Sheng, H.; Bai, Y.F.; Weng, Y.Y.; Fan, X.Y.; Lou, L.J.; Zhang, F. Neohesperidin enhances PGC-1α-mediated mitochondrial biogenesis and alleviates hepatic steatosis in high fat diet fed mice. Nutr. Diabetes 2020, 10, 27. [Google Scholar] [CrossRef]
- Azushima, K.; Ohki, K.; Wakui, H.; Uneda, K.; Haku, S.; Kobayashi, R.; Haruhara, K.; Kinguch, S.; Matsuda, M.; Maeda, A.; et al. Adipocyte-Specific Enhancement of Angiotensin II Type 1 Receptor-Associated Protein Ameliorates Diet-Induced Visceral Obesity and Insulin Resistance. J. Am. Heart Assoc. 2017, 6, e004488. [Google Scholar] [CrossRef] [Green Version]
- Kuwashiro, S.; Terai, S.; Oishi, T.; Fujisawa, K.; Matsumoto, T.; Nishinam, H.; Sakaida, I. Telmisartan improves nonalcoholic steatohepatitis in medaka (Oryzias latipes) by reducing macrophage infiltration and fat accumulation. Cell Tissue Res. 2011, 344, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Wang, H.; Li, X.; Liu, Y.; Mi, Y.; Kong, H.; Xi, D.; Yan, W.; Luo, X.; Ning, Q.; et al. Fibrinogen-like protein 2 aggravates nonalcoholic steatohepatitis via interaction with TLR4, eliciting inflammation in macrophages and inducing hepatic lipid metabolism disorder. Theranostics 2020, 10, 9702–9720. [Google Scholar] [CrossRef]
- Chen, J.W.; Kong, Z.L.; Tsai, M.L.; Lo, C.Y.; Ho, C.T.; Lai, C.S. Tetrahydrocurcumin ameliorates free fatty acid-induced hepatic steatosis and improves insulin resistance in HepG2 cells. J. Food Drug Anal. 2018, 26, 1075–1085. [Google Scholar] [CrossRef]
- Tardelli, M.; Bruschi, F.V.; Trauner, M. The role of metabolic lipases in the pathogenesis and management of liver disease. Hepatology 2020, 72, 1117–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nomura, K.; Yamanouchi, T. The role of fructose-enriched diets in mechanisms of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2012, 23, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Liu, L.; Li, F. Acetate alters the process of lipid metabolism in rabbits. Animal 2018, 12, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of dietary fructose and hepatic de novo lipogenesis in fatty liver disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, V.S.; Hvid, H.; Damgaard, J.; Nygaard, H.; Ingvorsen, C.; Wulff, E.M.; Lykkesfeldt, J.; Fledelius, C. Dietary fat stimulates development of NAFLD more potently than dietary fructose in Sprague-Dawley rats. Diabetol. Metab. Syndr. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Fielding, B. Tracing the fate of dietary fatty acids: Metabolic studies of postprandial lipaemia in human subjects. Proc. Nutr. Soc. 2011, 70, 342–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ameer, F.; Scandiuzzi, L.; Hasnain, S.; Kalbacher, H.; Zaidi, N. De novo lipogenesis in health and disease. Metabolism 2014, 63, 895–902. [Google Scholar] [CrossRef]
- Hirsova, P.; Ibrabim, S.H.; Gores, G.J.; Malhi, H. Lipotoxic lethal and sublethal stress signaling in hepatocytes: Relevance to NASH pathogenesis. J. Lipid Res. 2016, 57, 1758–1770. [Google Scholar] [CrossRef] [Green Version]
- Barreyro, F.J.; Kobayashi, S.; Bronk, S.F.; Werneburg, N.W.; Malhi, H.; Gores, G.J. Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J. Biol. Chem. 2007, 282, 27141–27154. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Chen, H.W.; Evankovich, J.; Yan, W.; Rosborough, B.R.; Nace, G.W.; Ding, Q.; Loughran, P.; Beer-Stolz, D.; Billiar, T.R.; et al. Histones activate the NLRP3 inflammasome in Kupffer cells during sterile inflammatory liver injury. J. Immunol. 2013, 191, 2665–2679. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Tsuchiya, K.; Kawamura, I.; Fang, R.; Hernandez-Cuellar, E.; Shen, Y.; Mizuguchi, J.; Schweighoffer, E.; Tybulewicz, V.; Mitsuyama, M. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 2013, 14, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.-H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Li, L.; Chen, L.; Hu, L.; Liu, Y.; Sun, H.Y.; Tang, J.; Hou, Y.J.; Chang, Y.X.; Tu, Q.Q.; Feng, G.S.; et al. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology 2011, 54, 1620–1630. [Google Scholar] [CrossRef] [Green Version]
- von Moltke, J.; Ayres, J.S.; Kofoed, E.M.; Chavarría-Smith, J.; Vance, R.E. Recognition of bacteria by inflammasomes. Annu. Rev. Immunol. 2013, 31, 73–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome complexes: Emerging mechanisms and effector functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signaling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, E.; Campbell, M.; Doyle, S.L. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: Current perspectives. J. Inflamm. 2015, 8, 15–27. [Google Scholar]
- Franchi, L.; Eigenbrod, T.; Munoz-Planillo, R.; Ozkurede, U.; Kim, Y.G.; Arindam, C.; Gale, M., Jr.; Silverman, R.H.; Colonna, M.; Akira, S.; et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J. Immunol. 2014, 193, 4214–4222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Lu, X.; Lu, C.; Shen, N.; Jiang, Y.; Chen, M.; Wu, H. Soluble uric acid increases PDZK1 and ABCG2 expression in human intestinal cell lines via the TLR4-NLRP3 inflammasome and PI3K/Akt signaling pathway. Arthritis Res. Ther. 2018, 20, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xing, R.; Wang, P.M.; Zhang, N.S.; Yin, S.J.; Li, X.C.; Zhang, L. NLRP1 and NLRP3 inflammasomes mediate LPS/ATPinduced pyroptosis in knee osteoarthritis. Mol. Med. Rep. 2018, 17, 5463–5469. [Google Scholar] [PubMed] [Green Version]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Kesavardhana, S.; Kanneganti, T.D. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int. Immunol. 2017, 29, 201–210. [Google Scholar] [CrossRef]
- Kepp, O.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Pyroptosis—A cell death modality of its kind? Eur. J. Immunol. 2010, 40, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Silveira, T.N.; Zamboni, D.S. Pore formation triggered by Legionella spp. is an Nlrc4 inflammasome-dependent host cell response that precedes pyroptosis. Infect. Immun 2010, 78, 1403–1413. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Liu, M.; Ji, Y.; Ma, M.; Chen, K.; Liang, T.; Liu, C. Genipin reverses HFD-induced liver damage and inhibits UCP2-mediated pyroptosis in mice. Cell Physiol. Biochem. 2018, 49, 1885–1897. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, S.; Wan, T.; Huang, Y.; Pang, N.; Jiang, X.; Gu, Y.; Zhang, Z.; Luo, J.; Yang, L. Cyanidin-3-O-β-glucoside inactivates NLRP3 inflammasome and alleviates alcoholic steatohepatitis via SirT1/NF-κB signaling pathway. Free Radic. Biol. Med. 2020, 160, 334–341. [Google Scholar] [CrossRef]
- Ioannou, G.N. The Role of Cholesterol in the pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95. [Google Scholar] [CrossRef]
- Guo, C.; Chi, Z.; Jiang, D.; Xu, T.; Yu, W.; Wang, Z.; Chen, S.; Zhang, L.; Liu, Q.; Guo, X.; et al. Cholesterol homeostatic regulator SCAP-SREBP2 integrates NLRP3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity 2018, 49, 842–856. [Google Scholar] [CrossRef] [Green Version]
- Fafián-Labora, J.; Carpintero-Fernández, P.; Jordan, S.J.D.; Shikh-Bahaei, T.; Abdullah, S.M.; Mahenthiran, M.; Rodríguez-Navarro, J.A.; Niklison-Chirou, M.V.; O’Loghlen, A. FASN activity is important for the initial stages of the induction of senescence. Cell Death Dis. 2019, 10, 318–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar] [PubMed]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system. From concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [CrossRef]
- Akazawa, Y.; Nakao, K. To die or not to die: Death signaling in nonalcoholic fatty liver disease. J. Gastroenterol. 2018, 53, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.; Son, M.; Lee, H.S.; Kim, H.S.; Jeon, Y.J.; Byun, K. Protective effect of pyrogallol-phloroglucinol-6,6-bieckol from Ecklonia cava on monocyte-associated vascular dysfunction. Mar. Drugs 2018, 16, 441. [Google Scholar] [CrossRef] [Green Version]
- Zhong, F.; Zhou, X.; Xu, J.; Gao, L. Rodent models of nonalcoholic fatty liver disease. Digestion 2020, 101, 522–535. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, S.; Son, M.; Byun, K.-A.; Jang, J.T.; Choi, C.H.; Son, K.H.; Byun, K. Attenuating Effects of Dieckol on High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease by Decreasing the NLRP3 Inflammasome and Pyroptosis. Mar. Drugs 2021, 19, 318. https://0-doi-org.brum.beds.ac.uk/10.3390/md19060318
Oh S, Son M, Byun K-A, Jang JT, Choi CH, Son KH, Byun K. Attenuating Effects of Dieckol on High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease by Decreasing the NLRP3 Inflammasome and Pyroptosis. Marine Drugs. 2021; 19(6):318. https://0-doi-org.brum.beds.ac.uk/10.3390/md19060318
Chicago/Turabian StyleOh, Seyeon, Myeongjoo Son, Kyung-A Byun, Ji Tae Jang, Chang Hu Choi, Kuk Hui Son, and Kyunghee Byun. 2021. "Attenuating Effects of Dieckol on High-Fat Diet-Induced Nonalcoholic Fatty Liver Disease by Decreasing the NLRP3 Inflammasome and Pyroptosis" Marine Drugs 19, no. 6: 318. https://0-doi-org.brum.beds.ac.uk/10.3390/md19060318