
Inferring the Phylogeny and Divergence of Chinese Curcuma (Zingiberaceae) in the Hengduan Mountains of the Qinghai–Tibet Plateau by Reduced Representation Sequencing


Abstract
:1. Introduction
2. Materials and Methods
2.1. Material and Sequencing
2.2. Clustering
2.3. Concatenation-Based Species Tree Inference
2.4. Population Structure and Divergence Time Inference
3. Results
3.1. Sequences Discovery and Characterization
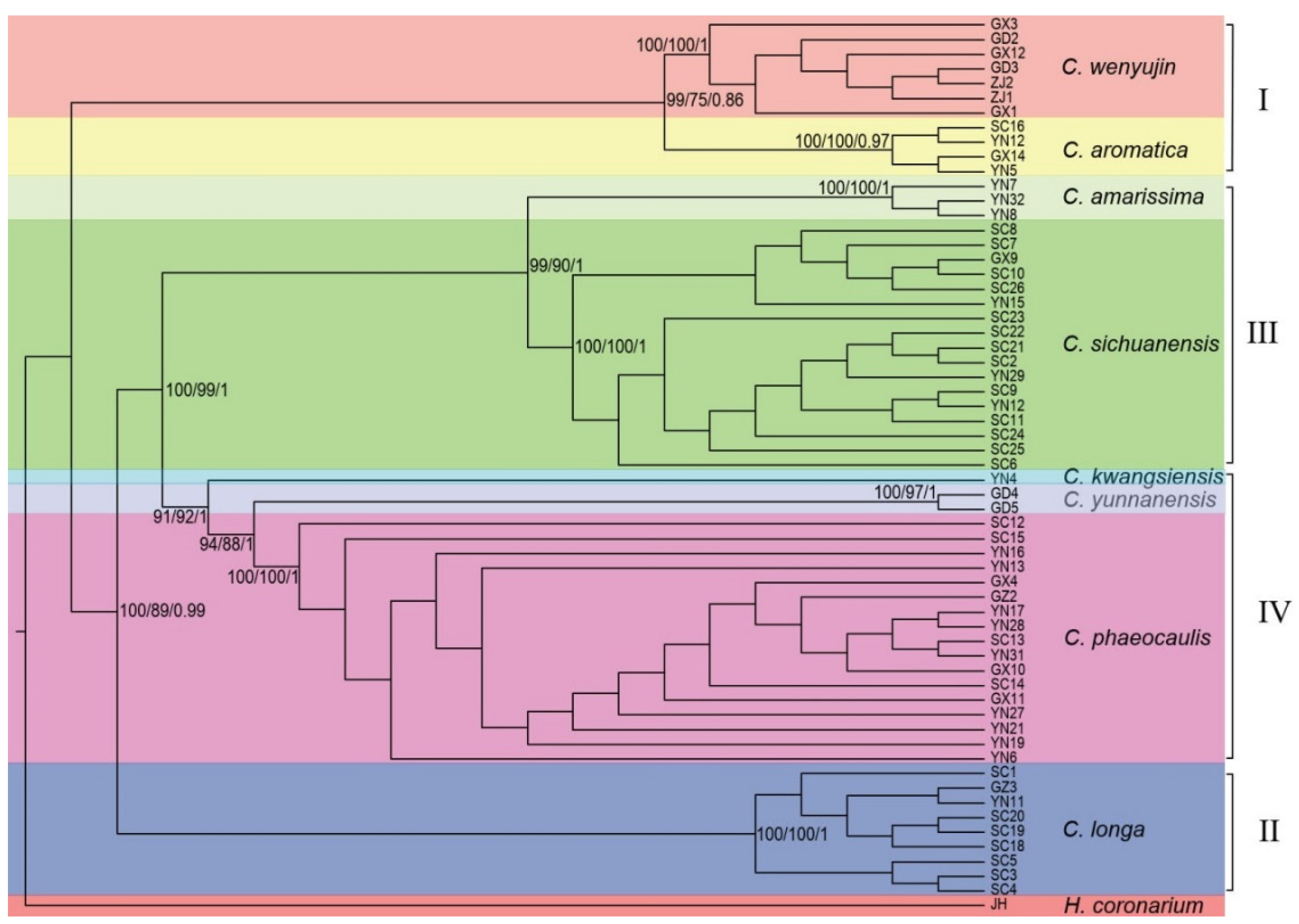
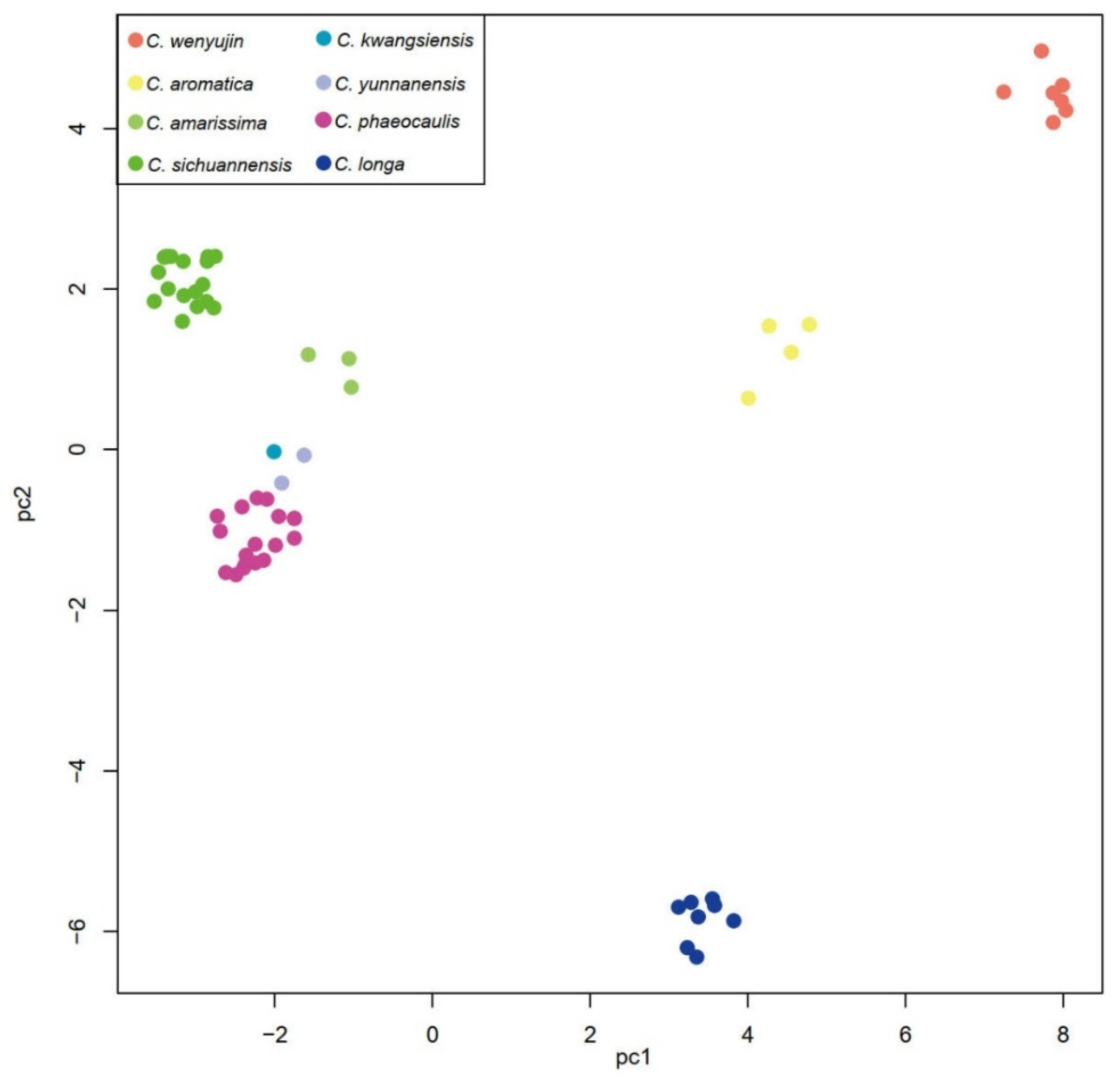
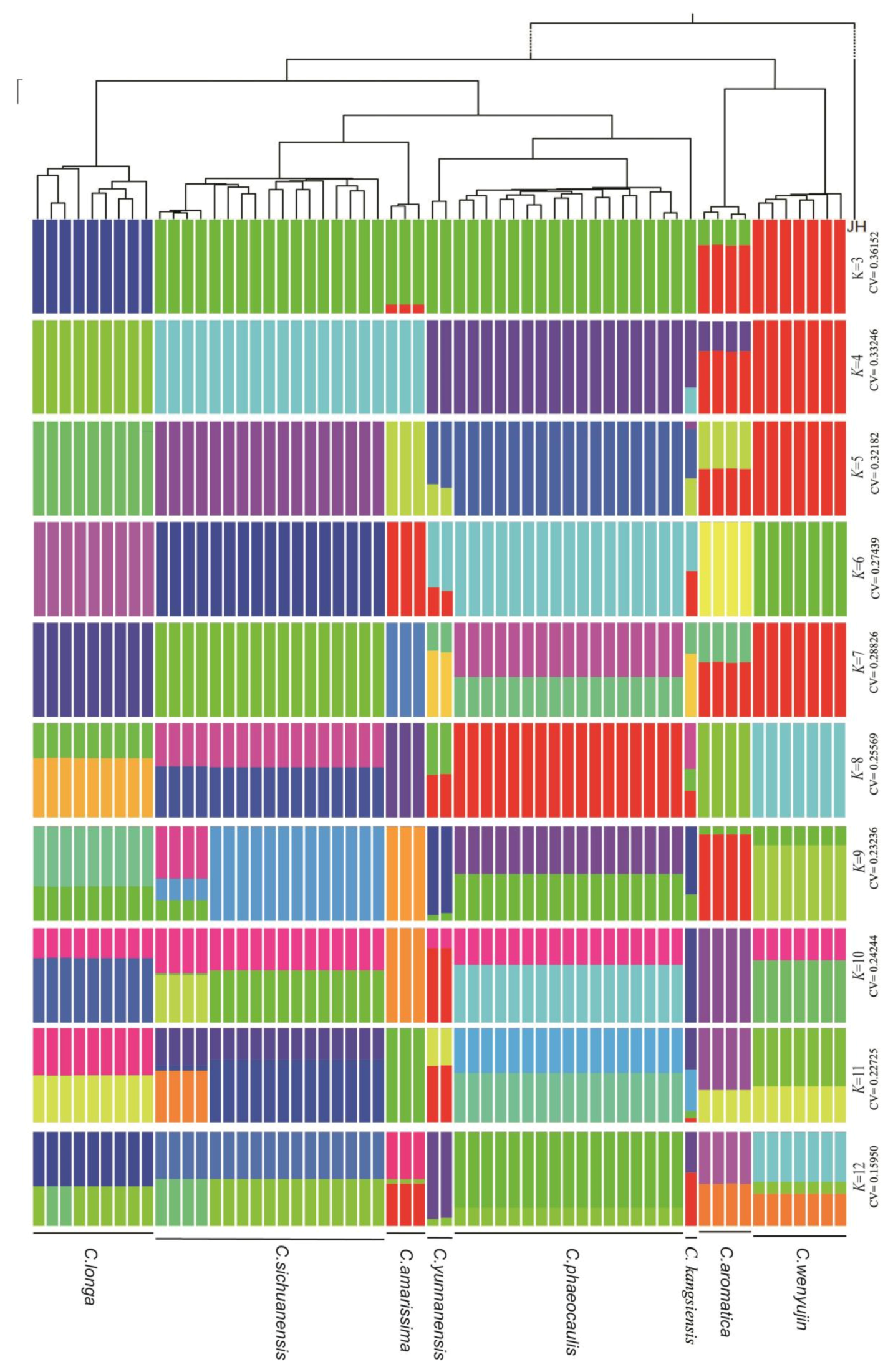
3.2. Phylogenetic Analyses
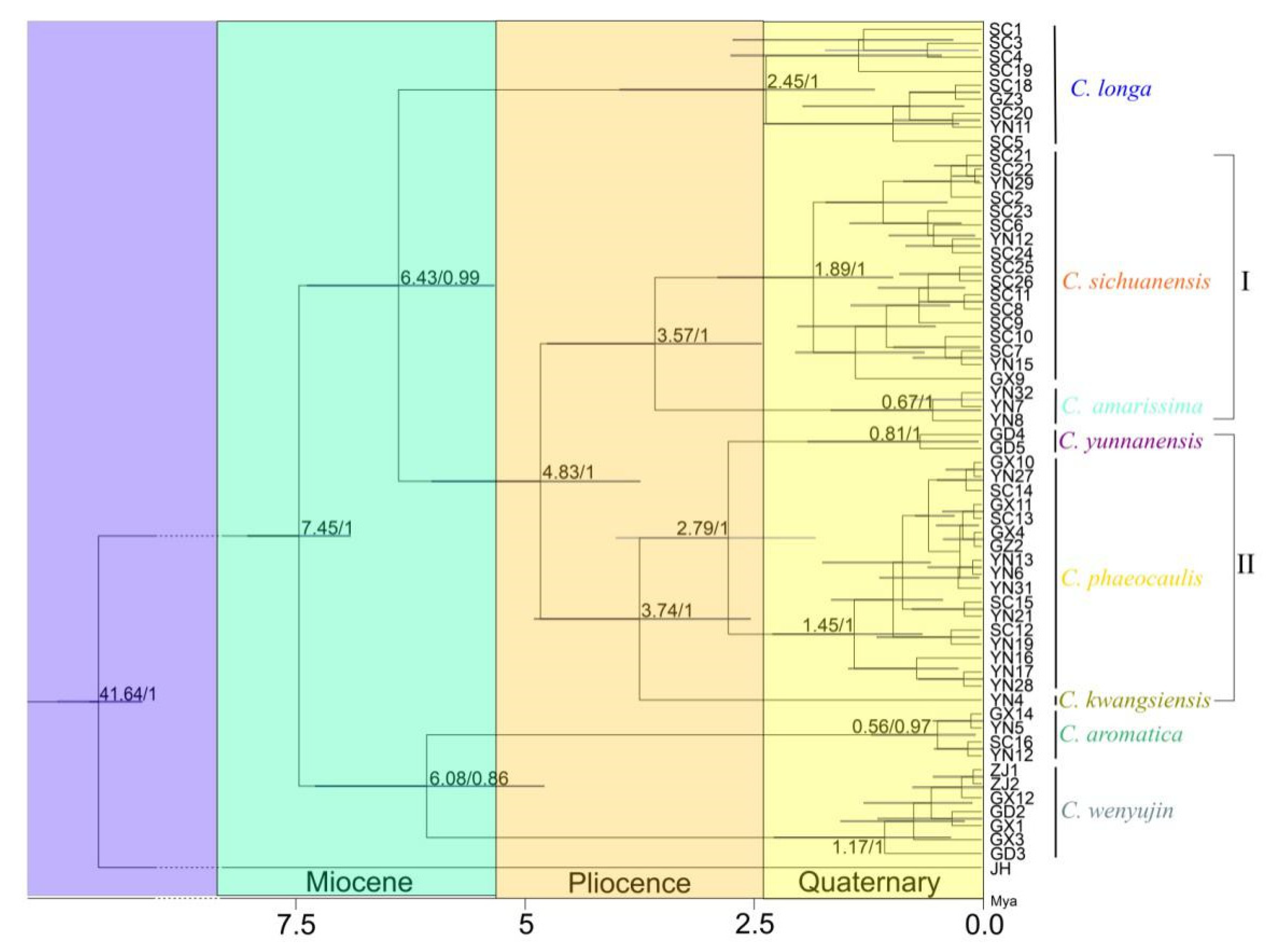
3.3. Divergence Time Inference
4. Discussion
4.1. Application of “Next-Generation” Sequencing in Estimating the Phylogeny and Biological Implication of Curcuma
4.2. Phylogeny Inference
4.3. Divergence Time
4.4. Future Directions
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deng, J.B.; Ding, C.B.; Zhang, L.; Zhou, Y.H.; Yang, R.W. Relationships among six herbal species (Curcuma) assessed by four isozymes. Phyton 2011, 80, 181–188. [Google Scholar]
- Zaveska, E.; Fer, T.; Sida, O.; Krak, K.; Marhold, K.; Leong-Skornickova, J. Phylogeny of Curcuma (Zingiberaceae) based on plastid and nuclear sequences: Proposal of the new subgenus Ecomata. Taxon 2012, 61, 747–763. [Google Scholar] [CrossRef]
- Deng, J.B.; Ding, C.; Zhang, L.; Yang, R.; Zhou, Y. Authentication of three related herbal species (Curcuma) by DNA barcoding. J. Med. Plants Res. 2011, 5, 6401–6406. [Google Scholar] [CrossRef]
- Nian, L.; Telin, W. Notes on Curcuma in China. J. Trop. Subtrop. Bot. 1999, 7, 146–150. [Google Scholar] [CrossRef]
- Deng, J.B.; Gao, G.; Ahmad, K.; Luo, X.; Zhang, F.; Li, S.; Yang, R. Evaluation on genetic relationships among China’s endemic Curcuma L. herbs by mtDNA. Phyton Int. J. Exp. Bot. 2018, 87, 156–161. [Google Scholar]
- Kress, W.J.; Prince, L.M.; Williams, K.J. The phylogeny and a new classification of the gingers (Zingiberaceae): Evidence from molecular data. Am. J. Bot. 2002, 89, 1682–1696. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, J.T.; Xia, N.H. Testing DNA barcodes in closely related species of Curcuma (Zingiberaceae) from Myanmar and China. Mol. Ecol. Resour. 2014, 2, 337–348. [Google Scholar]
- Zaveska, E.; Fer, T.; Sida, O.; Marhold, K.; Leong-Skornickova, J. Hybridization among distantly related species: Examples from the polyploid genus Curcuma (Zingiberaceae). Mol. Phylogenet. Evol. 2016, 100, 303–321. [Google Scholar] [CrossRef] [PubMed]
- Li, J. The Qinghai-Tibet Plateau Uplifting and Environmental Evolution in Asia: Article Collection of Academician; Science Press: Beijing, China, 2006. [Google Scholar]
- Liu, X.; Dong, B. Influence of the Tibetan Plateau uplift on the Asian monsoon-arid environment evolution. Chin. Sci. Bull. 2013, 58, 4277–4291. [Google Scholar] [CrossRef] [Green Version]
- Hoorn, C.; Mosbrugger, V.; Mulch, A.; Antonelli, A. Biodiversity from mountain building. Nat. Geosci. 2013, 6, 154. [Google Scholar] [CrossRef]
- Wen, J.; Zhang, J.Q.; Nie, Z.L.; Zhong, Y.; Sun, H. Evolutionary diversifications of plants on the Qinghai-Tibetan Plateau. Front. Genet. 2014, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Favre, A.; Päckert, M.; Pauls, S.U.; Jähnig, S.C.; Uhl, D.; Michalak, I.; Muellner-Riehl, A.N. The role of the uplift of the Qinghai-Tibetan Plateau for the evolution of Tibetan biotas. Biol. Rev. Camb. Philos. Soc. 2015, 90, 236–253. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.H.; Shu, G.M.; Li, L.Y.; Fang, Q.M.; Su, Z.W. Histological and morphological studies on the rhizomes of Curcuma. China J. Chin. Mater. Med. 2004, 20, 84–87. [Google Scholar]
- Xiao, X.H.; Zhao, Y.L.; Cheng, J.; Shu, G.M.; Shu, Z.W. Histological and morphological studies on leaves of Curcuma in China. China J. Chin. Mater. Med. 2004, 29, 203–207. [Google Scholar]
- Chen, J.; Xia, N.H. Pollen morphology of Chinese Curcuma L. and Boesenbergia Kuntz (Zingiberaceae): Taxonomic implications. Flora 2011, 206, 458–467. [Google Scholar] [CrossRef]
- Leong-Skornickova, J.; Šída, O.; Záveská, E.; Marhold, K. History of infrageneric classification, typification of supraspecific names and outstanding transfers in Curcuma (Zingiberaceae). Taxon 2015, 64, 362–373. [Google Scholar] [CrossRef]
- Leong-Škorničková, J.; Šída, O.; Jarolímová, V.; Sabu, M.; Fér, T.; Trávníček, P.; Suda, J. Chromosome Numbers and Genome Size Variation in Indian Species of Curcuma (Zingiberaceae). Ann. Bot. 2007, 100, 505–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Xia, N.; Zhao, J.; Chen, J.; Henny, R.J. Chromosome Numbers and Ploidy Levels of Chinese Curcuma Species. Hortscience 2013, 48, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.Y.; Huang, X.X.; Huang, S.F. A report on chromosome numbers on Chinese Zingiberaceae (5). Guihaia 1988, 8, 143–147. [Google Scholar]
- Chen, Z.Y.; Chen, Z.S.; Huang, F.S. A report on chromosome numbers on Chinese Zingiberaceae (2). Guihaia 1984, 4, 13–18. [Google Scholar]
- Sato, D. The karyotype analysis in Zingiberales with special reference to the protokaryotype and stable karyotype. Sci. Papers Coll. Gen. Educ. Univ. Tokyo 1960, 10, 225–243. [Google Scholar]
- Nian, L. The Taxonomic Study of Curcuma L. from China; South China Botanical Garden: Guangzhou, China, 1985. [Google Scholar]
- Sugiura, T. Studies on the chromosome numbers in higher plants. Cytologia 1936, 7, 544–595. [Google Scholar] [CrossRef] [Green Version]
- Raghavan, T.; Venkatasubban, K. Cytological studies in the family Zingiberace with special reference to chromosome number and Cyto-Taxonomy. In Proceedings of the Indian Academy of Sciences—Section B; Springer: New Delhi, India, 1943; Volume 17, pp. 118–132. [Google Scholar] [CrossRef]
- Ramachandran, K. Chromosome numbers in the genus Curcuma Linn. Curr. Sci. 1961, 30, 194–196. [Google Scholar]
- Liang, H.; Zhang, Y.; Deng, J.; Gao, G.; Ding, C.; Zhang, L.; Yang, R. The complete chloroplast genome sequences of 14 Curcuma species: Insights into genome evolution and phylogenetic relationships within Zingiberales. Front. Genet. 2020, 11, 802. [Google Scholar] [CrossRef]
- Cao, H.; Sasaki, Y.; Fushimi, H.; Komatsu, K. Molecular analysis of medicinally-used Chinese and Japanese Curcuma based on 18S rRNA gene and trnK gene sequences. Biol. Pharm. Bull. 2001, 24, 1389–1394. [Google Scholar] [CrossRef] [Green Version]
- Newmaster, S.G.; Subramanyam, R. Testing plant barcoding in a sister species complex of pantropical Acacia (Mimosoideae, Fabaceae). Mol. Ecol. Resour. 2009, 9, 172–180. [Google Scholar]
- Lemmon, E.M.; Lemmon, A.R. High-throughput identification of informative nuclear loci for shallow-scale phylogenetics and phylogeography. Syst. Biol. 2012, 61, 745–761. [Google Scholar] [CrossRef]
- Harvey, M.G.; Smith, B.T.; Glenn, T.C.; Faircloth, B.C.; Brumfield, R.T. Sequence capture versus restriction site associated DNA sequencing for shallow systematics. Syst. Biol. 2016, 65, 910–924. [Google Scholar] [CrossRef] [Green Version]
- Godden, G.T.; Jordon-Thaden, I.E.; Chamala, S.; Crowl, A.A.; Garcia, N.; Germain-Aubrey, C.C.; Heaney, J.M.; Latvis, M.; Qi, X.; Gitzendanner, M.A. Making next-generation sequencing work for you: Approaches and practical considerations for marker development and phylogenetics. Plant Ecol. Divers. 2012, 5, 427–450. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 2008, 3, e3376. [Google Scholar] [CrossRef]
- Poland, J.A.; Brown, P.J.; Sorrells, M.E.; Jannink, J.-L. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS ONE 2012, 7, e32253. [Google Scholar] [CrossRef] [Green Version]
- Nemati, Z.; Blattner, F.R.; Kerndorff, H.; Erol, O.; Harpke, D. Phylogeny of the saffron-crocus species group, Crocus series Crocus (Iridaceae). Mol. Phylogenet. Evol. 2018, 127, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E.; Orban, L. A robust, simple Genotyping-by-Sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Filipski, A.J.; Battistuzzi, F.U.; Pond, S.L.K.; Tamura, K. Statistics and Truth in Phylogenomics. Mol. Biol. Evol. 2012, 29, 457–472. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.A.; Bhangoo, J.; Fernández-Fernández, F.; Moore, P.; Swanson, J.D.; Viola, R.; Velasco, R.; Bassil, N.; Weber, C.A.; Sargent, D.J. Saturated linkage map construction in Rubus idaeus using genotyping by sequencing and genome-independent imputation. BMC Genom. 2013, 14, 1471–2164. [Google Scholar] [CrossRef] [Green Version]
- Bernhardt, N.; Brassac, J.; Dong, X.; Willing, E.M.; Poskar, C.H.; Kilian, B.; Blattner, F.R. Genome-wide sequence information reveals recurrent hybridization among diploid wheat wild relatives. Plant J. 2020, 102, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Perez-Escobar, O.A.; Bogarin, D.; Schley, R.; Bateman, R.M.; Gerlach, G.; Harpke, D.; Brassac, J.; Fernandez-Mazuecos, M.; Dodsworth, S.; Hagsater, E.; et al. Resolving relationships in an exceedingly young Neotropical orchid lineage using Genotyping-by-sequencing data. Mol. Phylogenet. Evol. 2020, 144, 12. [Google Scholar] [CrossRef]
- Deng, J.B.; Liu, J.; Ahmad, K.; Ding, C.; Zhang, L.; Zhou, Y.; Yang, R. Relationships evaluation on six herbal species (Curcuma) by dna barcoding. Pak. J. Bot. 2015, 47, 1103–1109. [Google Scholar]
- Myers, N.; Mittermeier, R.A.; Mittermeier, C.G.; da Fonseca, G.A.B.; Kent, J. Biodiversity hotspots for conservation priorities. Nature 2000, 403, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.-X.; Fu, C.-X.; Comes, H. Plant molecular phylogeography in China and adjacent regions: Tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogenet. Evol. 2011, 59, 225–244. [Google Scholar] [CrossRef]
- Fan, D.M.; Yue, J.P.; Nie, Z.L.; Li, Z.M.; Comes, H.P.; Sun, H. Phylogeography of Sophora davidii (Leguminosae) across the ‘Tanaka-Kaiyong Line’, an important phytogeographic boundary in Southwest China. Mol. Ecol. 2013, 22, 4270–4288. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Ree, R.H. Uplift-driven diversification in the Hengduan Mountains, a temperate biodiversity hotspot. Proc. Natl. Acad. Sci. USA 2017, 114, 3444–3451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Fang, X. Uplift of the Tibetan Plateau and environmental changes. Chin. Sci. Bull. 1999, 44, 2117–2124. [Google Scholar] [CrossRef]
- Yu, S.-H.; Wen, Q.-Z. Geochemical evolution and environmental changes of Qinghai—Xizang Plateau Since Late Cenozoic. Acta Geochim. 1998, 17, 258–264. [Google Scholar]
- Doyle, J. DNA Protocols for Plants-CTAB Total DNA Isolation; Springer: Berlin, Germany, 1991; Volume 283–293. [Google Scholar]
- Grabowski, P.P.; Morris, G.P.; Casler, M.D.; Borevitz, J.O. Population genomic variation reveals roles of history, adaptation and ploidy in switchgrass. Mol. Ecol. 2014, 23, 4059–4073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, D.A.; Overcast, I. ipyrad: Interactive assembly and analysis of RADseq datasets. Bioinformatics 2020, 36, 2592–2594. [Google Scholar] [CrossRef]
- Catchen, J.M.; Amores, A.; Hohenlohe, P.; Cresko, W.; Postlethwait, J.H.; De Koning, D.J. Stacks: Building and genotyping loci de novo from short-read sequences. G3 Genes Genomes Genet. 2011, 1, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Nylander, J. MrModeltest v2.(Program Distributed by the Author) Evolutionary Biology Centre; Uppsala University: Uppsala, Sweden, 2004; Available online: http://www.ebc.uu.se/systzoo/staff/nylander.html (accessed on 11 September 2018).
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]
- Drummond, A.J. Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 2010, 27, 570–580. [Google Scholar]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nature Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Durand, E.Y.; Patterson, N.; Reich, D.; Slatkin, M. Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 2011, 28, 2239–2252. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.L. A Phylogenetic Study on the Tribe Zingibereae (Zingiberaceae). Ph.D. Thesis, Xishuangbanna Tropical Botanical Garden Chinese Academy of Sciences, Xishuangbanna, China, 2010. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.Y.; Joseph, L.; Edwards, S.V. A species tree for the australo-papuan fairy-wrens and allies (Aves: Maluridae). Syst. Biol. 2012, 61, 253–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumaran, J.; Knowles, L.L. Multispecies coalescent delimits structure, not species. Proc. Natl. Acad. Sci. USA 2017, 114, 1607–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, C.H.; Cutler, J.S.; Friel, J.P.; Dening Touokong, C.; Coop, G.; Wainwright, P.C. Complex histories of repeated gene flow in Cameroon crater lake cichlids cast doubt on one of the clearest examples of sympatric speciation. Evolution 2015, 69, 1406–1422. [Google Scholar] [CrossRef] [Green Version]
- Meier, J.I.; Marques, D.A.; Mwaiko, S.; Wagner, C.E.; Excoffier, L.; Seehausen, O. Ancient hybridization fuels rapid cichlid fish adaptive radiations. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.J.; Soltis, P.S.; Bell, C.D.; Burleigh, J.G.; Soltis, D.E. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. USA 2010, 107, 4623–4628. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.Y.; Lei, Y.X.; Gao, G.; Zhang, Y.; Deng, J.B.; Tong, S.S.; Yang, R.W. Genetic Diversity of Chinese Curcuma based on RAMP Makers. Genom. Appl. Biol. 2015, 34, 1784–1790. [Google Scholar]
- Xiao, X.H.; Liu, F.Q.; Shi, C.H.; Li, L.Y.; Qiao, C.Z.; Su, Z.W. RAPD polymorphism and Authentication of Medicinal Plants from Turmeri (Curcuma L. ) in China. Chin. Tradit. Herb. Drugs 2000, 31, 209–212. [Google Scholar]
- Li, Y.Y.; Lei, Y.X.; Gao, G.; Zhang, Y.; Deng, J.B.; Tong, S.S.; Yang, R.W. Genetic diversity analysis of Chinese Curcuma based on ISSR makers. Mol. Plant Breed. 2016, 14, 1189–1194. [Google Scholar]
- Zhou, Z.; Huang, J.; Ding, W. The impact of major geological events on Chinese flora. Biodivers. Sci. 2017, 25, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Li, J.J.; Fang, X.M.; Pan, B.T.; Zhao, Z.J.; Song, Y.G. Late cenozoic intensive uplift of Qinghai-Xizang plateau and its impacts on environments in surrounding area. Quat. Sci. 2001, 21, 381–391. [Google Scholar]
- Yi, G.Z.; Ji, J.; Balsam, W.; Liu, L.; Chen, J. Mid-pliocene asian monsoon intensification and the onset of northern hemisphere glaciation. Geology 2009, 37, 599–602. [Google Scholar]
- Biasatti, D.; Yang, W.; Feng, G.; Xu, Y.; Flynn, L. Paleoecologies and paleoclimates of late cenozoic mammals from Southwest China: Evidence from stable carbon and oxygen isotopes. J. Asian Earth Sci. 2012, 44, 48–61. [Google Scholar] [CrossRef]
- Mallet, J. Hybrid speciation. Nature 2007, 446, 279–283. [Google Scholar] [CrossRef]
- Abbott, R.J.; Albach, D.; Ansell, S.; Arntzen, J.W.; Zinner, D. Hybridization and speciation. J. Evol. Biol. 2013, 26, 229–246. [Google Scholar] [CrossRef] [Green Version]
- Gui, L.; Jiang, S.; Xie, D.; Yu, L.; Huang, Y.; Zhang, Z.; Liu, Y. Analysis of complete chloroplast genomes of Curcuma and the contribution to phylogeny and adaptive evolution. Gene 2020, 732, 144355. [Google Scholar] [CrossRef]
- Doyle, J.J.; Flagel, L.E.; Paterson, A.H.; Rapp, R.A.; Soltis, D.E.; Soltis, P.S.; Wendel, J.F. Evolutionary genetics of genome merger and doubling in plants. Annu. Rev. Genet. 2008, 42, 443–461. [Google Scholar] [CrossRef] [Green Version]
- Chao, D.-Y.; Dilkes, B.; Luo, H.; Douglas, A.; Yakubova, E.; Lahner, B.; Salt, D.E. Polyploids exhibit higher potassium uptake and salinity tolerance in Arabidopsis. Science 2013, 341, 658–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selmecki, A.M.; Maruvka, Y.E.; Richmond, P.A.; Guillet, M.; Shoresh, N.; Sorenson, A.L.; De, S.; Kishony, R.; Michor, F.; Dowell, R. Polyploidy can drive rapid adaptation in yeast. Nature 2015, 519, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Yang, Z. Testing hybridization hypotheses based on incongruent gene trees. Syst. Biol. 2000, 48, 422–434. [Google Scholar]
- Sang, T.; Crawford, D.J.; Stuessy, T.F. Chloroplast DNA phylogeny, reticulate evolution, and biogeography of Paeonia (Paeoniaceae). Am. J. Bot. 1997, 84, 1120–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escallon, E.V.; Richardson, J.E.; Kidner, C.A.; Madriñán, S.; Stone, G.S. Transcriptome mining for phylogenetic markers in a recently radiated genus of tropical plants ( Renealmia L.f., Zingiberaceae). Mol. Phylogenet. Evol. 2017, 119, 13–24. [Google Scholar]
- Liang, H.; Zhang, Y.; Deng, J.; Gao, G.; Ding, C.; Zhang, L.; Yu, X.; Zhou, Y.; Yang, R. Application of genotyping-by-sequencing data on inferring the phylogeny of Curcuma (Zingiberaceae) from China. Res. Sq. 2019. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Species | Source Details | Total of Loci after Ipyrad |
|---|---|---|---|
| SC1 | Curcuma longa | Muchuan, Sichuan | 26,977 |
| SC3 | Curcuma longa | Fulu, Sichuan | 17,898 |
| SC4 | Curcuma longa | Cuiping, Sichuan | 39,772 |
| SC19 | Curcuma longa | Yibin, Sichuan | 30,974 |
| SC18 | Curcuma longa | Qianwe, Sichuan | 26,311 |
| GZ3 | Curcuma longa | Xingyi, Guizhou | 28,912 |
| SC20 | Curcuma longa | Shuangliu, Sichuan | 32,886 |
| YN11 | Curcuma longa | Menga, Yuannan | 20,606 |
| SC5 | Curcuma longa | Dayi, Sichuan | 34,053 |
| SC21 | Curcuma sichuanensis | Medicinal, Botanical Garden, Guangxi | 21,536 |
| SC22 | Curcuma sichuanensis | Chongzhou, Sichuan | 33,411 |
| YN29 | Curcuma sichuanensis | Yiwu, Yunnan | 19,503 |
| SC2 | Curcuma sichuanensis | Sanjiang, Sichuan | 26,973 |
| SC23 | Curcuma sichuanensis | Cuiping, Sichuan | 24,230 |
| SC6 | Curcuma sichuanensis | Ziyang, Sichuan | 27,677 |
| YN10 | Curcuma sichuanensis | Menga, Yuannan | 38,515 |
| SC24 | Curcuma sichuanensis | Chongzhou, Sichuan | 31,072 |
| SC25 | Curcuma sichuanensis | Qianwei, Sichuan | 20,902 |
| SC26 | Curcuma sichuanensis | Muchuan, Sichuan | 21,588 |
| SC11 | Curcuma sichuanensis | Weiyuan, Sichuan | 28,768 |
| SC8 | Curcuma sichuanensis | GAP Base, Sichuan | 30,753 |
| SC9 | Curcuma sichuanensis | Sanjiang, Sichuan | 28,621 |
| SC10 | Curcuma sichuanensis | Chongzhou, Sichuan | 23,043 |
| SC7 | Curcuma sichuanensis | Yibing, Sichuan | 28,068 |
| YN15 | Curcuma sichuanensis | Menglun, Yunnan | 13,089 |
| GX9 | Curcuma sichuanensis | Medicinal, Botanical Garden, Guangxi | 31,039 |
| YN32 | Curcuma amarissima | Mengkang, Yunnan | 16,865 |
| YN7 | Curcuma amarissima | Menga, Yunnan | 17,222 |
| YN8 | Curcuma amarissima | Menga, Yunnan | 17,923 |
| GD4 | Curcuma yunnanensis | Huaxian, Guangdong | 32,976 |
| GD5 | Curcuma yunnanensis | Huaxian, Guangdong | 24,819 |
| GX10 | Curcuma phaeocaulis | Medicinal, Botanical Garden, Guangxi | 23,145 |
| YN27 | Curcuma phaeocaulis | Yiwu, Yunnan | 22,636 |
| SC14 | Curcuma phaeocaulis | Shuangliu, Sichuan | 34,212 |
| GX11 | Curcuma phaeocaulis | Medicinal, Botanical Garden, Guangxi | 29,557 |
| SC13 | Curcuma phaeocaulis | Jianwei, Sichuan | 25,208 |
| GX4 | Curcuma phaeocaulis | Medicinal, Botanical Garden, Guangxi | 35,825 |
| GZ2 | Curcuma phaeocaulis | Anlong, Guizhou | 31,112 |
| YN13 | Curcuma phaeocaulis | Menga, Yunnan | 14,425 |
| YN6 | Curcuma phaeocaulis | Mengxing, Yunnan | 21,440 |
| YN31 | Curcuma phaeocaulis | Mengkang, Yunnan | 21,642 |
| SC15 | Curcuma phaeocaulis | Shuangliu, Sichuan | 13,089 |
| YN21 | Curcuma phaeocaulis | Xishuangbanna, Yunnan | 16,744 |
| SC12 | Curcuma phaeocaulis | Chongzhou, Sichuan | 18,289 |
| YN19 | Curcuma phaeocaulis | Menglun, Yunnan | 13,489 |
| YN16 | Curcuma phaeocaulis | Daluo, Yunnan | 26,946 |
| YN17 | Curcuma phaeocaulis | Xishuangbanna, Yunnan | 23,180 |
| YN28 | Curcuma phaeocaulis | Yiwu, Yunnan | 28,065 |
| YN4 | Curcuma kwangsiensis | Medicinal, Botanical Garden, Guangxi | 17,001 |
| GX14 | Curcuma aromatica | Medicinal, Botanical Garden, Guangxi | 26,739 |
| YN5 | Curcuma aromatica | Mengkang, Yunnan | 24,435 |
| SC16 | Curcuma aromatica | Jianyang, Sichuan | 26,817 |
| YN12 | Curcuma aromatica | Yiwu, Yunnan | 18,289 |
| ZJ1 | Curcuma wenyujin | Taoshan, Zhejiang, cultivated | 23,259 |
| ZJ2 | Curcuma wenyujin | Meiyu, Zhejiang | 27,430 |
| GX12 | Curcuma wenyujin | Medicinal, Botanical Garden, Guangxi | 18,273 |
| GD2 | Curcuma wenyujin | Sanshui, Guangdong | 17,080 |
| GX1 | Curcuma wenyujin | Medicinal, Botanical Garden, Guangxi | 22,593 |
| GX3 | Curcuma wenyujin | Medicinal, Botanical Garden, Guangxi | 28,736 |
| GD3 | Curcuma wenyujin | Huaxian, Guangdong | 35,582 |
| JH | Hedychium coronarium | Yaan, Sichuan | 13,383 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, H.; Deng, J.; Gao, G.; Ding, C.; Zhang, L.; Xu, K.; Wang, H.; Yang, R. Inferring the Phylogeny and Divergence of Chinese Curcuma (Zingiberaceae) in the Hengduan Mountains of the Qinghai–Tibet Plateau by Reduced Representation Sequencing. Forests 2021, 12, 520. https://0-doi-org.brum.beds.ac.uk/10.3390/f12050520
Liang H, Deng J, Gao G, Ding C, Zhang L, Xu K, Wang H, Yang R. Inferring the Phylogeny and Divergence of Chinese Curcuma (Zingiberaceae) in the Hengduan Mountains of the Qinghai–Tibet Plateau by Reduced Representation Sequencing. Forests. 2021; 12(5):520. https://0-doi-org.brum.beds.ac.uk/10.3390/f12050520
Chicago/Turabian StyleLiang, Heng, Jiabin Deng, Gang Gao, Chunbang Ding, Li Zhang, Ke Xu, Hong Wang, and Ruiwu Yang. 2021. "Inferring the Phylogeny and Divergence of Chinese Curcuma (Zingiberaceae) in the Hengduan Mountains of the Qinghai–Tibet Plateau by Reduced Representation Sequencing" Forests 12, no. 5: 520. https://0-doi-org.brum.beds.ac.uk/10.3390/f12050520