
Fish Collagen Peptides Protect against Cisplatin-Induced Cytotoxicity and Oxidative Injury by Inhibiting MAPK Signaling Pathways in Mouse Thymic Epithelial Cells

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
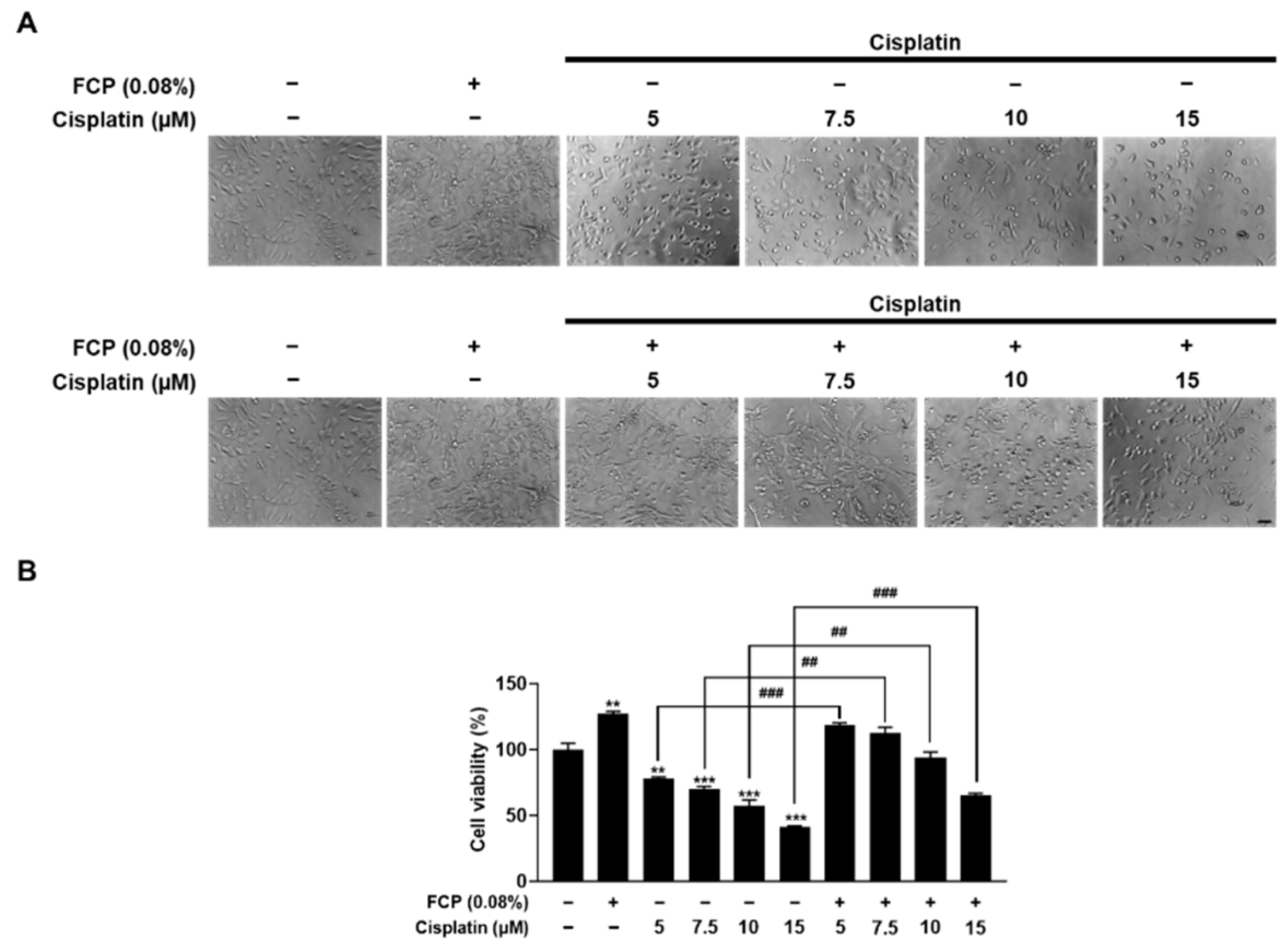
2.1. FCP Promote Cell Proliferation and Inhibit Cisplatin-Induced Cytotoxicity
2.2. FCP Attenuate Cisplatin-Induced ROS Generation
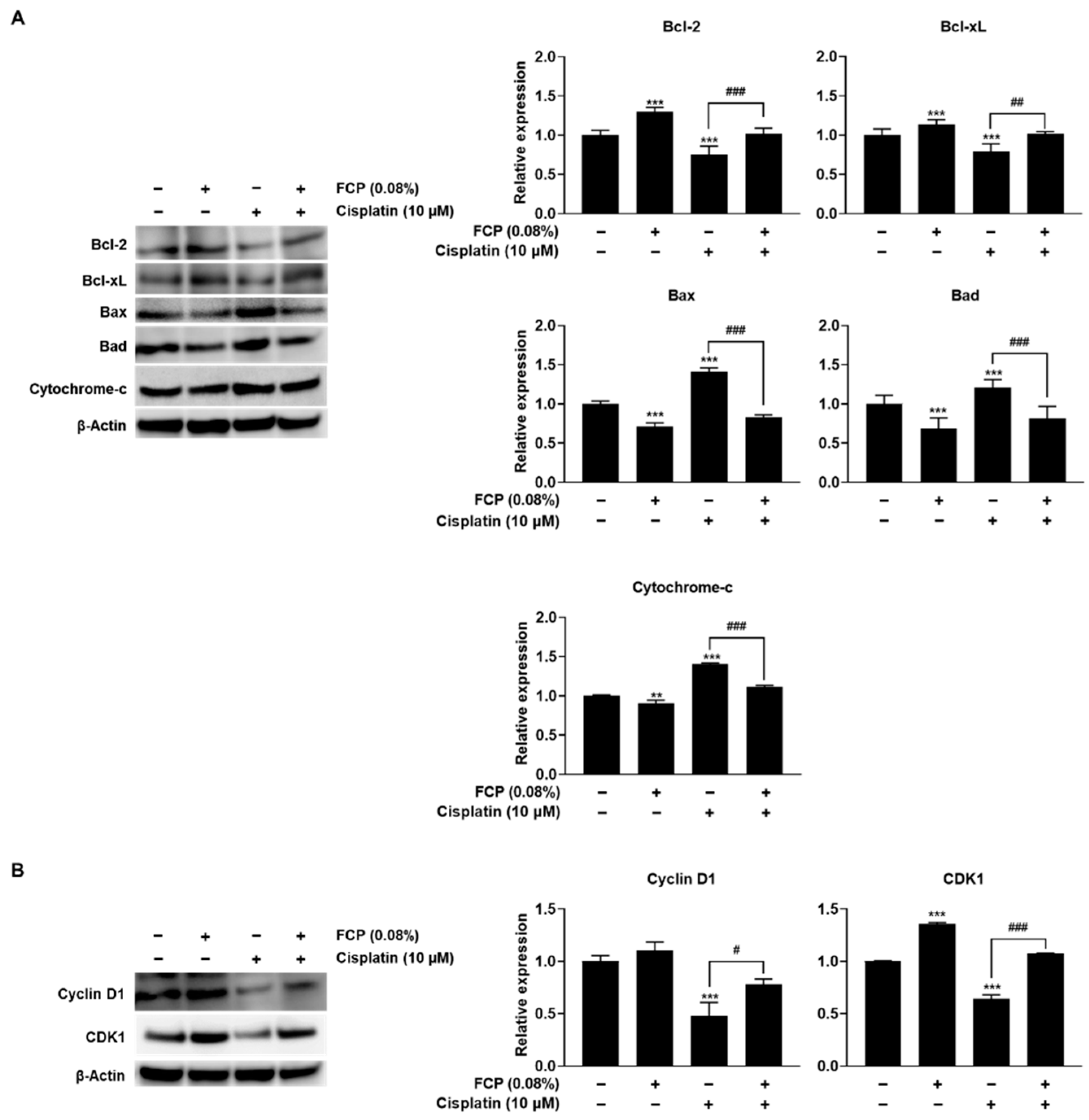
2.3. FCP Alleviate Cisplatin-Induced TEC Cytotoxicity through Suppression of MAPK Pathway
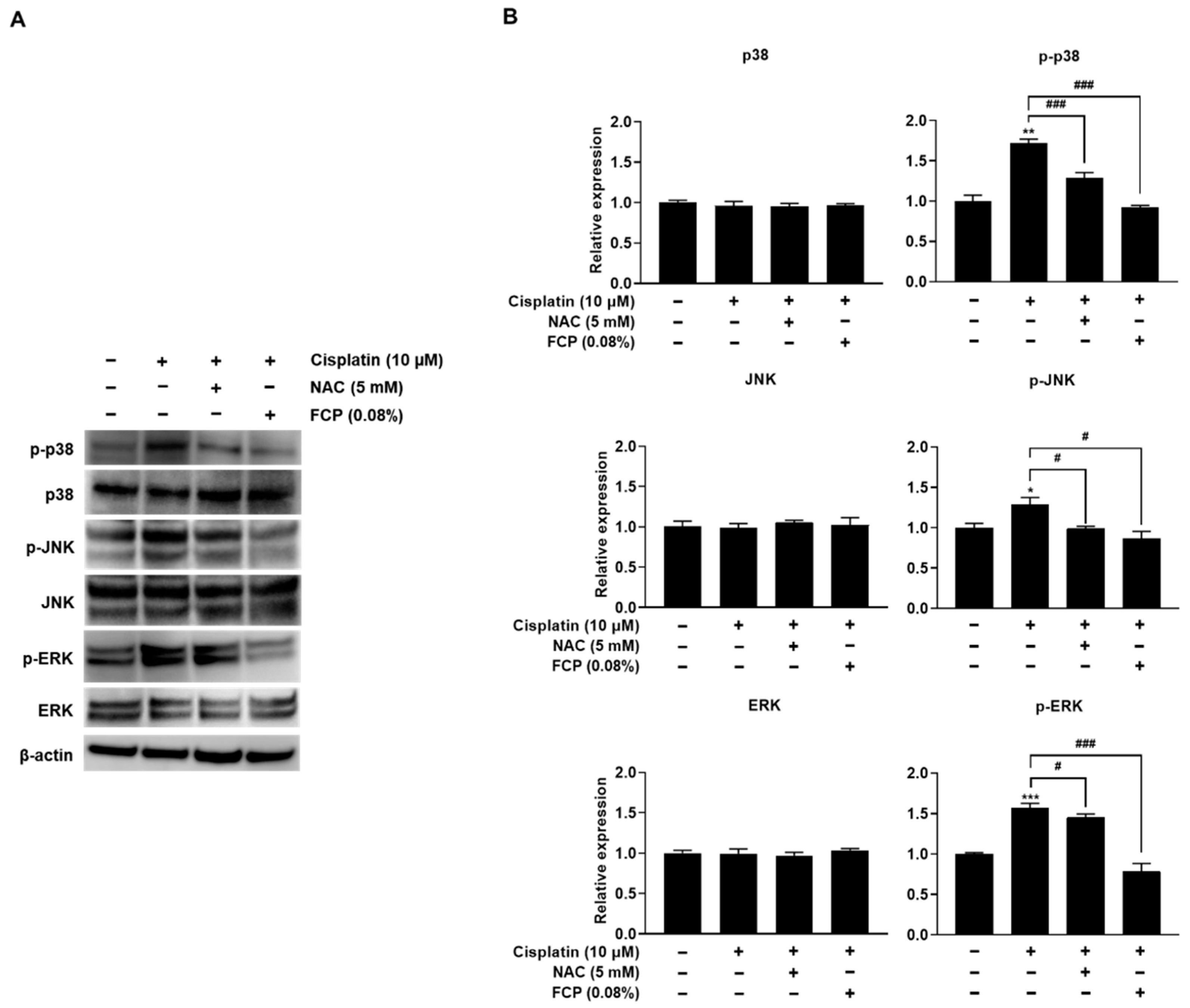
2.4. FCP Prevent Cisplatin-Induced ROS Generation by Inhibition of MAPK Signaling
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Cell Viability Assay
4.3. Measurement of ROS
4.4. Western Blot Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kadouri, N.; Nevo, S.; Goldfarb, Y.; Abramson, J. Thymic epithelial cell heterogeneity: TEC by TEC. Nat. Rev. Immunol. 2020, 20, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, S.; Dudakov, J.A. When the Damage Is Done: Injury and repair in thymus function. Front. Immunol. 2020, 11, 1745. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Xu, L.; Qian, Z.; Sun, X. Infection-associated thymic atrophy. Front. Immunol. 2021, 12, 652538. [Google Scholar] [CrossRef]
- Ménétrier-Caux, C.; Ray-Coquard, I.; Blay, J.Y.; Caux, C. Lymphopenia in cancer patients and its effects on response to immunotherapy: An opportunity for combination with cytokines? J. Immunother. Cancer 2019, 7, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Thomas, R.; Sizova, O.; Su, D.M. Thymic function associated with cancer development, relapse, and antitumor immunity—A Mini-Review. Front. Immunol. 2020, 11, 773. [Google Scholar] [CrossRef] [PubMed]
- Duah, M.; Li, L.; Shen, J.; Lan, Q.; Pan, B.; Xu, K. Thymus degeneration and regeneration. Front. Immunol. 2021, 12, 706244. [Google Scholar] [CrossRef]
- Lynch, H.E.; Goldberg, G.L.; Chidgey, A.; Van den Brink, M.R.; Boyd, R.; Sempowski, G.D. Thymic involution and immune reconstitution. Trends Immunol. 2009, 30, 366–373. [Google Scholar] [CrossRef] [Green Version]
- Ventevogel, M.S.; Sempowski, G.D. Thymic rejuvenation and aging. Curr. Opin. Immunol. 2013, 25, 516–522. [Google Scholar] [CrossRef] [Green Version]
- Ki, S.; Park, D.; Selden, H.J.; Seita, J.; Chung, H.; Kim, J.; Iyer, V.R.; Ehrlich, L.I.R. Global transcriptional profiling reveals distinct functions of thymic stromal subsets and agerelated changes during thymic involution. Cell Rep. 2014, 9, 402–415. [Google Scholar] [CrossRef] [Green Version]
- Barbouti, A.; Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.G.; Papoudou-Bai, A.; Gorgoulis, V.G.; Kanavaros, P. Implications of oxidative stress and cellular senescence in age-related thymus involution. Oxid. Med. Cell. Longev. 2020, 2020, 7986071. [Google Scholar] [CrossRef]
- Narendhirakannan, R.T.; Hannah, M.A. Oxidative stress and skin cancer: An overview. Indian J. Clin. Biochem. 2013, 28, 110–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, P.G.; Bundy, B.N.; Watkins, E.B.; Thigpen, J.T.; Deppe, G.; Maiman, M.A.; Clarke-Pearson, D.L.; Insalaco, S. Concurrent cisplatin-based radiotherapy and chemotherapy for locally advanced cervical cancer. N. Engl. J. Med. 1999, 340, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S.; Copeland, L.J.; Walker, J.L.; Burger, R.A. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Lu, Q.B. New combination chemotherapy of cisplatin with an electron-donating compound for treatment of multiple cancers. Sci. Rep. 2021, 11, 788. [Google Scholar] [CrossRef] [PubMed]
- Spanos, W.C.; Nowicki, P.; Lee, D.W.; Hoover, A.; Hostager, B.; Gupta, A.; Anderson, M.E.; Lee, J.H. Immune response during therapy with cisplatin or radiation for human papillomavirus-related head and neck cancer. Arch. Otolaryngol. Head Neck Surg. 2009, 135, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Fang, C.Y.; Lou, D.Y.; Zhou, L.Q.; Wang, J.C.; Yang, B.; He, Q.J.; Wang, J.J.; Weng, Q.J. Natural products: Potential treatments for cisplatin-induced nephrotoxicity. Acta Pharmacol. Sin. 2021, 42, 951–1969. [Google Scholar] [CrossRef]
- Rancoule, C.; Guy, J.B.; Vallard, A.; Mrad, M.B.; Rehailia, A.; Magne, N. 50th anniversary of cisplatin. Bull. Cancer 2017, 104, 167–176. [Google Scholar] [CrossRef]
- Rébé, C.; Demontoux, L.; Pilot, T.; Ghiringhelli, F. Platinum derivatives effects on anticancer immune response. Biomolecules 2020, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Rustad, T. Utilization of marine by-products. Electron. J. Environ. Agric. Food Technol. 2003, 2, 458–463. [Google Scholar]
- Kim, S.; Mendis, E. Bioactive compounds from marine processing byproducts-a review. Food Res. Int. 2006, 39, 383–393. [Google Scholar] [CrossRef]
- Wang, B.; Wang, Y.M.; Chi, C.F.; Luo, H.Y.; Deng, S.G.; Ma, J.Y. Isolation and characterization of collagen and antioxidant collagen peptides from scales of croceine croaker (Pseudosciaena crocea). Mar. Drugs 2013, 11, 4641–4661. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.H.; Wu, P.C.; Wu, C.H.; Shiau, C.Y. Studies on antioxidative activities of hydrolysates from fish scales collagen of tilapia. J. Taiwan Fish. Res. 2008, 15, 99–108. [Google Scholar]
- Wang, L.; An, X.; Yang, F.; Xin, Z.; Zhao, L.; Hu, Q. Isolation and characterisation of collagens from the skin, scale and bone of deep-sea redfish (Sebastes mentella). Food Chem. 2008, 108, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Cheung, R.C.; Ng, T.B.; Wong, J.H. Marine peptides: Bioactivities and applications. Mar. Drugs. 2015, 13, 4006–4043. [Google Scholar] [CrossRef]
- Asserin, J.; Lati, E.; Shioya, T.; Prawitt, J. The effect of oral collagen peptide supplementation on skin moisture and the dermal collagen network: Evidence from an ex vivo model and randomized, placebo-controlled clinical trials. J. Cosmet. Dermatol. 2015, 14, 291–301. [Google Scholar] [CrossRef] [Green Version]
- Ghorpade, V.S.; Yadav, A.V.; Dias, R.J. Citric acid crosslinked cyclodextrin/hydroxypropylmethylcellulose hydrogel films for hydrophobic drug delivery. Int. J. Biol. Macromol. 2016, 93, 75–86. [Google Scholar] [CrossRef]
- Ding, C.H.; Li, Q.; Xiong, Z.Y.; Zhou, A.W.; Jones, G.; Xu, S.Y. Oral administration of type II collagen suppresses pro-inflammatory mediator production by synoviocytes in rats with adjuvant arthritis. Clin. Exp. Immunol. 2003, 132, 416–423. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.F.; Li, G.Z.; Peng, H.B.; Zhang, F.; Chen, Y.; Li, Y. Treatment with marine collagen peptides modulates glucose and lipid metabolism in Chinese patients with type 2 diabetes mellitus. Appl. Physiol. Nutr. Metab. 2010, 35, 797–804. [Google Scholar] [CrossRef]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [Green Version]
- Brozovic, A.; Osmak, M. Activation of mitogen-activated protein kinases by cisplatin and their role in cisplatin-resistance. Cancer Lett. 2007, 251, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar]
- Mansour, H.H.; Hafez, H.F.; Fahmy, N.M. Silymarin modulates cisplatin-induced oxidative stress and hepatotoxicity in rats. J. Biochem. Mol. Biol. 2006, 39, 656–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afsar, T.; Razak, S.; Almajwal, A.; Khan, M.R. Acacia hydaspica R. Parker ameliorates cisplatin induced oxidative stress, DNA damage and morphological alterations in rat pulmonary tissue. BMC Complement. Altern. Med. 2018, 18, 49. [Google Scholar] [CrossRef] [Green Version]
- Soni, H.; Kaminski, D.; Gangaraju, R.; Adebiyi, A. Cisplatin-induced oxidative stress stimulates renal Fas ligand shedding. Ren. Fail. 2018, 40, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Rehman, M.U.; Rather, I.A. Myricetin abrogates cisplatin-induced oxidative stress, inflammatory response, and goblet cell disintegration in colon of wistar rats. Plants 2019, 9, 28. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.S.; Ok, Y.J.; Hwang, S.Y.; Kwak, J.Y.; Yoon, S. Marine Collagen as A Promising Biomaterial for Biomedical Applications. Mar. Drugs 2019, 17, 467. [Google Scholar] [CrossRef] [Green Version]
- Sivaraman, K.; Shanthi, C. Role of fish collagen hydrolysate in attenuating inflammation—An in vitro study. J. Food Biochem. 2021, 45, e13876. [Google Scholar] [CrossRef]
- Geahchan, S.; Baharlouei, P.; Rahman, A. Marine Collagen: A Promising Biomaterial for Wound Healing, Skin Anti-Aging, and Bone Regeneration. Mar. Drugs 2022, 20, 61. [Google Scholar] [CrossRef]
- Vijayan, D.K.; Sreerekha, P.R.; Dara, P.K.; Ganesan, B.; Mathew, S.; Anandan, R.; Ravisankar, C.N. Antioxidant defense of fish collagen peptides attenuates oxidative stress in gastric mucosa of experimentally ulcer-induced rats. Cell Stress Chaperones 2022, 27, 45–54. [Google Scholar] [CrossRef]
- Yoon, J.; Yoon, D.; Lee, H.; Lee, J.; Jo, S.; Kym, D.; Yim, H.; Hur, J.; Chun, W.; Kim, G.; et al. Wound healing ability of acellular fish skin and bovine collagen grafts for split-thickness donor sites in burn patients: Characterization of acellular grafts and clinical application. Int. J. Biol. Macromol. 2022, 14, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Aleman, A.; Martinez-Alvarez, O. Marine collagen as a source of bioactive molecules. A Review. Nat. Prod. J. 2013, 3, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Silva, T.H.; Moreira-Silva, J.; Marques, A.L.; Domingues, A.; Bayon, Y.; Reis, R.L. Marine origin collagens and its potential applications. Mar. Drugs 2014, 12, 5881–5901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.K.; Kim, Y.T.; Byun, H.G.; Nam, K.S.; Joo, D.S.; Shahidi, F. Isolation and characterization of antioxidative peptides from gelatin hydrolysate of Alaska pollack skin. J. Agric. Food Chem. 2001, 49, 1984–1989. [Google Scholar] [CrossRef] [PubMed]
- Mendis, E.; Rajapakse, N.; Byun, H.G.; Kim, S.K. Investigation of jumbo squid (Dosidicus gigas) skin gelatin peptides for their in vitro antioxidant effects. Life Sci. 2005, 77, 2166–2178. [Google Scholar] [CrossRef]
- Mendis, E.; Rajapakse, N.; Kim, S.K. Antioxidant properties of a radical-scavenging peptide purified from enzymatically prepared fish skin gelatin hydrolysate. J. Agric. Food Chem. 2005, 53, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Ngo, D.H.; Ryu, B.; Vo, T.S.; Himaya, S.W.; Wijesekara, I.; Kim, S.K. Free radical scavenging and angiotensin-I converting enzyme inhibitory peptides from Pacific cod (Gadus macrocephalus) skin gelatin. Int. J. Biol. Macromol. 2011, 49, 1110–1116. [Google Scholar] [CrossRef]
- Himaya, S.W.A.; Ngo, D.; Ryu, B.; Kim, S. An active peptide purified from gastrointestinal enzyme hydrolysate of Pacific cod skin gelatin attenuates angiotensin-1 converting enzyme (ACE) activity and cellular oxidative stress. Food Chem. 2012, 132, 1872–1882. [Google Scholar] [CrossRef]
- Subhan, F.; Kang, H.Y.; Lim, Y.; Ikram, M.; Baek, S.Y.; Jin, S.; Jeong, Y.H.; Kwak, J.Y.; Yoon, S. Fish scale collagen peptides protect against CoCl2/TNF-α-induced cytotoxicity and inflammation via inhibition of ROS, MAPK, and NF-κB pathways in HaCaT Cells. Oxid. Med. Cell. Longev. 2017, 2017, 9703609. [Google Scholar] [CrossRef] [Green Version]
- Kebede, M.; Admassu, S. Application of antioxidants in food processing industry: Options to improve the extraction yields and market value of natural products. Adv. Food Technol. Nutr. Sci. Open J. 2019, 5, 38–49. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, oxidative stress, and antioxidants: Back and forth in the pathophysiology of chronic diseases. Front. Physiol. 2020, 11, 694. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, S.C.; Moldão-Martins, M.; Alves, V.D. Antioxidants of natural plant origins: From sources to food industry applications. Molecules 2019, 24, 4132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peschel, W.; Sánchez-Rabaneda, F.; Diekmann, W.; Plescher, A.; Gartzía, I.; Jimenez, D.; Lamuela-Raventos, R.M.; Buxaderas, S.; Codina, C. An industrial approach in the search of natural antioxidants from vegetable and fruit wastes. Food Chem. 2006, 97, 137–150. [Google Scholar] [CrossRef]
- Mira-Sánchez, M.D.; Castillo-Sánchez, J.; Morillas-Ruiz, J.M. Comparative study of rosemary extracts and several synthetic and natural food antioxidants. Relevance of carnosic acid/carnosol ratio. Food Chem. 2020, 309, 125688. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Kim, H.P.; Hoetzel, A.; Park, J.W.; Nakahira, K.; Wang, X.; Choi, A.M. Mechanisms of cell death in oxidative stress. Antioxid. Redox Signal. 2007, 9, 49–89. [Google Scholar] [CrossRef]
- Liu, C.; Sun, J. Potential application of hydrolyzed fish collagen for inducing the multidirectional differentiation of rat bone marrow mesenchymal stem cells. Biomacromolecules 2014, 15, 436–443. [Google Scholar] [CrossRef]
- Choi, D.J.; Choi, S.M.; Kang, H.Y.; Min, H.J.; Lee, R.; Ikram, M.; Subhan, F.; Jin, S.W.; Jeong, Y.H.; Kwak, J.Y.; et al. Bioactive fish collagen/polycaprolactone composite nanofibrous scaffolds fabricated by electrospinning for 3D cell culture. J. Biotechnol. 2015, 205, 47–58. [Google Scholar] [CrossRef]
- Liu, C.; Xue, Y.; Sun, J. Hydrolyzed fish collagen inhibits inflammatory cytokines secretion in lipopolysaccharide-induced HUVECs. Adv. Mater. Res. 2014, 1025–1026, 570–573. [Google Scholar] [CrossRef]
- Koul, H.K.; Pal, M.; Koul, S. Role of p38 MAP kinase signal transduction in solid tumors. Genes Cancer 2013, 4, 342–359. [Google Scholar] [CrossRef]
- Marchesi, E.; Rota, C.; Fann, Y.C.; Chignell, C.F.; Mason, R.P. Photoreduction of the fluorescent dye 2′-7′-dichlorofluorescein: A spin trapping and direct electron spin resonance study with implications for oxidative stress measurements. Free Radic. Biol. Med. 1999, 26, 148–161. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, W.H.; Kim, H.-Y.; Lim, Y.S.; Hwang, S.Y.; Lee, C.; Lee, D.Y.; Moon, Y.; Song, Y.J.; Yoon, S. Fish Collagen Peptides Protect against Cisplatin-Induced Cytotoxicity and Oxidative Injury by Inhibiting MAPK Signaling Pathways in Mouse Thymic Epithelial Cells. Mar. Drugs 2022, 20, 232. https://0-doi-org.brum.beds.ac.uk/10.3390/md20040232
Song WH, Kim H-Y, Lim YS, Hwang SY, Lee C, Lee DY, Moon Y, Song YJ, Yoon S. Fish Collagen Peptides Protect against Cisplatin-Induced Cytotoxicity and Oxidative Injury by Inhibiting MAPK Signaling Pathways in Mouse Thymic Epithelial Cells. Marine Drugs. 2022; 20(4):232. https://0-doi-org.brum.beds.ac.uk/10.3390/md20040232
Chicago/Turabian StyleSong, Won Hoon, Hye-Yoon Kim, Ye Seon Lim, Seon Yeong Hwang, Changyong Lee, Do Young Lee, Yuseok Moon, Yong Jung Song, and Sik Yoon. 2022. "Fish Collagen Peptides Protect against Cisplatin-Induced Cytotoxicity and Oxidative Injury by Inhibiting MAPK Signaling Pathways in Mouse Thymic Epithelial Cells" Marine Drugs 20, no. 4: 232. https://0-doi-org.brum.beds.ac.uk/10.3390/md20040232