The Evolution of Our Understanding of Immunoproliferative Small Intestinal Disease (IPSID) over Time

 ,
,
Abstract
:1. Introduction
2. Epidemiology
3. Etiology and Pathophysiology
4. Clinical Features and Prognosis
5. Differential Diagnoses
6. Diagnostics and Investigations
6.1. Radiological Findings

{kind=link}
{kind=link}
{kind=link}
| Chemistry | Electrolyte imbalances (hypokalemia, hyponatremia, hypocalcemia, non-anion gap metabolic acidosis) [1,8,21]. |
| Low albumin. | |
| Elevated alkaline phosphatase. | |
| Hypocholesteremia [5]. | |
| Vitamin deficiencies (vitamin B12 and folate) [5]. | |
| Elevated lactate dehydrogenase (LDH), reported in advanced stages of lymphoma [11]. | |
| Hematology [1,8] | Complete blood count: mild anemia and leukocytosis. |
| Peripheral blood morphology can demonstrate plasmacytic infiltrates. | |
| Bone marrow biopsy rarely is involved and can show plasmacytosis and features of plasma cell leukemia [20]. | |
| Microbiology [1,8,12,21] | Stool cultures were stated in different reports to be positive for various organisms including Campylobacter jejuni, Vibrio fluvialis, Giardia. |
| Tissue culture was positive for E. coli in one case report. | |
| Immunology and Electrophoresis | Low or normal IgA levels with normal-to-high IgG and IgM levels [4]. |
| No Bence-Jones proteins in the urine [4]. | |
| Decreased or absent cellular and humoral responses. | |
| Immuno-electrophoresis and immunoselection detect α-heavy chain proteins in serum and body fluids, it is considered the most sensitive and specific method and is detected in almost 70% of cases [4,11]. | |
| Other | Positive stool occult [1]. |
| Sudan III stain of the stool positive, suggestive of malabsorption [18]. |
| Radiological Stage | Description |
|---|---|
| Stage I | Focal lymphoma involving the mucosa or submucosa or mesenteric lymph node. |
| Stage II | Lymphoma extension to the transmural layer and lymphadenopathy in several regions. |
| Stage III | Lymphoma involvement of the bowel and massive mesenteric lymph nodes. |
| Stage IV | Lymphoma involving the bowel and extra-mesenteric lymph nodes or parenchymatous organs. |
6.2. Esophagogastroduodenoscopy and Laparotomy
6.3. Histopathology and Immunohistochemistry Stains
- Diffuse infiltration by plasma cells solely or mixed with lymphocytes in the intestine’s mucosa at sites other than the neoplastic mass if present. The development of immunoblastic malignant lymphoma with detectable α-HC proteins in body fluids and tissues is commonly seen in association with this variant.
- Diffuse follicular lymphoid hyperplasia in the mucosa of the small intestine. This variant is commonly associated with diffuse undifferentiated malignant lymphomas and undetectable α-HC proteins.
6.4. Molecular and Cytogenetics
7. Atypical IPSID Entities
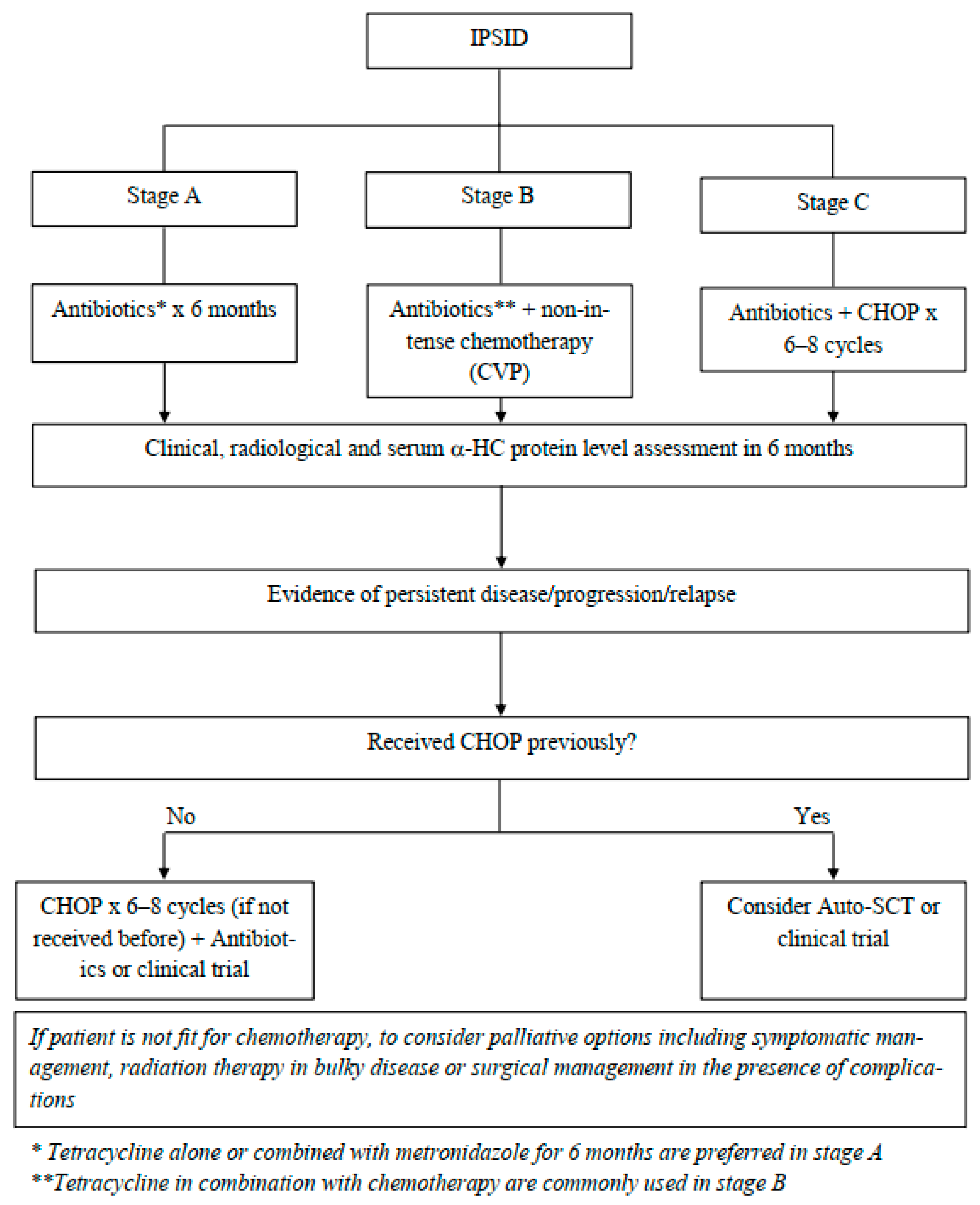
8. Management and Outcomes
8.1. Supportive Measurements
8.2. Pharmacological Therapy
8.3. Radiation Therapy
8.4. Surgical Intervention
8.5. Stem Cell Transplantation
8.6. Response to Therapy and Maintenance Therapy
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Evangelista-Leite, D.; Madaloso, B.A.; Yamashita, B.S.; Aloise, F.E.; Verdasca, L.P.; de Mello, M.L.; Chehter, E.Z. Treating chronic diarrhea: A systemic review on Immunoproliferative Small Intestinal Disease (IPSID). PLoS ONE 2021, 16, e0253695. [Google Scholar] [CrossRef] [PubMed]
- Pervez, S.; Mumtaz, K.; Siddiq Ullah, S.; Akhtar, N.; Ali, N.; Aaqil, H. Immunoproliferative Small Intestinal Disease (IPSID). J. Coll. Physicians Surg. Pak. 2011, 21, 57–58. [Google Scholar] [PubMed]
- AlSaleem, T.; AlMondhiry, H. Immunoproliferative small intestinal disease (IPSID): A model for mature B-cell neoplasms. Blood 2005, 105, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Gani, A.S.; AlMahtab, M.; Hossain, M.M.; Kamal, M.; Rahman, S. Immunoproliferative Small Intestinal Disease: First Case Report from Bangladesh. Euroasian J. Hepato-Gastroenterol. 2012, 2, 47–50. [Google Scholar]
- WHO Meeting Report. Alpha-chain Disease and Related Small-intestinal Lymphoma: A Memorandum. Bull. World Health Organ. 1976, 65, 615–624. [Google Scholar]
- AlJurf, M.D.; Owaidah, T.W.; Ezzat, A.; Ibrahim, E.; Tbakhi, A. Antigen- and/or immune-driven lymphoproliferative disorders. Ann. Oncol. 2003, 14, 1595–1606. [Google Scholar] [CrossRef]
- Salem, P.; ElHashmi, L.; Anassie, E.; Geha, S.; Habboubi, N.; Ibrahim, N.; Allam, C. Primary Small Intestinal Lymphoma in Adults: A comparative study of IPSID versus Non-IPSID in the Middle East. Cancer 1987, 59, 1670–1676. [Google Scholar] [CrossRef]
- Ewers, E.C.; Sheffler, R.L.; Wang, J.; Ngauy, V. Case Report: Immunoproliferative Small Intestinal Disease Associated with Overwhelming Polymicrobial Gastrointestinal Infection with Transformation to Diffuse Large B-cell Lymphoma. Am. J. Trop. Med. Hyg. 2016, 94, 1177–1181. [Google Scholar] [CrossRef] [Green Version]
- Lankarani, K.B.; Masoompour, S.M.; Malekzadeh, R.; Tabei, S.Z.; Haghshenas, M. Changing epidemiology of IPSID in southern Iran. Gut 2005, 54, 311–312. [Google Scholar] [CrossRef] [Green Version]
- Nath, L.C.; Bhattacharya, C.S.; Bharadwaj, C.R.; Chatterjee, C.T. Immunoproliferative small intestinal disease—a rare extranodal marginal zone lymphoma of mucosa associated lymphoid tissue in the Indian subcontinent. MJAFI 2011, 67, 277–279. [Google Scholar] [CrossRef] [Green Version]
- Malik, I.A. Immunoproliferative Small Intestinal Disease and Primary Small Intestinal Lymphoma: Review of Literature. J. Pak. Med. Assoc. 1997, 47, 215–218. [Google Scholar] [PubMed]
- Smith, W.J.; Price, S.K.; Isaacson, P.G. Immunoglobulin gene rearrangement in immunoproliferative small intestinal disease (IPSID). J. Clin. Pathol. 1987, 40, 1291–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecuit, M. Immunoproliferative small intestinal disease associated with Campylobacter jejuni. Haematol. Rep. 2005, 1, 66–69. [Google Scholar]
- Tommaso, N.D.; Gasbarrini, A.; Ponziani, F.R. Intestinal Barrier in Human Health and Disease. Int. J. Environ. Res. Public Health 2021, 18, 12836. [Google Scholar] [CrossRef] [PubMed]
- Wlodarska, M.; Kostic, A.D.; Xavier, R.J. An integrative view of microbiome-host interactions in inflammatory bowel diseases. Cell Host Microbe 2015, 17, 577–591. [Google Scholar] [CrossRef] [Green Version]
- Biswas, B.; Sharma, A.; Makharia, G.K.; Thulkar, S.; Arava, S.; Bahl, A.; Chaudhary, S. Immunoproliferative small intestinal disease: A report of 6 cases. Trop. Gastroenterol. 2014, 35, 269–272. [Google Scholar] [CrossRef]
- Afroza, S.; Zabin, M.; Uddin, M.M.; Ahmed, S.U. Immunoproliferative Small Intestinal Disease—A Case Report. Bangladesh Oncol. J. 2009, 4, 90–92. [Google Scholar]
- Ali, M.A.; Atiyeh, M.; Lewall, D.; Godwin, J.T.; Akhtar, M. Clinicopathologic conference case presentation. Ann. Saudi Med. 1983, 3. [Google Scholar] [CrossRef] [Green Version]
- Vessal, K.; Dutz, W.; Kohout, E.; Rezvani, L. Immunoproliferative Small Intestinal Disease with Duodenojejunal Lymphoma: Radiologic Changes. Am. J. Roentgenol. 1980, 135, 491–497. [Google Scholar] [CrossRef] [Green Version]
- Blumstein, M.; Bank, S.; Greenberg, R.E.; Abrol, R.P.; Kahn, L.; Siegal, F. Immunoproliferative Small Intestinal Disease in an American Patient with Lymphoma and Macroamylasemia. Gastroenterology 1992, 103, 1071–1074. [Google Scholar] [CrossRef]
- Imanzade, F.; Sayarri, A.; Tajik, P. Immunoproliferative Small Intestine Disease (IPSID): A Case Report. Int. J. Pediatr. 2015, 3, 89–92. [Google Scholar]
- Nassar, V.H.; Salem, P.A.; Shahid, M.J. Mediterranean abdominal lymphoma or immunoproliferative small intestinal disease. Part II: Pathological aspects. Cancer 1978, 41, 1340. [Google Scholar] [CrossRef] [Green Version]
- Galian, A.; Lecestre, M.J.; Scotto, J.; Bognel, C.; Matuchansky, C.; Rambaud, J.C. Pathological study of alpha-chain disease, with special emphasis on evolution. Cancer 1977, 39, 2081–2101. [Google Scholar] [CrossRef]
- Ben Ayed, F.; Halphen, M.; Najjar, T.; Boussene, H.; Jaafoura, H.; Bouguerra, A.; Tufrali, G. Treatment of alpha chain disease. Result of a prospective study in 21 Tunisian patients by the Tunisian-French Intestinal Lymphoma Study Group. Cancer 1989, 63, 1251–1256. [Google Scholar] [CrossRef]
- Nasir, U.M.; Paer, J.M.; Srialluri, N.; Panchal, D.; Ghavimi, S.; Wang, W. A Rare Presentation of Immunoproliferative Small Intestinal Disease. Am. J. Gastroenterol. 2020, 115, 1500–1501. [Google Scholar] [CrossRef]



| Stage | Type of Cellular Infiltrate and Histopathological Description | Mesenteric Nodal Involvement |
|---|---|---|
| Stage A (Benign) | Heavy infiltrations of lamina propria with typical lymphocytes with few dysplastic plasma cells infiltrate with variable atrophic villi in the small intestine. | Few CD20-positive marginal zone B cells with plasmacytic infiltration of mesenteric or other abdominal and retroperitoneal lymph nodes with limited disorganization of histological structure. |
| Stage B (Intermediate) | Infiltration extending beyond the mucosa with atypical lymphoplasmacytic cells, immunoblastic-like cells with the areas remote from the mucosa, containing many dysplastic cells with total or subtotal villous atrophy. | Atypical plasmacytic and immunoblastic dense infiltrations causing structural changes of the mesenteric and abdominal lymph node construction. |
| Stage C (Malignant) | Immunoblastic high-grade B-cell lymphomas, some with strong CD20-positivity with plasmacytoid differentiation and proliferative histocytes extending into all layers of the intestinal wall and some forming confined large tumors of malignant formations. | Mesenteric and abdominal lymph nodes with sarcomatous proliferation alter the entire structure. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AlYamany, R.; Kharfan-Dabaja, M.A.; Hamadani, M.; Alshaibani, A.; Aljurf, M. The Evolution of Our Understanding of Immunoproliferative Small Intestinal Disease (IPSID) over Time. Curr. Oncol. 2022, 29, 3759-3769. https://0-doi-org.brum.beds.ac.uk/10.3390/curroncol29050301
AlYamany R, Kharfan-Dabaja MA, Hamadani M, Alshaibani A, Aljurf M. The Evolution of Our Understanding of Immunoproliferative Small Intestinal Disease (IPSID) over Time. Current Oncology. 2022; 29(5):3759-3769. https://0-doi-org.brum.beds.ac.uk/10.3390/curroncol29050301
Chicago/Turabian StyleAlYamany, Ruah, Mohamed A. Kharfan-Dabaja, Mehdi Hamadani, Alfadel Alshaibani, and Mahmoud Aljurf. 2022. "The Evolution of Our Understanding of Immunoproliferative Small Intestinal Disease (IPSID) over Time" Current Oncology 29, no. 5: 3759-3769. https://0-doi-org.brum.beds.ac.uk/10.3390/curroncol29050301