Transcriptomic Identification of Floral Transition and Development-Associated Genes in Styrax japonicus


College of Landscape Architecture and Forestry, Qingdao Agricultural University, Qingdao 266109, China
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Forests 2020, 11(1), 10; https://0-doi-org.brum.beds.ac.uk/10.3390/f11010010
Submission received: 24 October 2019
/
Revised: 3 December 2019
/
Accepted: 16 December 2019
/
Published: 19 December 2019
(This article belongs to the Section Forest Ecophysiology and Biology)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Styrax japonicus (S. japonicus) is an important flowering tree species in temperate regions, and it is regarded as a nectariferous plant. However, there have been few studies to date analyzing floral development in this species. In order to understand gene expression dynamics during S. japonicus flower development, we; therefore, prepared cDNA libraries from three distinct stages of S. japonicus. Illumina sequencing generated 31,471 differentially expressed unigenes during flower development. We additionally conducted pathway enrichment analyses using the GO and KEGG database in order to assess the functions of genes differentially expressed during different stages of the floral development process, revealing these genes to be associated with pathways including phytohormone signaling, Transcription factor, protein kinase, and circadian rhythms. In total, 4828 TF genes, 8402 protein kinase genes, and 78 DEGs related to hormone pathways were identified in flower development stages. Six genes were selected for confirmation of expression levels using quantitative real-time PCR. The gene expression data presented herein represent the most comprehensive dataset available regarding the flowering of S. japonicus, thus offering a reference for future studies of the flowering of this and other Styracaceae species.
1. Introduction
Styrax japonicus (S. japonicus) is a deciduous species within the Styracaceae family that is distributed from the Qinling Mountains to the south of the Yellow River in China [1]. S. japonicus is an important flowering tree species in temperate regions, where it regarded as a nectariferous plant. Owing to its ornamental value and practical characteristics, S. japonicus is widely used in city greening efforts. However, there have been relatively few studies conducted to date regarding the basic biology and genetics of this species, with previous molecular biology studies on this plant only focused on the development of simple sequence repeat markers [2].
In higher plants, flower development is a key life cycle stage, especially in ornamental plants, and it is influenced by both endogenous and exogenous factors that modulate gene expression [3]. A great deal of research has been conducted examining the flowering mechanisms of model plants, but comparatively little research has focused on woody flowering plants [4]. Unlike annual plants, perennial woody plants bloom only under favorable conditions, with some meristematic tissues transforming into flower organs, while other meristematic tissues remain vegetative. In temperate regions, most perennial plants flower seasonally, including S. japonicus, which blooms in the spring. In Arabidopsis, there are six key pathways regulating floral development: The photoperiod, autonomous, vernalization, gibberellin (GA), ambient temperature, and age pathways [5]. Several key transcription factors (TFs) regulate floral development, including FLOWERING LOCUST (FT), CONSTANS (CO), SUPPRESSOR OF OVEREXPRESSION OF CO1 (SOC1), and FLOWERING LOCUS C (FLC). These floral integrators trigger floral meristem identity genes, including LEAFY (LFY), APETALA1 (AP1), and CAULIFLOWER (CAL) [6,7,8,9], which then determine whether a given tissue will undergo the transition from vegetative to reproductive growth.
Vernalization is the process whereby plants are exposed to cold for extended periods of time in order to complete their floral transformation [10]. Photoperiod regulation and vernalization are the primary means of preventing early flowering. GAs can promote cell growth, but studies have shown that GA can also induce flowering in many plants [11,12]. In the woody flowering plant Amygdalus persica, GA treatment can inhibit flower bud differentiation [13]. In sugar apples, GA, abscisic acid (ABA), and cytokinin (CK) have all been found to play key roles in flower development [14]. In Agapanthus praecox, GAs can promote early flowering, while IAA can delay this flowering [15]. Studies of aspen trees have found that CO mRNA accumulation peaks at the end of the day, and that CO is able to bind to the FT promoter and to thereby induce its expression, resulting in a circadian pattern of FT expression [16]. Diurnal CO and FT expression have significant effects on the growth and development of certain plants [17]. Indeed, many similar studies have been carried out in species including Cymbidium sinense, Citrus limon, and Vernicia fordii [18,19,20].
In the present study, we used RNA-seq technology to construct cDNA libraries corresponding to the three different flowering stages in S. japonicus, yielding a dataset that was sufficiently comprehensive to allow us to identify all flowering-related genes and major metabolic pathways encoded in the transcriptome of these plants. Overall our annotation of the S. japonicus transcriptome and analysis of differential gene expression profiles in these plants serve as a valuable genomic resource for future studies of the flowering mechanisms of S. japonicus and related species.
2. Materials and Methods
Samples of S. japonicus were obtained from the Rongcheng Donglin Seedling planting cooperative (37°25′12″ N, 122°17′40″ E). Samples obtained included small buds (1–2 mm) (C1), flower buds (7–9 mm) (C2), and open flowers (C3). Three groups of samples were taken for each of these three periods and from three plants. The samples were snap frozen prior to storage at −80 °C. Paraffin sections preparation and RNA extraction were performed at the same time (Figure 1). Samples from the C1, C2, and C3 stages were collected on 12 March 2017, 10 April 2017, and 25 April 2017, respectively.
Total RNA was extracted from these developing flower tissues using TRIzol (Promega, Beijing, China). Equal quantities of three RNA samples for each stage of floral development were mixed prior to treatment with DNase I (Takara, Dalian, China) for 30 min. Agarose gel electrophoresis was used to assess RNA quality, while a 2100 Bioanalyzer device was used for assessing RNA quality and concentrations. High-quality samples with 260/280 nm values between 1.8 and 2.0 were used for library preparation. Briefly, oligo (dT) was used to separate total RNA, which was then fragmented into −200 nt fragments in fragmentation buffer. These mRNA fragments were then used as templates for first-strand cDNA synthesis using random primers and reverse transcriptase. The second cDNA strand was synthesized from this first cDNA strand. AMPure XP beads were then used to purify double-stranded cDNA, and these cDNA fragments were then subjected to purification, end pairing, and a single adenine residue was added to the end of each fragment prior to Illumina adapter ligation. Following ligation, these fragments were separated via agarose gel electrophoresis, with appropriately sized fragments being collected for PCR amplification prior to sequencing on an Illumina HiSeq 2500 platform, with sequencing being performed by Gene De-novo Co. (Guangzhou, China). The transcriptomic sequencing dataset for S. japonicus produced in the present study is available from the NCBI Short Read Archive (SRA) with the accession number SRR7888585-7888593.
Raw high throughput sequencing data were filtered to yield high-quality clean data. Initial filtering focused on the removal of reads that contained adapters, were of low quality (>20% base mass value < 10), had >5% unknown bases, or were <60 nucleotides long. The remaining sequences were then assembled into contigs using the Trinity application [21], yielding a transcriptome reference database. Clean reads next underwent de novo splicing for transcript assembly, and the longest sequence of a given transcript was considered to be a unique gene (“Unigene”). The BLASTX alignment algorithm was then used to align these unigenes to proteins with an E-value ≤ 1 × 10−5 [22]. Differentially expressed genes were analyzed based upon fragments per million bases (FPKM) values, with the Benjamini and Hochberg method used for p-value correction (false discovery rate, FDR) [23].
A cuffdiff analysis was used to analyze levels of differentially expressed genes (DEG) in these samples, with levels being used to map DEG tags, after which the transcriptomic data were filtered to remove tags that had only a single copy, tags that were of low quality, tags with unknown sequences, and empty tags corresponding only to adapter sequences. The number of mapped clean reads for each unigene was then determined and normalized based upon FPKM numbers in order to calculate unigene expression [24].
RT-qPCR was then used to identify the relative expression levels of the candidate genes during each floral developmental stage. RT-qPCR was conducted using the SYBR Green (TaKaRa, Beijing, China) fluorescent dye on an ABI 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The RT-qPCR primers were designed by Primer Premier 5.0 software. GAPDH was selected as the reference gene. Each sample was analyzed in three technical replicates. Relative expression levels were calculated using the 2−ΔΔCt method [25].
3. Results
3.1. Differential Gene Expression during Floral Development
The basic transcriptome information of S. japonicus have been reported in a previous study [2]. By comparing unigene transcript levels in samples from these three stages of floral development, we were able to explore the transcriptomic dynamics of this flowering process. We used a false discovery rate (FDR) ≤ 0.05 and an absolute log2 ratio value ≥ 1 as criteria to identify DEGs. Using this approach, a total of 14,763 unigenes were differentially expressed between the C1 and C2 stages (7879 up- and 6884 down-regulated, respectively), while 21,617 unigenes were differentially expressed between the C1 and C3 stages (9967 up- and 11,650 down-regulated, respectively) and 15,401 genes were differentially expressed between the C2 and C3 stages (6569 up- and 8830 down-regulated, respectively) (Figure 2). There were; thus, the fewest differentially expressed genes between stages C1 and C2, suggesting that of the three analyzed stages of floral development, stages C1 and C2 are the most similar.
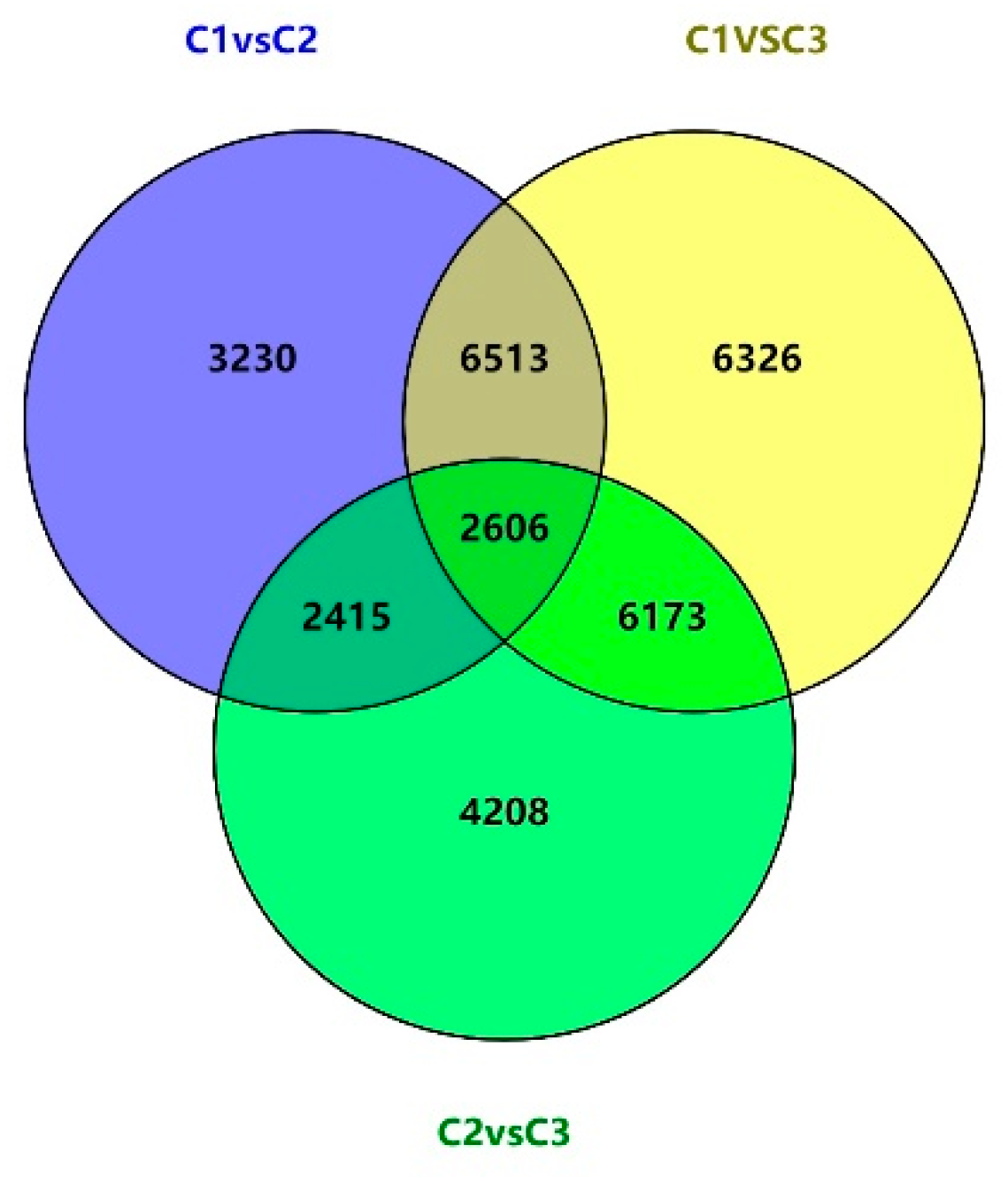
Gene expression differences in different flowering stages were next compared using a Venn diagram (Figure 3). Among the 31,471 total differentially expressed unigenes, 17,707 (56.26%) were shared across at least two comparisons, and 2606 (8.3%) were differentially expressed across all three comparisons. The overlap between the C1 vs. C2 and C1 vs. C3 comparisons consisted of 9119 (28.9%) unigenes, while there were 5021 (15.9%) unigenes overlapping between C1 vs. C2 and C2 vs. C3, and 8779 (27.8%) unigenes overlapping between C1 vs. C3 and C2 vs. C3. During these three stages of floral development, 957 unigenes were progressively up-regulated, while 598 were progressively down-regulated. This suggests that these genes may play a key role in regulating the growth and reproduction of this species.
3.2. GO and KEGG Annotation of DEGs
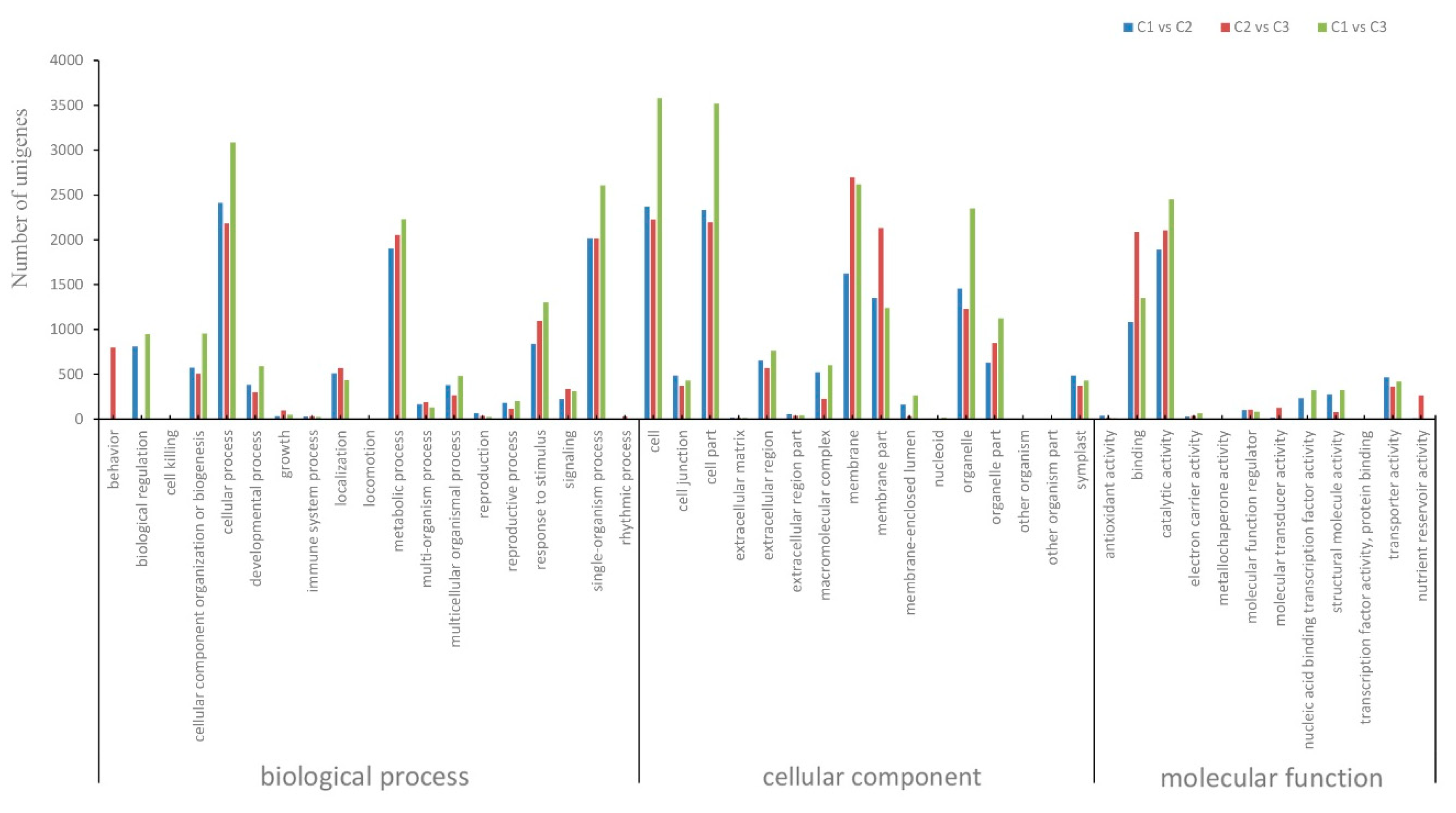
Further GO annotation of DEGs in the present study allowed for their annotation into three categories, including cellular components (15 subgroups), molecular functions (15 subgroups), and biological processes (21 subgroups) (Figure 4, Table S1). The five largest subgroups of DEGs up-regulated between stages C1 and C3 were cellular processes (1197, 3.8%), membrane (1150, 3.2%), membrane part (1106, 3.1%), catalytic activity (1256, 3.5%), and single-organism processes (1102, 3.1%). The five subgroups with the most significant down-regulation between these stages were cellular processed (1889, 5.3%), metabolic processes (1583, 4.5%), cell (2851, 8.0%), cell part (1197, 3.3%), and organelles (1978, 5.6%).
The KEGG Orthology-Based Annotation System (KOBAS, Vienna, Austria) [26] was used for the further identification of biosynthetic and other pathways and to explore the functions of these DEGs in-depth. KEGG annotation of these DEGs indicated that they were involved in 82 pathways (Table S2). KEGG enrichment analysis further revealed that, when comparing the initial and final stages of flower development (C1 vs. C3), DEGs differed significantly with respect to their enrichment in the Terpenoid backbone biosynthesis (p = 0.042), ubiquitin mediated proteolysis (p = 0.041), ABC transporter (p = 0.041), pentose and glucuronate interconversion (p = 0.037), and glyoxylate and dicarboxylate metabolism pathways (p = 0.035).
3.3. Differential Expression of Transcription Factors and Protein Kinases
Transcription factors (TFs) and protein kinases are known to play major roles in regulating flower development. Therefore, we studied the expression dynamics of TF genes in S. japonicus. In total, 4828 TF genes and 8402 protein kinase genes were identified in flower development stages. The 10 most predominant TF families identified during floral development included the basic helix-loop-helix, NAC, B3, MYB-related, bZIP, WRKY, ERF, FAR1, C2H2, and MYB TF families, with 626, 479, 45, 10, 58, 357, 347, 428, 25, and 422 members, respectively, identified via de novo transcriptome assembly (Figure 5A). Among these, some TF families were significantly up-regulated. For example, the bHLH, MYB, and bZIP families were significantly up-regulated after the C1 stage, while the NAC and C2H2 families were up-regulated after the C2 stage. In contrast, other TF families, such as WRKY, ERF, and FAR, were significantly down-regulated at these same time points. A heat map depicting the trends in TF family gene expression during flower development was constructed using the MeV software (Dana-Farber Cancer Institute and Harvard School of Public Health, Boston, USA). These results suggested that complex transcriptional reprogramming is involved in S. japonicus flower development (Figure S1).
The most abundant group of protein kinases in these sequenced samples were members of the serine/threonine-protein kinase family (3387, 40%). Other common protein kinases in these plant samples included receptor-like protein kinases (1673, 20%), LRR receptor-like serine/threonine-protein kinases (1093, 13%), and CBL-interacting protein kinases (306, 4%). We also found that CBL-interacting serine/threonine-protein kinases (305, 4%) and Wall-associated receptor kinases (227, 3%), which have been reported to involved in flower development and maturation [27,28], were also highly abundant in our transcriptomic dataset (Figure 5B). These results suggested that complex transcriptional reprogramming is involved in S. japonicus floral growth and development.
3.4. Transcriptome Analysis of Phytohormone Pathway Genes during Flower Development
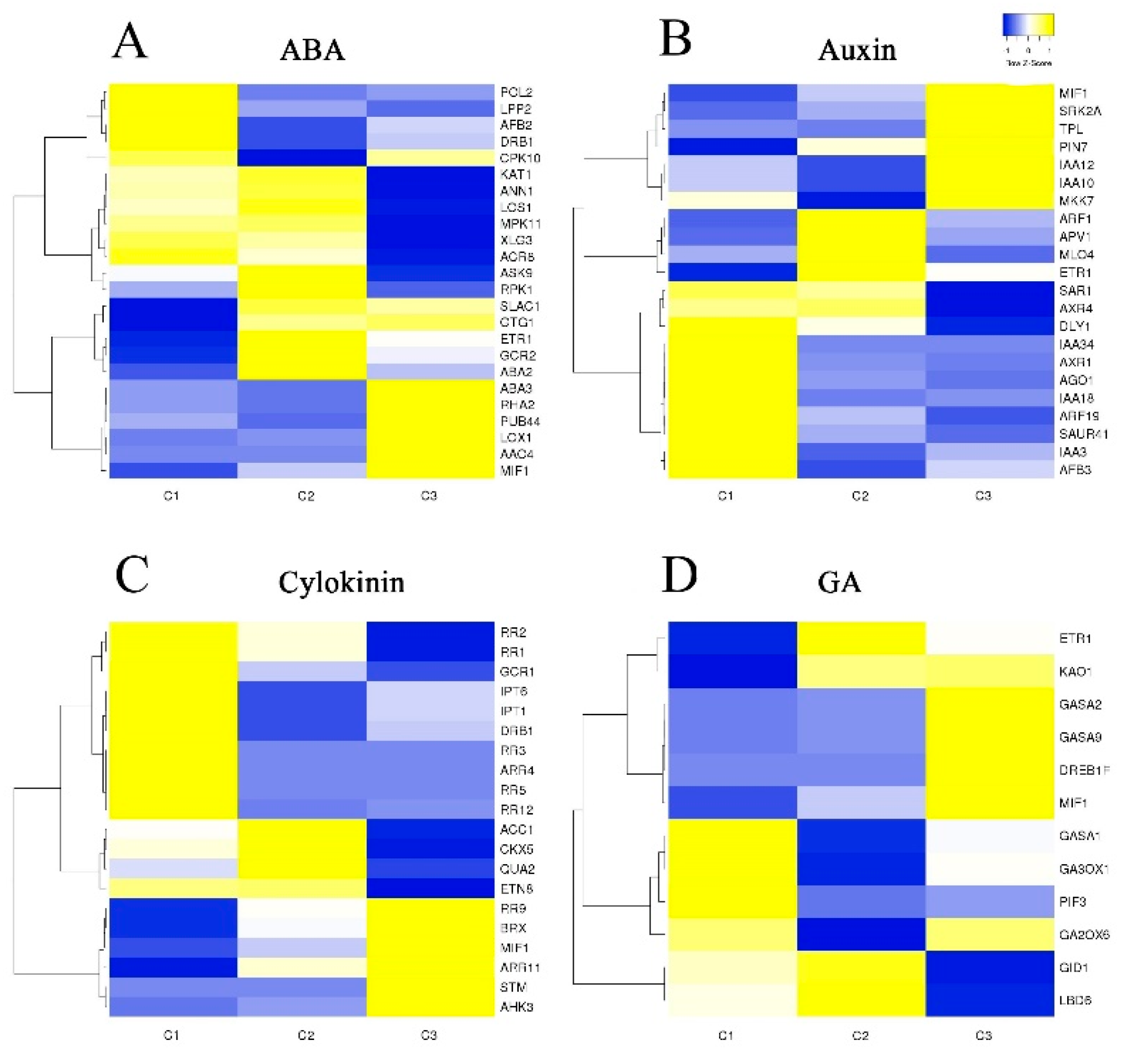
Several phytohormones, including auxin (AUX), ABA, GA, and cytokinin (CTK), are closely associated with flower transition and development. In order to investigate the relationship between phytohormones and flower development in S. japonicus, changes in the expression of unigenes involved in phytohormone signaling pathways were next analyzed (Figure 6). Hormones including AUX, ABA, CTK, and GA serve as internal cues that govern petal expansion and flower development. In our transcriptomic dataset, we identified 78 DEGs related to these hormone pathways which were arranged into a heat map (Figure 6). A total of 24 floral unigenes were annotated as being associated with the ABA signaling pathway, of which 15 were expressed at higher levels in stages C2 and C3. In addition, we identified 22 DEGs that were involved in the AUX signaling pathway, 20 that were involved in the CTK signaling pathway, and 12 that were involved in the GA signaling pathway. These genes exhibited strong and specific expression patterns during flower development. MIF1 is involved in the ABA, GA, CTK, and AUX hormone pathways. Four genes in the ABA signaling pathway were also involved in the AUX signaling pathway (MIF1, DRB1, ETR1, ABA3). There were additionally two overlapping genes between the AUX and GA signaling pathways (MIF1, ETR1), and two overlapping genes between the ABA and CTK signaling pathways (DRB1, MIF1).
3.5. RT-qPCR Validation of Selected Flowering-Related Genes
To confirm the reliability of our RNA-Seq data, we additionally performed RT-qPCR assays in order to analyze gene expression in independent samples collected from these S. japonicus flowers during different developmental stages (C1, C2, and C3). Six flowering-related genes were selected for these RT-qPCR analyses. We found that the expression levels of these selected genes were strongly correlated with our RNA-seq results, verifying the reliability of our RNA-Seq data (Figure 7).
4. Discussion
4.1. Transcription Factors Associated with Floral Development
TFs are key regulators of flower development; therefore, we assessed their expression dynamics during floral development in S. japonicus. MYB genes contain DNA binding domains that can control plant growth and development [29]. In Arabidopsis, stamen maturation is regulated by several MYB genes, with MYB108 and MYB24 acting similarly downstream of MYB21 to facilitate transcriptional events leading to stamen and pollen maturation [30]. Recently, the apple R2R3-MYB transcription factor MdMYB1 was found to be a master regulator of anthocyanin biosynthesis and fruit coloration [31]. C2H2 zinc finger (C2H2-ZF) proteins are a large gene family in plants that participate in various aspects of plant growth and development. C2H2 genes can be expressed in carpel primordium of flower buds and regulate stamen development [32]. Members of the WRKY family modulate many biological processes, including responses to biotic and abiotic stress, dormancy, and development [33]. With respect to the regulation of flower development, WRKY71 accelerates flowering in A. thaliana via its activation of FT and the floral meristem identity gene LEAFY [34]. During S. japonicus floral development, we found certain TFs to be substantially up-regulated. For example, the bHLH, MYB, and bZIP TF families were significantly up-regulated between stages C2 and C3, whereas the NAC and C2H2 family members were expressed reached to the highest in stage C3. In contrast, the WRKY, ERF, and ARF family members were down-regulated.
4.2. Time-Associated Gene Expression during S. japonicus Flowering
In woody flowering plants, photoperiod regulation is the most important pathway governing the flowering process, and it consists of three modules: Photoreceptors, a circadian clock, and a flowering-specific signaling pathway that is linked to the activity of this circadian clock [30]. Light signals are received by phytochromes (PHYA, PHYB, PHYC, PHYD, PHYE) and cryptochromes (CRYl, CRY2), creating an internal circadian clock that coordinates plant activity [35,36]. In Dimocarpus longan, the circadian clock can measure changes in day length so as to regulate the expression of FKF1 and GI [37], which work together to drive the up-regulation of the TF GO that in turn drives FT and SOC1 expression. ESD4 further regulates SUMO conjugation, thereby controlling the time of flowering [38]. The synthesis of the protein kinase WNK1 is also regulated by circadian rhythms [39]. The FVE, FPA, and FY genes all affect plant circadian rhythms and function in a manner similar to the FCA genes [40,41]. LD serves primarily to repress FLC expression, while FVE, FPA, and FY can all repress FLC and serve as positive regulators of the integration pathway [42]. These genes are redundant in the context of voluntary floral development. As experiments have shown that FLC insertion mutations do not change flowering time [43], FLC has been determined not to be the master regulator of this process. In the present study, we identified genes homologous to LD, FRI, FPA, and FLC in our S. japonicus transcriptome database. We also detected many unigenes associated with the circadian rhythm process, which is a key regulator of flowering. During the C1 stage, we observed the highest expression of the CK2α, CK2β, COP1, CHE, and FT, genes, whereas the genes encoding PHYA, CRY, PRR3, EARLY FLOWERING 3, and TOC1, were most highly expressed during stage C2, and those encoding PHYB, LHY, PRR9, and GI were most highly expressed during stage C3.
4.3. Identification of Hormone Unigenes Related to Flowering Stage
Through KEGG pathway enrichment analyses, we determined that genes differentially expressed during S. japonicus flowering were significantly enriched in hormone metabolism and signal transduction pathways. Auxin plays an important role in flower development, especially in regulating the formation of flower primordium and the transformation of flower organs [44]. During the flowering of S. japonicus, auxin-related unigene levels changed significantly, with several genes linked to ARFs and Aux/IAAs being differentially expressed at different time points. IAA10 is an early mediator of auxin regulation [45]. In S. japonicus, peak IAA10 expression was observed during the C3 stage of flower development. This suggests that IAA10 may play a role in flower maturation. Many ARFs are known to be involved in floral development. For example, in Arabidopsis, ARF3 integrates AGAMOUS and APETALA2 functions during floral meristem determinacy [46]. Two other members of the ARF family, ARF6 and ARF8, have been shown to play some role in controlling vegetative and floral organ growth and development. We found that in S. japonicus, ARF1 was highly expressed primarily during the C2 stage, while ARF19 was highly expressed during the C1 stage. This suggests that ARF19 and ARF6 play different roles in flower development.
Studies have shown that certain auxin transporter genes are important mediators of floral development [47,48]. The regulation of auxin flux by ATPIN1, for example, is known to impact male and female gamete development in Arabidopsis [49]. ATPIN1 expression is controlled by ATMLO4, which encodes a heptahelical protein found on the plasma membrane [50]. In this study, we identified the PIN7 and MLO4 auxin transporter genes. Interestingly, the expression levels of PIN7 increased during flower development in S. japonicus, while MLO4 expression exhibited a trend of first increasing and then decreasing.
GA, ABA, and cytokinin are also key mediators of floral development [51]. GA acts primarily through the DELLA proteins GIBBERELLIC ACID INSENSITIVE (GAI), REPRESSOR of ga1-3 (RGA), RGA-LIKE1 (RGL1), RGL2, and RGL3 [52]. ABA can promote flower bud differentiation in some woody plants [53]. Cytokinin drives the flowering of Arabidopsis via regulating the expression of TSF, which is paralogous to FT [54]. GA2OX family genes, encoding the 2oxoglutarate-dependent dioxygenases that catalyze later steps in the biosynthetic pathway of GA, have been identified in different plant species [55]. Interestingly, the expression of the GA biosynthesis gene GA3OX1 was down-regulated in this study, while that of GA2OX6 was initially down-and then up-regulated during the flowering process in this study, reaching peak expression at stage C3. These differential expression dynamics of these GA2OX family members suggest that diverse GA biosynthesis mechanisms are engaged during floral development in S. japonicus. As further research regarding the mechanisms of flowering is required, trends in hormone gene expression and the specific functions of these genes in S. japonicus flowering remain to be further clarified. Furthermore, the MIF1 and ETR1 regulators are involved in the auxin and ABA signaling pathways. ETR1 is regulated by both GA and auxin signaling, while MIF1 is controlled by ABA and cytokinin signaling.
5. Conclusions
In conclusion, by generating and sequencing transcriptomic libraries for three different stages of S. japonicus flowering, we were able to identify patterns of differential gene expression during the flowering process. Based on the results of GO and KEGG pathway analyses, we were able to highlight particular patterns of gene expression that were linked to this floral development process. We further focused on the identification of particular TFs, circadian rhythm-associated genes, genes linked with flowering time, and hormone-related genes that were engaged during the flowering process. Some of these key genes were further selected for RT-qPCR-mediated confirmation of their expression levels. Together these identified genes will help to support future efforts to more thoroughly understand the molecular basis for the flowering of S. japonicus.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/1999-4907/11/1/10/s1. Figure S1: Identification and analysis of floral transition and flower development-associated transcription factor genes, Table S1: GO annotation results of differentially expressed genes in three stages, Table S2: KEGG annotation results of differentially expressed genes in three stages.
Author Contributions
K.W. and C.Z. conceived and designed the experiments; W.L. performed the experiments, conducted the data analysis. and Z.X. wrote the manuscript; X.J. revised the manuscript and contributed the plant material. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Collection and Protection of Featured, Rare and Endangered Forest Tree Germplasm Resources [2016LZGC038].
Acknowledgments
We thank the National Forest Genetic Resources Platform (NFGRP) for providing the Styrax japonicus resources.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wang, X.Y.; Yu, S.; Liu, M.; Yang, Q.-S.; Chen, X.-Y. Twenty-three microsatellite loci for Styrax confusus and Styrax japonicus (Styracaceae). Conserv. Genet. Resour. 2010, 2, 51–54. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.P.; Jiang, X.Q.; Liu, Q.C.; Liu, Q.H.; Wang, K. De Novo Transcriptomic Analysis and Development of EST–SSRs for Styrax japonicus. Forests 2018, 9, 748. [Google Scholar] [CrossRef] [Green Version]
- Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, J.; Schemske, D.W. Pathways, Mechanisms, and Rates of Polyploid Formation in Flowering Plants. Annu. Rev. Ecol. Syst. 1998, 29, 467–501. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, J.; Jie, Z.; Zhao, J.; Ren, X. Function of protein ubiquitination and SUMOylation in regulating flowering time of plants: A review. J. Zhejiang Univ. 2015, 41, 371–384. [Google Scholar]
- Mizoguchi, T. Distinct Roles of GIGANTEA in Promoting Flowering and Regulating Circadian Rhythms in Arabidopsis. Plant Cell 2005, 17, 2255–2270. [Google Scholar] [CrossRef] [Green Version]
- Imaizumi, T.; Tran, H.G.; Swartz, T.E.; Briggs, W.R.; Kay, S.A. FKF1 is essential for photoperiodic-specific light signalling in Arabidopsis. Nature 2003, 426, 302–306. [Google Scholar] [CrossRef]
- Morris, K.; Jackson, S.P. DAY NEUTRAL FLOWERING does not act through GIGANTEA and FKF1 to regulate CONSTANS expression and flowering time. Plant Signal. Behav. 2010, 5, 1105–1107. [Google Scholar] [CrossRef] [Green Version]
- Han, P.; García-Ponce, B.; Fonseca-Salazar, G.; Alvarez-Buylla, E.R.; Yu, H. AGAMOUS-LIKE 17, a novel flowering promoter, acts in a FT-independent photoperiod pathway. Plant J. 2008, 55, 253. [Google Scholar] [CrossRef]
- Kim, D.H.; Doyle, M.R.; Sung, S.; Amasino, R.M. Vernalization: Winter and the Timing of Flowering in Plants. Annu. Rev. Cell Dev. Biol. 2009, 25, 277–299. [Google Scholar] [CrossRef] [Green Version]
- Mutasa-Gottgens, E.; Qi, A.; Mathews, A.; Thomas, S.; Phillips, A.; Hedden, P. Modification of gibberellin signalling (metabolism & signal transduction) in sugar beet: Analysis of potential targets for crop improvement. Transgenic Res. 2009, 18, 301–308. [Google Scholar] [PubMed]
- Foley, M.E.; Anderson, J.V.; Horvath, D.P. The effects of temperature, photoperiod, and vernalization on regrowth and flowering competence in Euphorbia esula (Euphorbiaceae) crown buds. Botany 2009, 87, 986–992. [Google Scholar] [CrossRef] [Green Version]
- An, L.; Jin, L.; Yang, C.; Li, T. Effect and Functional Mechanism of Exogenous Gibberellin on Flowering of Peach. Sci. Agric. Sin. 2009, 42, 605–611. [Google Scholar]
- Liu, K.; Feng, S.; Pan, Y.; Zhong, J.; Chen, Y.; Yuan, C.; Li, H. Transcriptome Analysis and Identification of Genes Associated with Floral Transition and Flower Development in Sugar Apple (Annona squamosa L.). Front. Plant Sci. 2016, 7, 1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Ren, L.; Yue, J.H.; Wang, L.; Shen, X.H. GA4 and IAA were involved in the morphogenesis and development of flowers in Agapanthus praecox ssp. orientalis. J. Plant Physiol. 2014, 171, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Bohlenius, H. CO/FT Regulatory Module Controls Timing of Flowering and Seasonal Growth Cessation in Trees. Science 2007, 312, 1040–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.F. Overexpression of COL9, a CONSTANS-LIKE gene, delays flowering by reducing expression of CO and FT in Arabidopsis thaliana. Plant J. 2010, 43, 758–768. [Google Scholar] [CrossRef]
- Li, J.X.; Hou, X.J.; Zhu, J.; Zhou, J.J.; Huang, H.B.; Yue, J.Q.; Gao, J.Y.; Du, Y.X.; Hu, C.X.; Hu, C.G.; et al. Identification of Genes Associated with Lemon Floral Transition and Flower Development during Floral Inductive Water Deficits: A Hypothetical Model. Front. Plant Sci. 2017, 8, 1013. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Liu, W.; Chen, X.; Xu, Y.; Lu, W.; Hou, J.; Ni, J.; Wang, Y.; Wu, L. Flower Development and Sex Determination between Male and Female Flowers in Vernicia fordii. Front. Plant Sci. 2017, 8, 1291. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Fu, J.; Du, H.; Hu, J.; Wuyun, T. De novo sequencing of Eucommia ulmoides flower bud transcriptomes for identification of genes related to floral development. Genom. Data 2016, 9, 105–110. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langdon, B.W. Performance of genetic programming optimised Bowtie2 on genome comparison and analytic testing (GCAT) benchmarks. Biodata Min. 2015, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainer, H.; Sireci, S.G.; Thissen, D. Differential Testlet Functioning: Definitions and Detection. J. Educ. Meas. 1991, 28, 197–219. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.; Schmittgen, T. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−△△Ct Method. Methods 2000, 25, 402–408. [Google Scholar] [CrossRef]
- Mao, X.; Tao, C.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]
- Kato, R.; Fujii, T. Increase in the Activities of Protein Kinases under a Flower-Inducing Condition in Lemna paucicostata. Plant Cell Physiol. 1988, 29, 85–88. [Google Scholar]
- Lease, K.A.; Lau, N.Y.; Schuster, R.A.; Torii, K.U.; Walker, J.C. Receptor serine/threonine protein kinases in signalling: Analysis of the erecta receptor-like kinase of Arabidopsis thaliana. New Phytol. 2001, 151, 133–143. [Google Scholar] [CrossRef]
- Vimolmangkang, S.; Han, Y.; Wei, G.; Korban, S.S. An apple MYB transcription factor, MdMYB3, is involved in regulation of anthocyanin biosynthesis and flower development. BMC Plant Biol. 2013, 13, 176. [Google Scholar] [CrossRef] [Green Version]
- Browse, M.J. MyB108 Acts Together with MyB24 to Regulate Jasmonate-Mediated Stamen Maturation in Arabidopsis. Plant Physiol. 2009, 149, 851–862. [Google Scholar]
- Takos, A.M.; Jaffe, F.W.; Jacob, S.R.; Bogs, J.; Robinson, S.P.; Walker, A.R. Light-Induced Expression of a MYB Gene Regulates Anthocyanin Biosynthesis in Red Apples. Plant Physiol. 2006, 142, 1216–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zhanchao, W.; Xuemei, X.; Haizhen, Z.; Chenghao, L.; Manoj, P. Genome-Wide Analysis of C2H2 Zinc-Finger Family Transcription Factors and Their Responses to Abiotic Stresses in Poplar (Populus trichocarpa). PLoS ONE 2015, 10, e0134753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.N.; Liu, Y.; Xin, Z.-Z.; Zhang, D.-Z.; Ge, B.-M.; Yang, R.-P.; Wang, Z.-F.; Yang, L.; Tang, B.-P.; Zhou, C.-L. Genome-wide identification and characterization of the WRKY gene family in potato (Solanum tuberosum). Biochem. Syst. Ecol. 2017, 71, 212–218. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, Z.; Wang, L.; Kim, S.G.; Xiang, F. WRKY71 accelerates flowering via the direct activation of FLOWERING LOCUS T and LEAFY in Arabidopsis thaliana. Plant J. 2015, 85, 96–106. [Google Scholar] [CrossRef] [Green Version]
- Halliday, K.J.; Praekelt, U.M.; Salter, M.G.; Whitelam, G.C. Functions and Actions of Arabidopsis Phytochromes; Springer: Boston, MA, USA, 2001. [Google Scholar]
- Imaizumi, T. Arabidopsis circadian clock and photoperiodism: Time to think about location. Curr. Opin. Plant Biol. 2010, 13, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Tiyayon, P.; Hegele, M.; Wünsche, J.N.; Pongsriwat, K.; Samach, A. Studies on the molecular basis of flowering in longan (Dimocarpus longan). Acta Hortic. 2011, 903, 979–985. [Google Scholar] [CrossRef]
- Zhu, L.; Doyle, T.J.; Kim, K.H. Retinoic Acid Modulates the Subcellular Localization of Small Ubiquitin-Related Modifier-2/3 (SUMO-2/3) in the Testis. J. Androl. 2010, 31, 406–418. [Google Scholar] [CrossRef]
- Murakami-Kojima, M. The APRR3 Component of the Clock-Associated APRR1/TOC1 Quintet is Phosphorylated by a Novel Protein Kinase Belonging to the WNK Family, the Gene for which is also Transcribed Rhythmically in Arabidopsis thaliana. Plant Cell Physiol. 2002, 43, 675–683. [Google Scholar] [CrossRef] [Green Version]
- Marquardt, S.; Boss, P.; Hadfield, J.; Dean, C. Additional targets of the Arabidopsis autonomous pathway members, FCA and FY. J. Exp. Bot. 2006, 57, 3379–3386. [Google Scholar] [CrossRef]
- Hong, J.K.; Kim, J.A.; Lee, S.I.; Suh, E.J.; Chang, A.; Koo, B.S.; Lee, Y.-H. Flowering Time Genes and Application in Crops. Korean J. Breed. Sci. 2013, 45, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Domagalska, M.A.; Schomburg, F.M.; Amasino, R.M.; Vierstra, R.D.; Nagy, F.; Davis, S.J. Attenuation of brassinosteroid signaling enhances FLC expression and delays flowering. Development 2007, 134, 2841–2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Yan, L.; Harsh, R.; Zou, X.; Jing, W.; Dai, S.; Xiao, Q.; Cong, L.; Fan, L.; Liu, B. A Tourist -like MITE insertion in the upstream region of the BnFLC.A10 gene is associated with vernalization requirement in rapeseed (Brassica napus L). BMC Plant Biol. 2012, 12, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alabadí, D.; Blázquez, M.A. Molecular interactions between light and hormone signaling to control plant growth. Plant Mol. Biol. 2009, 69, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Bartolome, J.; Minguet, E.G.; Grau-Enguix, F.; Abbas, M.; Locascio, A.; Thomas, S.G.; Alabadi, D.; Blazquez, M.A. Molecular mechanism for the interaction between gibberellin and brassinosteroid signaling pathways in Arabidopsis. Proc. Natl. Acad. Sci. USA 2012, 109, 13446–13451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Kim, Y.J.; Dinh, T.T.; Chen, X. miR172 regulates stem cell fate and defines the inner boundary of APETALA3 and PISTILLATA expression domain in Arabidopsis floral meristems. Plant J. 2007, 51, 840–849. [Google Scholar] [CrossRef] [Green Version]
- Krizek, B.A. Aintegumenta and Aintegumenta-Like6 regulate auxin-mediated flower development in Arabidopsis. BMC Res. Notes 2011, 4, 176. [Google Scholar] [CrossRef] [Green Version]
- Aloni, R.; Aloni, E.; Langhans, M.; Ullrich, C.I. Role of auxin in regulating Arabidopsis flower development. Planta 2005, 223, 315–328. [Google Scholar] [CrossRef]
- Luca, C.; Simona, M.; Dola, S.R.; Stefano, B.; Irma, R.-V.; Anicet, D.F.; Klaus, P.; Rüdiger, S.; Lucia, C.; Markus, G. Maternal Control of PIN1 Is Required for Female Gametophyte Development in Arabidopsis. PLoS ONE 2013, 8, e66148. [Google Scholar]
- Ottenschläger. Gravity Regulated Differential Auxin Transport in Arabidopsis Roots and the Search for Interaction Partners of AtPIN1. Ph.D. Thesis, University of Cologne, Cologne, Germany, 2002.
- Naor, V.; Kigel, J.; Ziv, M. effect of Gibberellin and cytokinin on floral development in Zantedeschia spp. in vivo and in vitro. Acta Hortic. 2005, 673, 255–263. [Google Scholar] [CrossRef]
- Sheerin, D.J.; Buchanan, J.; Kirk, C.; Harvey, D.; Spagnuolo, J. Inter- and intra-molecular interactions of Arabidopsis thalianaDELLA protein RGL1. Biochem. J. 2011, 435, 629–639. [Google Scholar] [CrossRef] [Green Version]
- Su, W.R.; Huang, K.-L.; Shen, R.-S.; Chen, W.-S. Abscisic acid affects floral initiation in Polianthes tuberosa. J. Plant Physiol. 2002, 159, 557–559. [Google Scholar] [CrossRef]
- D’Aloia, M.; Bonhomme, D.; Bouché, F.; Tamseddak, K.; Périlleux, C. Cytokinin promotes flowering of Arabidopsis via transcriptional activation of the FT paralogue TSF. Plant J. 2011, 65, 972–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, S.; Huttly, A.K.; Prosser, I.M.; Li, Y.D.; Phillips, A.L. Heterologous expression and transcript analysis of gibberellin biosynthetic genes of grasses reveals novel functionality in the GA3ox family. BMC Plant Biol. 2015, 15, 130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Pictures of the three stages of flower development and corresponding paraffin sections. Small buds, flower buds and open flowers are designated C1, C2 and C3, respectively.
Figure 1.
Pictures of the three stages of flower development and corresponding paraffin sections. Small buds, flower buds and open flowers are designated C1, C2 and C3, respectively.

Figure 2.
Numbers of differentially expressed unigenes for the indicated comparisons, with up- and down-regulated unigenes marked in blue and yellow, respectively.
Figure 2.
Numbers of differentially expressed unigenes for the indicated comparisons, with up- and down-regulated unigenes marked in blue and yellow, respectively.

Figure 3.
A comparison of the overlapping differentially expressed gene profiles across the different stages of floral development.
Figure 3.
A comparison of the overlapping differentially expressed gene profiles across the different stages of floral development.

Figure 4.
GO classification of DEGs of the progression from C1 vs. C2, C2 vs. C3 and C2 vs. C3.

Figure 5.
Number of unique unigenes annotated as transcription factor (A) and protein kinase (B) in S. japonicus transcriptome sequences.
Figure 5.
Number of unique unigenes annotated as transcription factor (A) and protein kinase (B) in S. japonicus transcriptome sequences.

Figure 6.
Genetic identification of hormone-related DEGs associated with flower development. (A) ABA-related gene expression patterns during flowering. (B) Auxin-related gene expression patterns during flowering. (C) Cytokinin-related gene expression patterns during flowering. (D) GA-related gene expression patterns during flowering.
Figure 6.
Genetic identification of hormone-related DEGs associated with flower development. (A) ABA-related gene expression patterns during flowering. (B) Auxin-related gene expression patterns during flowering. (C) Cytokinin-related gene expression patterns during flowering. (D) GA-related gene expression patterns during flowering.

Figure 7.
Real-time PCR validation of important genes related to S. japonicus flowering.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Li, W.; Xu, Z.; Zhang, C.; Jiang, X.; Wang, K. Transcriptomic Identification of Floral Transition and Development-Associated Genes in Styrax japonicus. Forests 2020, 11, 10. https://0-doi-org.brum.beds.ac.uk/10.3390/f11010010
AMA Style
Li W, Xu Z, Zhang C, Jiang X, Wang K. Transcriptomic Identification of Floral Transition and Development-Associated Genes in Styrax japonicus. Forests. 2020; 11(1):10. https://0-doi-org.brum.beds.ac.uk/10.3390/f11010010
Chicago/Turabian StyleLi, Wei, Zhengzhao Xu, Cuiping Zhang, Xinqiang Jiang, and Kuiling Wang. 2020. "Transcriptomic Identification of Floral Transition and Development-Associated Genes in Styrax japonicus" Forests 11, no. 1: 10. https://0-doi-org.brum.beds.ac.uk/10.3390/f11010010
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.