
SNP Detection in Pinus pinaster Transcriptome and Association with Resistance to Pinewood Nematode

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. PWN Inoculum
2.3. Inoculation Assays and Sample Collection
2.4. RNA-Seq
2.5. SNP Calling and Analysis
2.6. SNP Validation
2.7. SNP Genotyping and Sequence Analysis
2.8. Association Analysis
3. Results
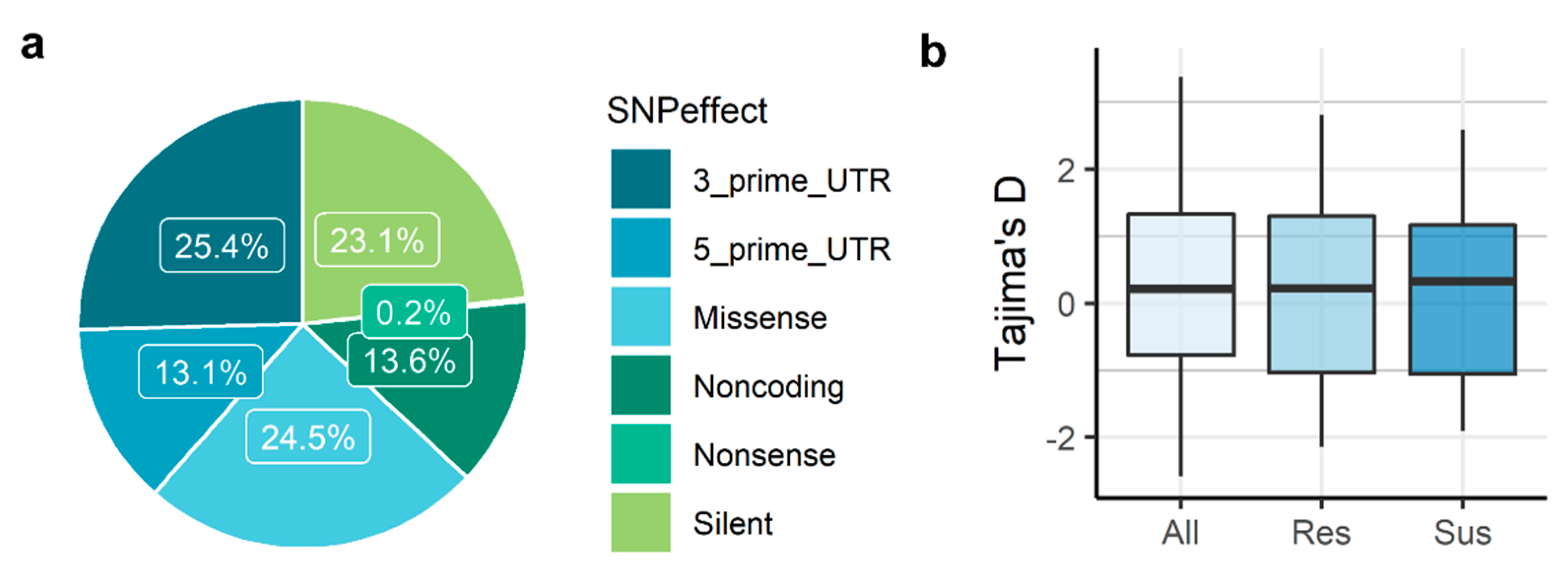
3.1. RNA-Seq, SNP Discovery and SNP Annotation
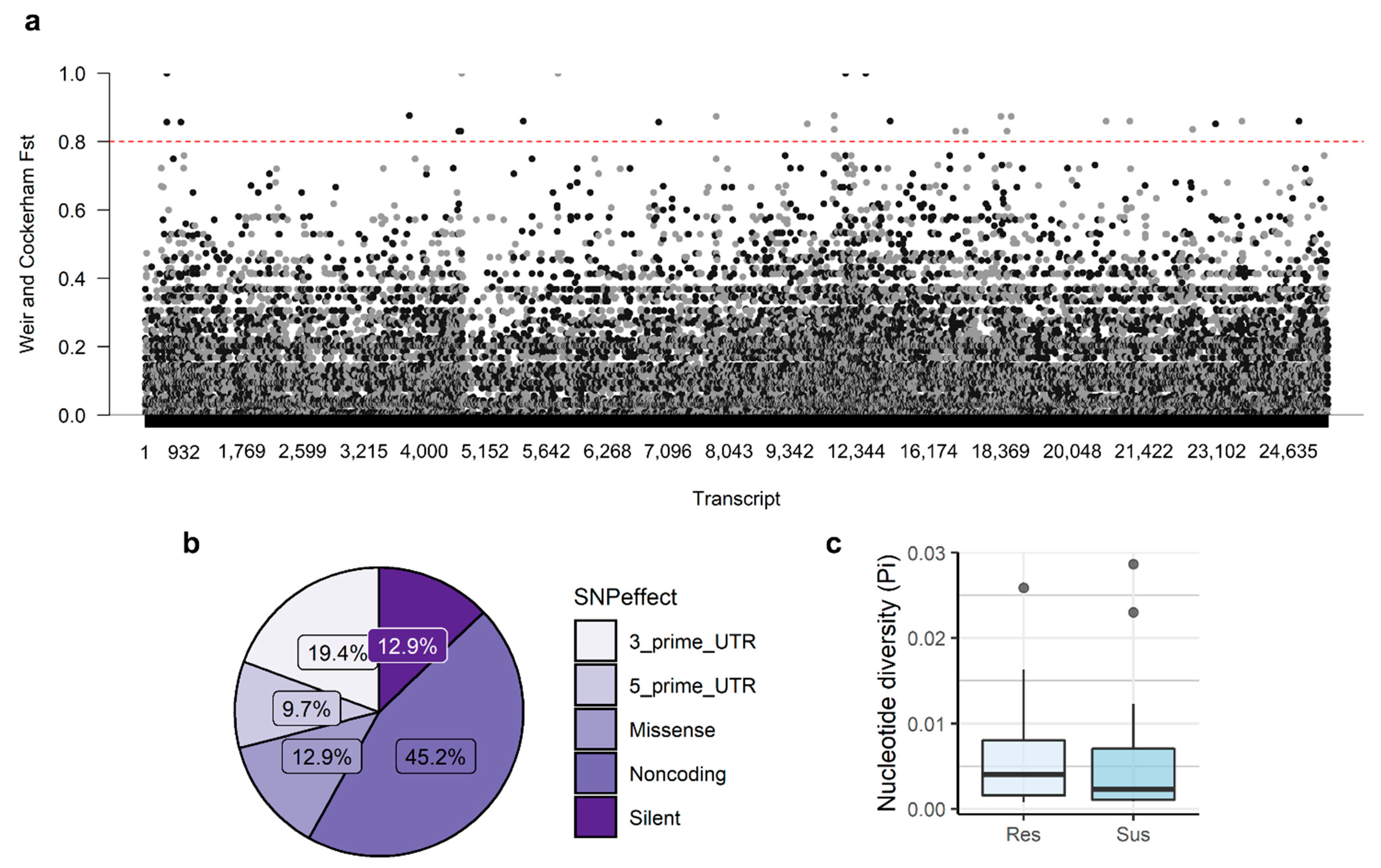
3.2. Genetic Diversity and Differentiation
3.3. SNP Validation through Sanger Sequencing
3.4. Inoculation Assay, Genotyping, and Sequence Analysis
3.5. Association Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Webster, J.; Mota, M. PineWilt Disease: Global Issues, Trade and Economic Impact. In Pine Wilt Disease: A Worldwide Threat to Forest Ecosystems; Mota, M., Vieira, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 1–4. [Google Scholar]
- Vicente, C.; Espada, M.; Vieira, P.; Mota, M. Pine Wilt Disease: A Threat to European Forestry. Eur. J. Plant Pathol. 2012, 133, 89–99. [Google Scholar] [CrossRef]
- Jones, J.T.; Moens, M.; Mota, M.; Li, H.; Kikuchi, T. Bursaphelenchus xylophilus: Opportunities in Comparative Genomics and Molecular Host-Parasite Interactions. Mol. Plant Pathol. 2008, 9, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Mota, M.; Braasch, H.; Bravo, M.A.; Penas, A.C.; Burgermeister, W.; Metge, K.; Sousa, E. First Report of Bursaphelenchus xylophilus in Portugal and in Europe. Nematology 1999, 1, 727–734. [Google Scholar] [CrossRef]
- Eveno, E.; Collada, C.; Guevara, M.A.; Léger, V.; Soto, A.; Díaz, L.; Léger, P.; González-Martínez, S.C.; Cervera, M.T.; Plomion, C.; et al. Contrasting Patterns of Selection at Pinus pinaster Ait. Drought Stress Candidate Genes as Revealed by Genetic Differentiation Analyses. Mol. Biol. Evol. 2008, 25, 417–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasquinho, I.; Lisboa, A.; Inácio, M.L.; Gonçalves, E. Genetic Variation in Susceptibility to Pine Wilt Disease of Maritime Pine Half-Sib Families. Ann. For. Sci. 2018, 75, 85. [Google Scholar] [CrossRef] [Green Version]
- Menéndez-Gutiérrez, M.; Alonso, M.; Toval, G.; Díaz, R. Testing of Selected Pinus pinaster Half-Sib Families for Tolerance to Pinewood Nematode (Bursaphelenchus xylophilus). Forestry 2017, 91, 38–48. [Google Scholar] [CrossRef]
- Menéndez-Gutiérrez, M.; Alonso, M.; Toval, G.; Díaz, R. Variation in Pinewood Nematode Susceptibility among Pinus pinaster Ait. Provenances from the Iberian Peninsula and France. Ann. For. Sci. 2017, 74, 76. [Google Scholar] [CrossRef] [Green Version]
- Toda, T.; Kurinobu, S. Realized Genetic Gains Observed in Progeny Tolerance of Selected Red Pine (Pinus densiflora) and Black Pine (P. thunbergii) to Pine Wilt Disease. Silvae Genet. 2002, 51, 42–44. [Google Scholar]
- Xu, L.-Y.; Zhang, J.; Gao, J.-B.; Chen, X.-L.; Jiang, C.-W.; Hao, Y.-P. Study on the Disease Resistance of Candidate Clones in Pinus massoniana to Bursaphelenchus xylophilus. China For. Sci. Technol. 2012, 26, 27–30. [Google Scholar]
- Nose, M.; Shiraishi, S. Breeding for Resistance to Pine Wilt Disease. In Pine Wilt Disease; Zhao, B.G., Futai, K., Sutherland, J.R., Takeuchi, Y., Eds.; Springer: Tokyo, Japan, 2008; pp. 334–350. [Google Scholar]
- Sniezko, R.A.; Koch, J. Breeding Trees Resistant to Insects and Diseases: Putting Theory into Application. Biol. Invasions 2017, 19, 3377–3400. [Google Scholar] [CrossRef]
- Naidoo, S.; Slippers, B.; Plett, J.M.; Coles, D.; Oates, C.N. The Road to Resistance in Forest Trees. Front. Plant Sci. 2019, 10, 273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirao, T.; Matsunaga, K.; Hirakawa, H.; Shirasawa, K.; Isoda, K.; Mishima, K.; Tamura, M.; Watanabe, A. Construction of Genetic Linkage Map and Identification of a Novel Major Locus for Resistance to Pine Wood Nematode in Japanese Black Pine (Pinus thunbergii). BMC Plant Biol. 2019, 19, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.-J.; Sniezko, R.A.; Sturrock, R.N.; Chen, H. Western White Pine SNP Discovery and High-Throughput Genotyping for Breeding and Conservation Applications. BMC Plant Biol. 2014, 14, 380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plomion, C.; Bartholomé, J.; Lesur, I.; Boury, C.; Rodríguez-Quilón, I.; Lagraulet, H.; Ehrenmann, F.; Bouffier, L.; Gion, J.M.; Grivet, D.; et al. High-Density SNP Assay Development for Genetic Analysis in Maritime Pine (Pinus pinaster). Mol. Ecol. Resour. 2016, 16, 574–587. [Google Scholar] [CrossRef]
- Guo, Y.; Su, B.; Tang, J.; Zhou, F.; Qiu, L.J. Gene-Based SNP Identification and Validation in Soybean Using next-Generation Transcriptome Sequencing. Mol. Genet. Genom. 2018, 293, 623–633. [Google Scholar] [CrossRef]
- Liu, J.-J.; Sniezko, R.; Murray, M.; Wang, N.; Chen, H.; Zamany, A.; Sturrock, R.N.; Savin, D.; Kegley, A. Genetic Diversity and Population Structure of Whitebark Pine (Pinus albicaulis Engelm.) in Western North America. PLoS ONE 2016, 11, e0167986. [Google Scholar] [CrossRef] [Green Version]
- Modesto, I.; Sterck, L.; Arbona, V.; Gómez-Cadenas, A.; Carrasquinho, I.; Van de Peer, Y.; Miguel, C.M. Insights Into the Mechanisms Implicated in Pinus pinaster Resistance to Pinewood Nematode. Front. Plant Sci. 2021, 12, 1102. [Google Scholar] [CrossRef]
- Rodrigues, A.M.; Carrasquinho, I.; António, C. Primary Metabolite Adjustments Associated With Pinewood Nematode Resistance in Pinus pinaster. Front. Plant Sci. 2021, 12, 777681. [Google Scholar] [CrossRef]
- Ribeiro, B.; Espada, M.; Vu, T.; Nóbrega, F.; Mota, M.; Carrasquinho, I. Pine Wilt Disease: Detection of the Pinewood Nematode (Bursaphelenchus xylophilus) as a Tool for a Pine Breeding Programme. For. Pathol. 2012, 42, 521–525. [Google Scholar] [CrossRef]
- Rodrigues, A.M.; Langer, S.; Carrasquinho, I.; Bergström, E.; Larson, T.; Thomas-Oates, J.; António, C. Pinus pinaster Early Hormonal Defence Responses to Pinewood Nematode (Bursaphelenchus xylophilus) Infection. Metabolites 2021, 11, 227. [Google Scholar] [CrossRef]
- Whitehead, A.G.; Hemming, J.R. A Comparison of Some Quantitative Methods of Extracting Small Vermiform Nematodes from Soil. Ann. Appl. Biol. 1965, 55, 25–38. [Google Scholar] [CrossRef]
- Futai, K.; Furuno, T. The Variety of Resistances among Pine Species to Pine Wood Nematode, Bursaphelenchus lignicolus. Bull. Kyoto Univ. 1979, 51, 23–36. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 13 June 2022).
- Cañas, R.A.; Li, Z.; Pascual, M.B.; Castro-Rodríguez, V.; Ávila, C.; Sterck, L.; Van de Peer, Y.; Cánovas, F.M. The Gene Expression Landscape of Pine Seedling Tissues. Plant J. 2017, 91, 1064–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, T.; Cotton, J.A.; Dalzell, J.J.; Hasegawa, K.; Kanzaki, N.; McVeigh, P.; Takanashi, T.; Tsai, I.J.; Assefa, S.A.; Cock, P.J.A.; et al. Genomic Insights into the Origin of Parasitism in the Emerging Plant Pathogen Bursaphelenchus xylophilus. PLoS Pathog. 2011, 7, e1002219. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud; O’Reilly Media: Sebastopol, CA, USA, 2020; ISBN 9781491975190. [Google Scholar]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila melanogaster Strain W1118; Iso-2; Iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Marshall, O.J. PerlPrimer: Cross-Platform, Graphical Primer Design for Standard, Bisulphite and Real-Time PCR. Bioinformatics 2004, 20, 2471–2472. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J. DNA Protocols for Plants. In Molecular Techniques in Taxonomy; Hewitt, G.M., Johnston, A.W.B., Young, J.P.W., Eds.; Springer: Berlin/Heidelberg, Germany, 1991; pp. 283–293. ISBN 978-3-642-83962-7. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, M.; Smith, N.J.; Donnelly, P. A New Statistical Method for Haplotype Reconstruction from Population Data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephens, M.; Scheet, P. Accounting for Decay of Linkage Disequilibrium in Haplotype Inference and Missing-Data Imputation. Am. J. Hum. Genet. 2005, 76, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- González, J.R.; Armengol, L.; Solé, X.; Guinó, E.; Mercader, J.M.; Estivill, X.; Moreno, V. SNPassoc: An R Package to Perform Whole Genome Association Studies. Bioinformatics 2007, 23, 644–645. [Google Scholar] [CrossRef] [Green Version]
- Tajima, F. Statistical Method for Testing the Neutral Mutation Hypothesis by DNA Polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Madur, D.; Kumar, S.; Krier, C.; Bou-Dagher-Kharrat, M.; Bertocchi, E.; Brach, J.; Plomion, C. A high density genetic map of maritime pine based on AFLPs. Ann. For. Sci. 2002, 59, 627–636. [Google Scholar] [CrossRef] [Green Version]
- Hurel, A.; de Miguel, M.; Dutech, C.; Desprez-Loustau, M.L.; Plomion, C.; Rodríguez-Quilón, I.; Cyrille, A.; Guzman, T.; Alía, R.; González-Martínez, S.C.; et al. Genetic Basis of Growth, Spring Phenology, and Susceptibility to Biotic Stressors in Maritime Pine. Evol. Appl. 2021, 14, 2750–2772. [Google Scholar] [CrossRef]
- Vogt, T. Phenylpropanoid Biosynthesis. Mol. Plant 2010, 3, 2–20. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Zhang, J.; Tschaplinski, T.J.; Tuskan, G.A.; Chen, J.G.; Muchero, W. Regulation of Lignin Biosynthesis and Its Role in Growth-Defense Tradeoffs. Front. Plant Sci. 2018, 9, 1427. [Google Scholar] [CrossRef] [Green Version]
- Mageroy, M.H.; Jancsik, S.; Yuen, M.M.S.; Fischer, M.; Withers, S.G.; Paetz, C.; Schneider, B.; MacKay, J.J.; Bohlmann, J. A Conifer UDP-Sugar Dependent Glycosyltransferase Contributes to Acetophenone Metabolism and Defense against Insects. Plant Physiol. 2017, 175, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Pagnussat, G.C.; Yu, H.J.; Ngo, Q.A.; Rajani, S.; Mayalagu, S.; Johnson, C.S.; Capron, A.; Xie, L.F.; Ye, D.; Sundaresan, V. Genetic and Molecular Identification of Genes Required for Female Gametophyte Development and Function in Arabidopsis. Development 2005, 132, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Li, H.J.; Shi, D.Q.; Yuan, L.; Liu, J.; Sreenivasan, R.; Baskar, R.; Grossniklaus, U.; Yang, W.C. The Central Cell Plays a Critical Role in Pollen Tube Guidance in Arabidopsis. Plant Cell 2007, 19, 3563–3577. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.X.; Wang, Y.; Liu, Z.Y.; Wang, X.; Lu, Q.; Jia, X.Z.; Zhang, X.Y. Functional Analysis of the Venom Allergen-like Protein Gene from Pine Wood Nematode Bursaphelenchus xylophilus Using a Baculovirus Expression System. Physiol. Mol. Plant Pathol. 2016, 93, 58–66. [Google Scholar] [CrossRef]
- Huibers, R.P.; De Jong, M.; Dekter, R.W.; Van Den Ackerveken, G. Disease-Specific Expression of Host Genes during Downy Mildew Infection of Arabidopsis. Mol. Plant-Microbe Interact. 2009, 22, 1104–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Durrant, W.E.; Song, J.; Spivey, N.W.; Dong, X. Arabidopsis BRCA2 and RAD51 Proteins Are Specifically Involved in Defense Gene Transcription during Plant Immune Responses. Proc. Natl. Acad. Sci. USA 2010, 107, 22716–22721. [Google Scholar] [CrossRef] [Green Version]
- Qin, T.; Zhao, P.; Sun, J.; Zhao, Y.; Zhang, Y.; Yang, Q.; Wang, W.; Chen, Z.; Mai, T.; Zou, Y.; et al. Research Progress of PPR Proteins in RNA Editing, Stress Response, Plant Growth and Development. Front. Genet. 2021, 12, 765580. [Google Scholar] [CrossRef]
- Saha, D.; Prasad, A.M.; Srinivasan, R. Pentatricopeptide Repeat Proteins and Their Emerging Roles in Plants. Plant Physiol. Biochem. 2007, 45, 521–534. [Google Scholar] [CrossRef]
- Hayes, M.L.; Dang, K.N.; Diaz, M.F.; Mulligan, R.M. A Conserved Glutamate Residue in the C-Terminal Deaminase Domain of Pentatricopeptide Repeat Proteins Is Required for RNA Editing Activity. J. Biol. Chem. 2015, 290, 10136–10142. [Google Scholar] [CrossRef] [Green Version]
- Ichinose, M.; Tasaki, E.; Sugita, C.; Sugita, M. A PPR-DYW Protein Is Required for Splicing of a Group II Intron of Cox1 Pre-MRNA in Physcomitrella patens. Plant J. 2012, 70, 271–278. [Google Scholar] [CrossRef]
- Hao, Y.; Wang, Y.; Wu, M.; Zhu, X.; Teng, X.; Sun, Y.; Zhu, J.; Zhang, Y.; Jing, R.; Lei, J.; et al. The Nuclear-Localized PPR Protein OsNPPR1 Is Important for Mitochondrial Function and Endosperm Development in Rice. J. Exp. Bot. 2019, 70, 4705–4720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koussevitzky, S.; Nott, A.; Mockler, T.C.; Hong, F.; Sachetto-Martins, G.; Surpin, M.; Lim, J.; Mittler, R.; Chory, J. Signals from Chloroplasts Converge to Regulate Nuclear Gene Expression. Science 2007, 316, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Zsigmond, L.; Rigó, G.; Szarka, A.; Székely, G.; Otvös, K.; Darula, Z.; Medzihradszky, K.F.; Koncz, C.; Koncz, Z.; Szabados, L. Arabidopsis PPR40 Connects Abiotic Stress Responses to Mitochondrial Electron Transport. Plant Physiol. 2008, 146, 1721–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laluk, K.; Abuqamar, S.; Mengiste, T. The Arabidopsis Mitochondria-Localized Pentatricopeptide Repeat Protein PGN Functions in Defense against Necrotrophic Fungi and Abiotic Stress Tolerance. Plant Physiol. 2011, 156, 2053–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, H.; Fu, X.; Yang, C.; Tang, X.; Guo, L.; Li, C.; Xu, C.; Luo, K. Genome-Wide Investigation of Pentatricopeptide Repeat Gene Family in Poplar and Their Expression Analysis in Response to Biotic and Abiotic Stresses. Sci. Rep. 2018, 8, 2817. [Google Scholar] [CrossRef] [Green Version]
- De Abreu-Neto, J.B.; Turchetto-Zolet, A.C.; De Oliveira, L.F.V.; Bodanese Zanettini, M.H.; Margis-Pinheiro, M. Heavy Metal-Associated Isoprenylated Plant Protein (HIPP): Characterization of a Family of Proteins Exclusive to Plants. FEBS J. 2013, 280, 1604–1616. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Liu, J.; Niu, Y.; Chen, Y.; Hao, Y.; Zhao, J.; Sun, L.; Wang, H.; Xiao, J.; et al. Characterization of the Heavy-Metal-Associated Isoprenylated Plant Protein (Hipp) Gene Family from Triticeae species. Int. J. Mol. Sci. 2020, 21, 6191. [Google Scholar] [CrossRef]
- Radakovic, Z.S.; Anjam, M.S.; Escobar, E.; Chopra, D.; Cabrera, J.; Silva, A.C.; Escobar, C.; Sobczak, M.; Grundler, F.M.W.; Siddique, S. Arabidopsis HIPP27 Is a Host Susceptibility Gene for the Beet Cyst Nematode Heterodera schachtii. Mol. Plant Pathol. 2018, 19, 1917–1928. [Google Scholar] [CrossRef] [Green Version]
- Tiffin, P.; Moeller, D.A. Molecular Evolution of Plant Immune System Genes. Trends Genet. 2006, 22, 662–670. [Google Scholar] [CrossRef]
- Keith, R.; Mitchell-Olds, T. Genetic Variation for Resistance to Herbivores and Plant Pathogens: Hypotheses, Mechanisms and Evolutionary Implications. Plant Pathol. 2013, 62, 122–132. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| N | SNPs | Transcripts | Ts/Tv | Syn | NonSyn | π | MAF | |
|---|---|---|---|---|---|---|---|---|
| All samples | 9 | 186,506 | 25,857 | 1.41 | 48,992 | 52,882 | 0.003282 (±0.036491) | 0.274 (±0.147) |
| Susceptible | 4 | 164,416 | 24,206 | 1.41 | 43,312 | 46,784 | 0.003396 (±0.039505) | 0.304 (±0.140) |
| Resistant | 5 | 166,979 | 24,514 | 1.40 | 43,809 | 47,685 | 0.003250 (±0.037789) | 0.294 (±0.142) |
| Transcript | SNP Pos. | SNP Annotation | Gene Annotation | Geno. RNA-Seq | Geno. Sanger | Val. | Gen. | Additional SNPs | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Sus | Res | Sus | Res | |||||||
| isotig67703 | 386 | 3′-UTR | pentatricopeptide repeat-containing protein At2g27610 [Quercus suber] (PCMP) | AA | GG | AA | GG | ✓ | ✓ | 304CT; 320CT |
| isotig30230 | 197 | 5′-UTR | maternal effect embryo arrest 12 [Arabidopsis thaliana] (MEE12) | AA | CC | AA | CC | ✓ | ✓ | - |
| isotig42428 | 236 | 3′-UTR | protein PHOSPHATE STARVATION RESPONSE 1 [Quercus suber] (PHR1) | AA | GG | AA | GG | ✓ | ✓ | - |
| isotig53013 | 453 | Syn | pentatricopeptide repeat-containing protein At4g21065 [Elaeis guineensis] (PCMP-H28) | CC | GG | CC | GG | ✓ | ✓ | 594TA; 651AC |
| unigene161 | 348 | Syn | kinesin-like protein KIN-12F [Nelumbo nucifera] (KIN12) | AA | GG | AA | GG | ✓ | ✓ | - |
| unigene8832 | 646 | Syn | heavy metal-associated isoprenylated plant protein 41-like [Elaeis guineensis] (HIPP41) | TT | CC | TT | CC | ✓ | ✓ | - |
| unigene52225 | 105 | Noncoding | unknown [Picea sitchensis] (ung52225) | CC | TT | CC | TT | ✓ | ✓ | 145CT; 171GA |
| isotig37698 | 586 | NonSyn | UDP-glycosyltransferase UGT5 [Picea glauca] (UGT5) | GG | CC | GC | CC | ✓ | × | 505CT; 577AT; 739TG; 745TC |
| unigene58419 | 178 | NonSyn | pentatricopeptide repeat-containing protein At3g16610 [Prunus mume] (PCMP-E91) | GG | AA | GG | AG | ✓ | × | - |
| unigene188104 | 297 | Noncoding | no annotation (ung188104) | CC | GG | CC | GG | ✓ | ✓ | - |
| 298 | Noncoding | GG | CC | GG | CC | ✓ | ✓ | - | ||
| 305 | Noncoding | TT | GG | TT | GG | ✓ | ✓ | - | ||
| isotig09645 | 590 | 5′-UTR | Guanine nucleotide-binding protein, beta subunit [Parasponia andersonii] (GB1) | AA | GG | AA | GG | ✓ | ✓ | 780AG; 804CT |
| 620 | 5′-UTR | TT | AA | TT | AA | ✓ | ✓ | |||
| isotig46969 | 1304 | NonSyn | hypothetical protein PHAVU_003G104100g [Phaseolus vulgaris] (HP) | GG | CC | CC | CC | × | × | - |
| Gene | Frag. Size (bp) | Group | Seq | Regions | SNPs | SNP Effect | π (±SD) | πS | πN | Hap. | H (±SD) | Tajima’s D | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Noncoding | Syn | NonSyn | ||||||||||||
| HIPP41 | 673 | All | 80 | 3 exons; 2 introns | 6 | 2 | 2 | 2 | 0.00227 (±0.00018) | 0.01095 | 0.00173 | 9 | 0.757 (±0.028) | 0.61044 |
| Res | 40 | - | 6 | 2 | 2 | 2 | 0.00230 (±0.00039) | 0.01020 | 0.00176 | 9 | 0.676 (±0.074) | 0.26373 | ||
| Sus | 40 | - | 4 | 1 | 2 | 1 | 0.00204 (±0.00014) | 0.01014 | 0.00167 | 6 | 0.762 (±0.031) | 1.10499 | ||
| KIN12 | 395 | All | 80 | exon | 1 | - | 1 | - | 0.00108 (±0.00011) | 0.00575 | 0.00000 | 2 | 0.425 (±0.042) | 1.32948 |
| Res | 40 | - | 1 | - | 1 | - | 0.00091 (±0.00019) | 0.00483 | 0.00000 | 2 | 0.358 (±0.073) | 0.74452 | ||
| Sus | 40 | - | 1 | - | 1 | - | 0.00122 (±0.00011) | 0.00650 | 0 | 2 | 0.481 (±0.042) | 1.49197 | ||
| MEE12 | 384 | All | 80 | 5′-UTR | 1 | 1 | - | - | 0.00130 (±0.00002) | - | - | 2 | 0.506 (±0.008) | 1.81156 |
| Res | 40 | - | 1 | 1 | - | - | 0.00124 (±0.00011) | - | - | 2 | 0.481 (±0.042) | 1.49197 | ||
| Sus | 40 | - | 1 | 1 | - | - | 0.00124 (±0.00011) | - | - | 2 | 0.481 (±0.042) | 1.49197 | ||
| PCMP-E91 | 124 | All | 68 | exon | 4 | - | - | 4 | 0.00910 (±0.00065) | 0.00000 | 0.01149 | 5 | 0.667 (±0.033) | 0.74798 |
| Res | 30 | - | 2 | - | - | 2 | 0.00803 (±0.00053) | 0.00000 | 0.01013 | 4 | 0.683 (±0.053) | 1.99045 | ||
| Sus | 38 | - | 4 | - | - | 4 | 0.00984 (±0.00102) | 0.00000 | 0.01242 | 5 | 0.653 (±0.047) | 0.68160 | ||
| PHR1 | 486 | All | 80 | 3′-UTR | 3 | 3 | - | - | 0.00216 (±0.00012) | - | - | 5 | 0.578 (±0.036) | 1.36955 |
| Res | 40 | - | 2 | 2 | - | - | 0.00206 (±0.00012) | - | - | 3 | 0.549 (±0.041) | 2.12756 * | ||
| Sus | 40 | - | 3 | 3 | - | - | 0.00230 (±0.00022) | - | - | 5 | 0.614 (±0.059) | 1.2714 | ||
| UGT5 | 297 | All | 78 | exon | 5 | - | 4 | 1 | 0.00845 (±0.00020) | 0.03037 | 0.00216 | 3 | 0.558 (±0.027) | 3.27745 ** |
| Res | 40 | - | 5 | - | 4 | 1 | 0.00856 (±0.00036) | 0.02959 | 0.00205 | 3 | 0.528 (±0.051) | 2.72844 ** | ||
| Sus | 38 | - | 5 | - | 4 | 1 | 0.00856 (±0.00036) | 0.03053 | 0.00225 | 3 | 0.585 (±0.038) | 2.92830 ** | ||
| SNP | Genetic Model | Genotypes | Sus n = 20 n (%) | Res n = 20 n (%) | OR (95% CI) | p-Value | AIC | OR (95% CI) adj. | p-Value adj. | AIC adj. |
|---|---|---|---|---|---|---|---|---|---|---|
| MEE12 SNP197 | Dominant | A/A | 8 (40%) | 2 (10%) | 1.00 | 0.0244 * | 54.4 | 1.00 | 0.0168 * | 54.7 |
| A/C-C/C | 12 (60%) | 18 (90%) | 6.00 (1.08–33.27) | 7.40 (1.20–45.67) | ||||||
| log-Additive | 0,1,2 | 20 (50%) | 20 (50%) | 3.00 (1.09–8.25) | 0.0222 * | 54.2 | 3.69 (1.23–11.09) | 0.0109 * | 53.9 | |
| PCMP-E91 SNP178 | Recessive | A/A-A/G | 10 (52.6%) | 13 (86.7%) | 1.00 | 0.0295 * | 45.9 | 1.00 | 0.0074 ** | 44.1 |
| G/G | 9 (47.4%) | 2 (13.3%) | 0.17 (0.03–0.97) | 0.07 (0.01–0.69) |
| Gene | Haplotype | Haplotype Freq. | OR (95% CI) | p-Value | |
|---|---|---|---|---|---|
| HIPP41 | 1 | CAG | 0.3868 | 1.00 | - |
| 2 | TAA | 0.1018 | 1.06 (0.19–5.91) | 0.9459 | |
| 3 | TAG | 0.2232 | 0.22 (0.06–0.84) | 0.0263 * | |
| 4 | TGA | 0.2475 | 0.34 (0.12–0.97) | 0.0441 * | |
| genoH.rare | 0.0407 | 1.00 (0.07–14.61) | 0.9995 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Modesto, I.; Inácio, V.; Novikova, P.; Carrasquinho, I.; Van de Peer, Y.; Miguel, C.M. SNP Detection in Pinus pinaster Transcriptome and Association with Resistance to Pinewood Nematode. Forests 2022, 13, 946. https://0-doi-org.brum.beds.ac.uk/10.3390/f13060946
Modesto I, Inácio V, Novikova P, Carrasquinho I, Van de Peer Y, Miguel CM. SNP Detection in Pinus pinaster Transcriptome and Association with Resistance to Pinewood Nematode. Forests. 2022; 13(6):946. https://0-doi-org.brum.beds.ac.uk/10.3390/f13060946
Chicago/Turabian StyleModesto, Inês, Vera Inácio, Polina Novikova, Isabel Carrasquinho, Yves Van de Peer, and Célia M. Miguel. 2022. "SNP Detection in Pinus pinaster Transcriptome and Association with Resistance to Pinewood Nematode" Forests 13, no. 6: 946. https://0-doi-org.brum.beds.ac.uk/10.3390/f13060946