
Multi-Organ Involvement of Immunoglobulin G4-Related Disease

 ,
,
Abstract
:1. Introduction
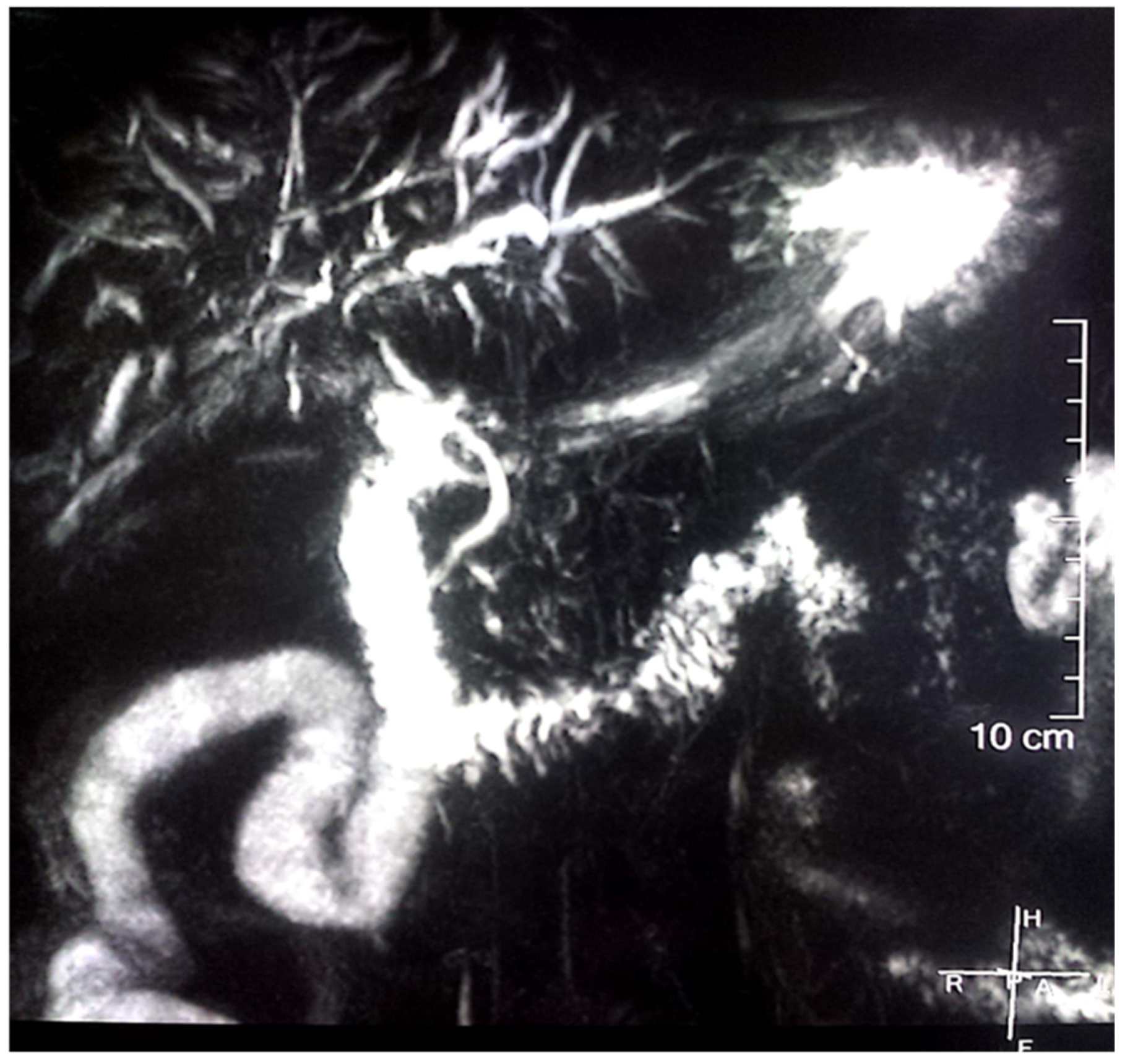
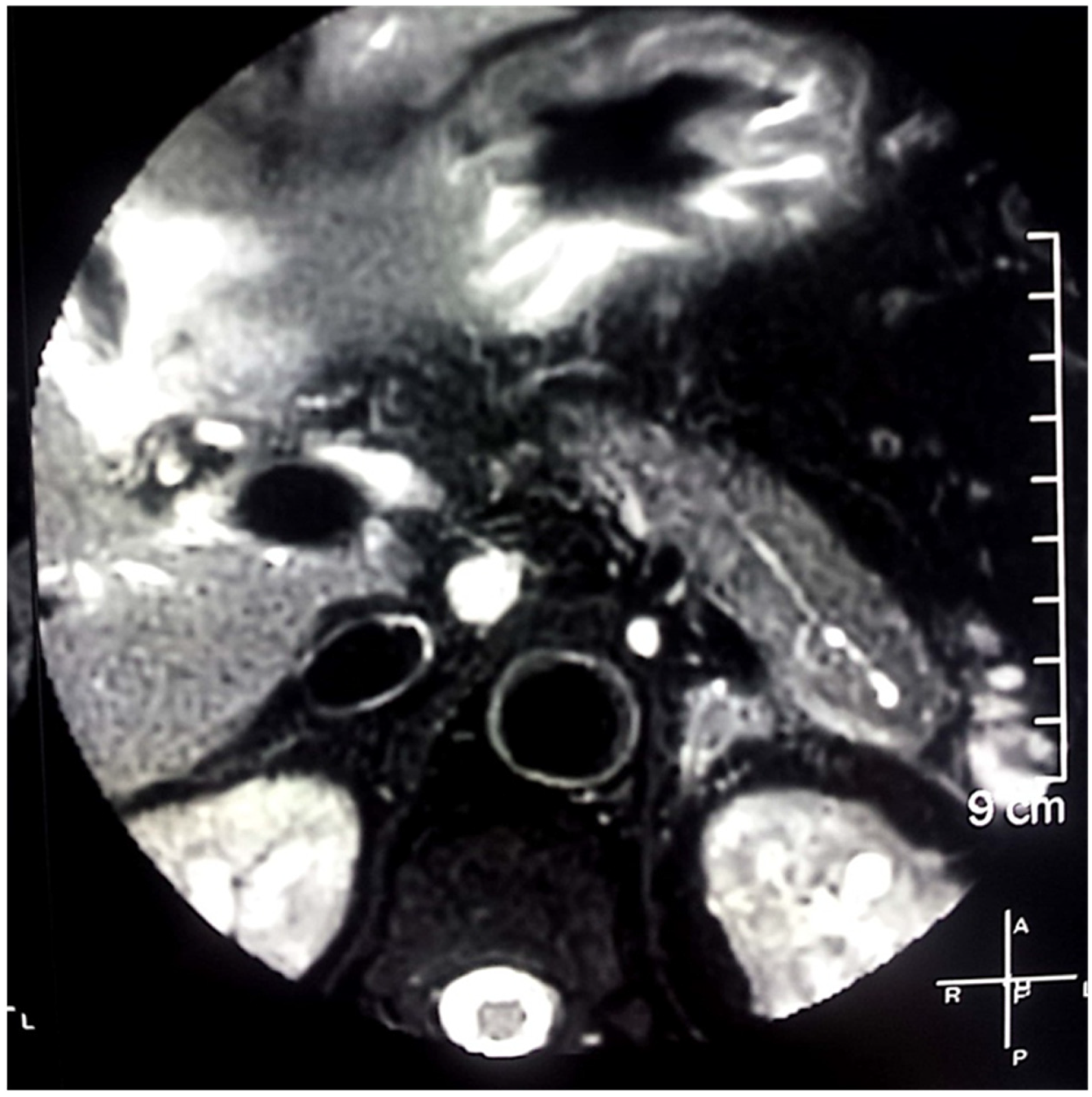
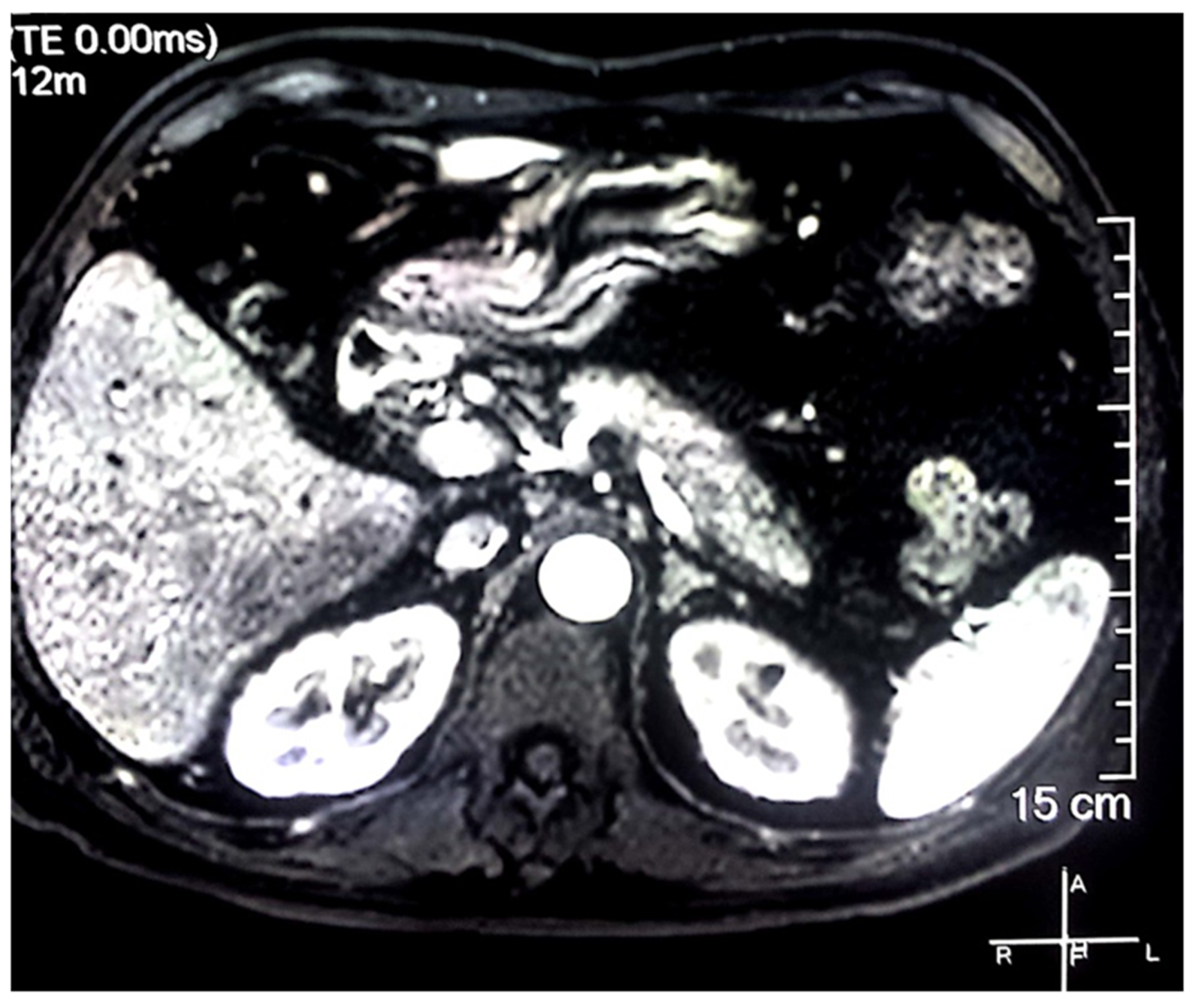
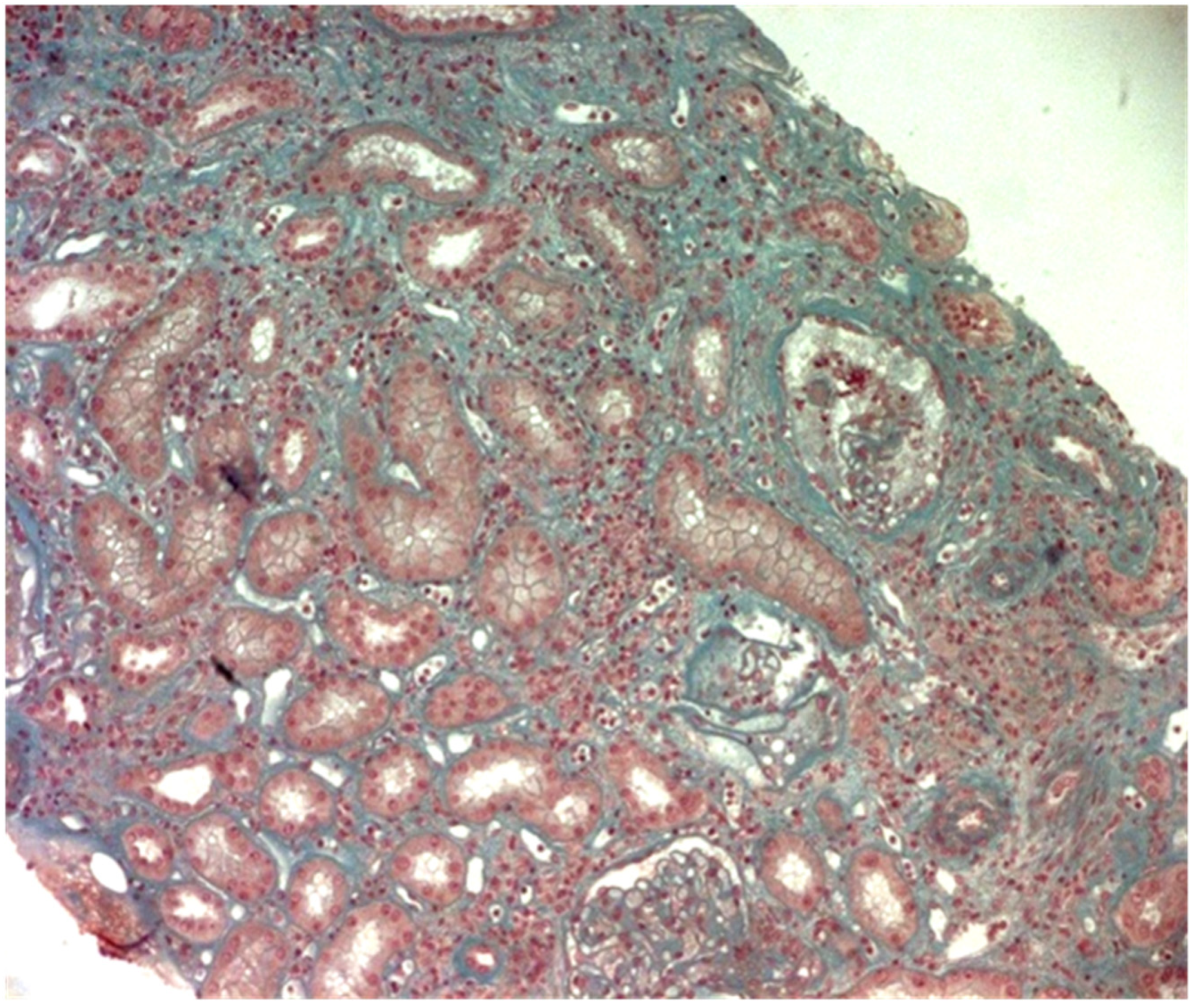
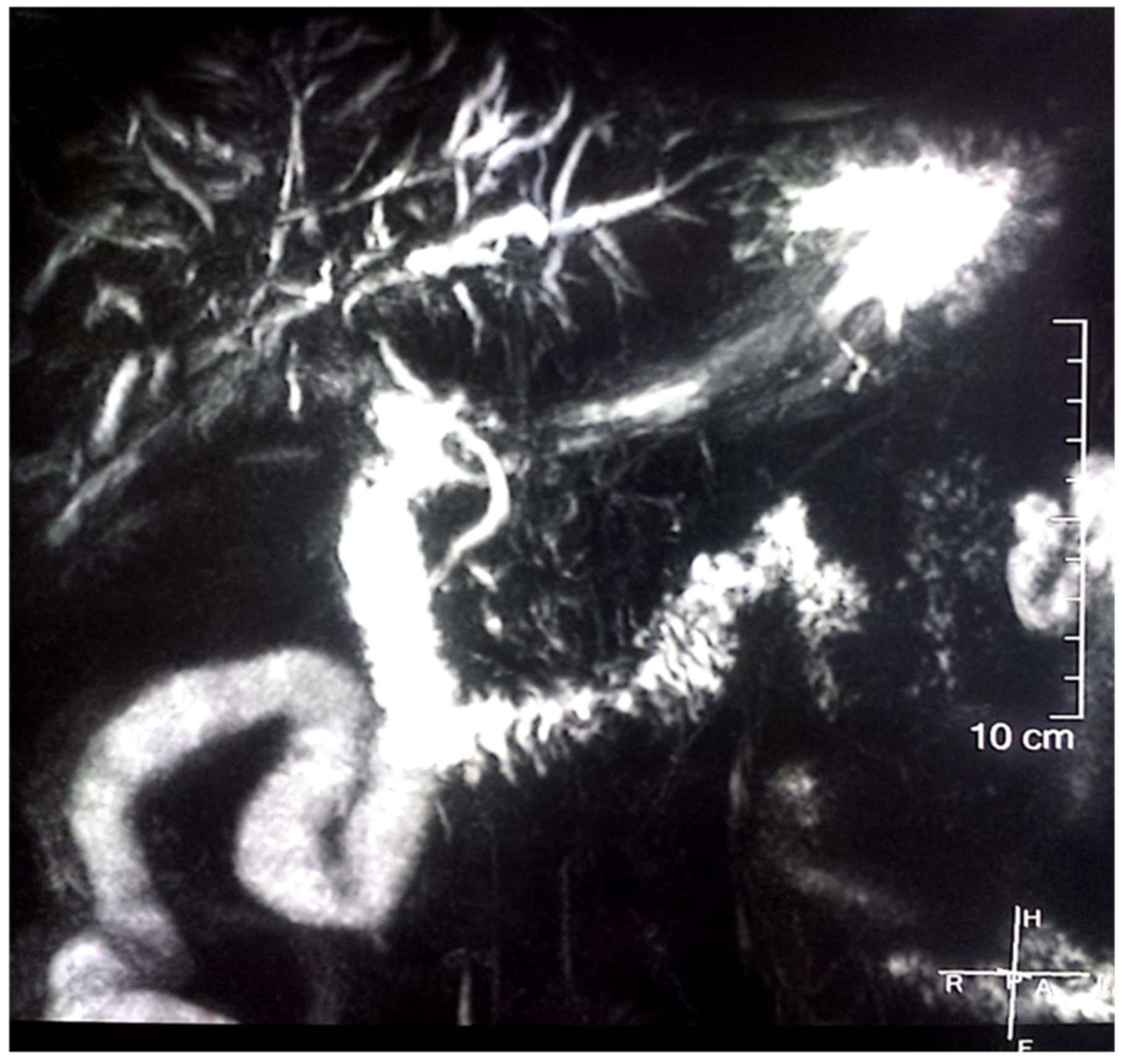
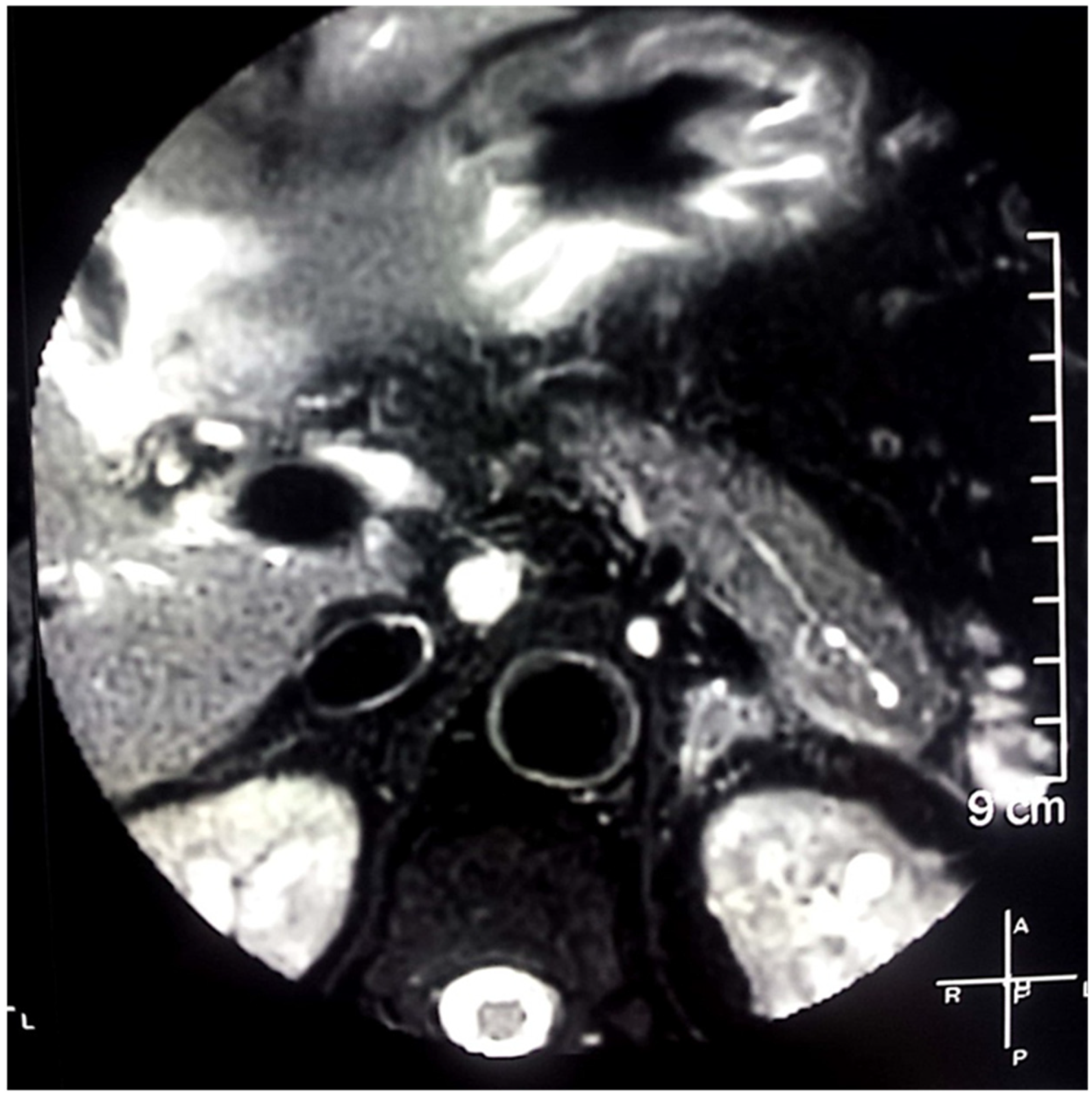
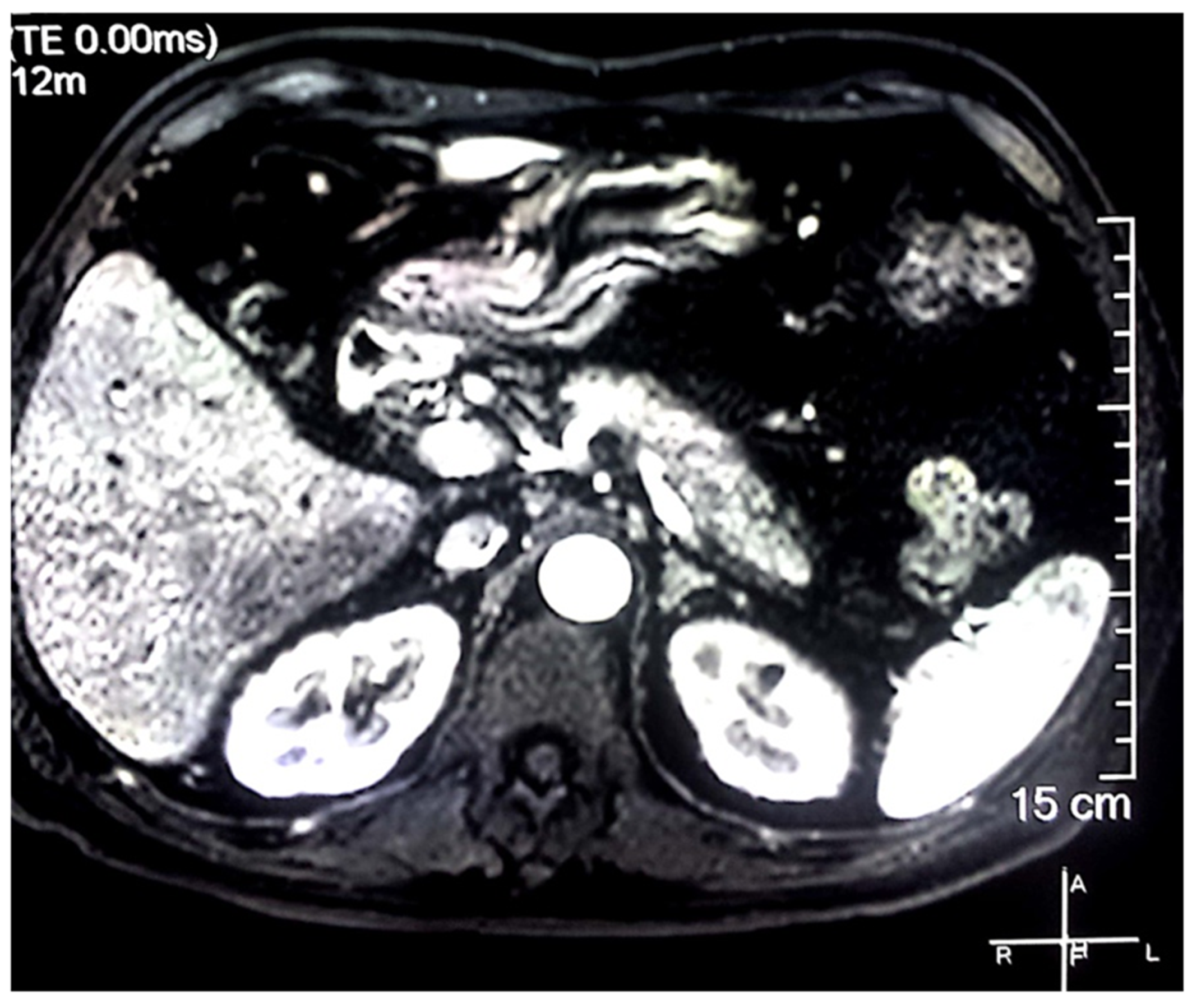
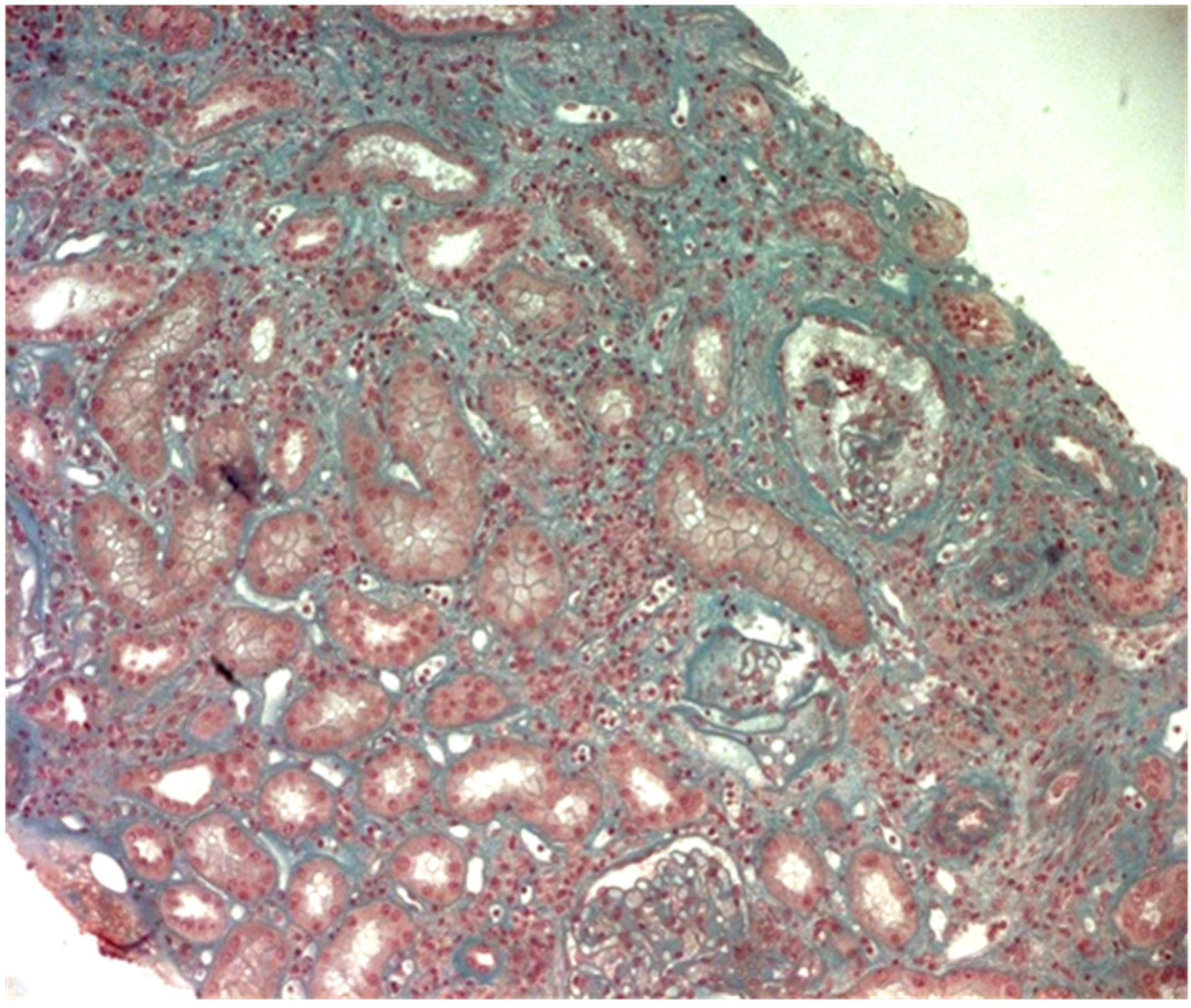
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| IgG4-RD | Immunoglobulin G4-related disease |
| AIP | IgG4-related pancreatitis |
| CD | comprehensive diagnostic |
| IgG4-SC | IgG4-sclerosing cholangitis |
References
- Umehara, H.; Okazaki, K.; Masaki, Y.; Kawano, M.; Yamamoto, M.; Saeki, T.; Matsui, S.; Sumida, T.; Mimori, T.; Tanaka, Y.; et al. A novel clinical entity, IgG4- related disease (IgG4RD): General concept and details. Mod. Rheumatol. 2012, 22, 1–14. [Google Scholar] [CrossRef]
- Nakazawa, T.; Ohara, H.; Sano, H.; Ando, T.; Aoki, S.; Kobayashi, S.; Okamoto, T.; Nomura, T.; Joh, T.; Itoh, M. Clinical differences between primary sclerosing cholangitis and sclerosing cholangitis with autoimmune pancreatitis. Pancreas 2005, 30, 20–25. [Google Scholar]
- Ghazale, A.; Chari, S.T.; Zhang, L.; Smyrk, T.C.; Takahashi, N.; Levy, M.J.; Topazian, M.D.; Clain, J.E.; Pearson, R.K.; Petersen, B.T.; et al. Immunoglobulin G4-associated cholangitis: Clinical profile and response to therapy. Gastroenterology 2008, 134, 706–715. [Google Scholar] [CrossRef]
- Stone, J.H.; Brito-Zeron, P.; Bosch, X.; Ramos-Casals, M. Diagnostic approach to the complexity of IgG4-related disease. Mayo Clin. Proc. 2015, 90, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Hamano, H.; Kawa, S.; Horiuchi, A.; Unno, H.; Furuya, N.; Akamatsu, T.; Fukushima, M.; Nikaido, T.; Nakayama, K.; Usuda, N.; et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N. Engl. J. Med. 2001, 344, 732–738. [Google Scholar] [CrossRef]
- Kamisawa, T.; Funata, N.; Hayashi, Y.; Eishi, Y.; Koike, M.; Tsuruta, K.; Okamoto, A.; Egawa, N.; Nakajima, H. A new clinicopathological entity of IgG4-related autoimmune disease. J. Gastroenterol. 2003, 38, 982–984. [Google Scholar] [CrossRef] [PubMed]
- Webster, G.J.; Yamamoto, M.; Zamboni, G.; Umehara, H.; Stone, J.H. Consensus statement on the pathology of IgG4-related disease. Mod. Pathol. 2012, 25, 1181–1192. [Google Scholar]
- Umehara, H.; Okazaki, K.; Masaki, Y.; Kawano, M.; Yamamoto, M.; Saeki, T.; Matsui, S.; Yoshino, T.; Nakamura, S.; Kawa, S.; et al. Comprehensive diagnostic criteria for IgG4-related disease. (IgG4-RD). Mod. Rheumatol. 2012, 22, 21–30. [Google Scholar] [CrossRef]
- Stone, J.H.; Zen, Y.; Deshpande, V. IgG4-related disease. N. Engl. J. Med. 2012, 366, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Sato, Y.; Ohara, N.; Hashimoto, K.; Kobashi, H.; Koyama, Y.; Yoshino, T. Systemic IgG4-related disease with extensive peripheral nearve involvement that progressed from localized IgG4-related lymphadenopathy: An autopsy case. Diagn. Pathol. 2014, 9, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbajal, H.; Waters, L.; Popovich, J.; Boniuk, M.; Chevez-Barrios, P.; Marcus, D.M.; Sessoms, S. IgG4 related cardiac disease. Methodist DeBakey Cardiovasc. J. 2013, 9, 230–232. [Google Scholar] [CrossRef] [Green Version]
- Ise, M.; Yasuda, F.; Suzaki, R.; Kurihara, S.; Konohana, I. Skin lesions in a patient with IgG4-related disease. Clin. Exp. Dermatol. 2014, 39, 713–716. [Google Scholar] [CrossRef]
- Andrew, N.; Kearney, D.; Sladden, N.; Goss, A.; Selva, D. Immunoglobulin G4-related disease of the hard palate. J. Oral Maxillofac. Surg. 2014, 72, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.; Woo, J.Y.; Kim, J.W.; Hong, H.S.; Yang, I.; Lee, Y.; Hwang, D.; Min, S.J. An immunoglobulin G4-related sclerosing disease of the small bowel: CT and small bowel series findings. Korean J. Radiol. 2013, 14, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Dumas-Campagna, M.; Bouchard, S.; Soucy, G.; Bouin, M. IgG4-related esophageal disease presenting as esophagitis dissecanssuperficialis with chronic strictures. J. Clin. Med. Res. 2014, 6, 295–298. [Google Scholar] [PubMed] [Green Version]
- Wimmer, D.B.; Ro, J.Y.; Lewis, A.; Schwartz, M.R.; Caplan, R.; Schwarz, P.; Ayala, A.G. ExtranodalRosai-Dorfman disease associated with increased numbers of immunoglobulin G4 plasma cells involving the colon: Case report with literature review. Arch. Pathol. Lab. Med. 2013, 137, 999–1004. [Google Scholar] [CrossRef]
- Wallace, Z.S.; Perugino, C.; Matza, M.; Deshpande, V.; Sharma, A.; Stone, J.H. Immunoglobulin G4-related Disease. Clin. Chest Med. 2019, 40, 583–597. [Google Scholar] [CrossRef]
- Umeharaa, H.; Okazakib, K.; Nakamuraa, T.; Satoh-Nakamuraa, T.; Nakajimac, A.; Kawanod, M.; Mimorie, T.; Chibaf, T. Current approach to the diagnosis of IgG4-related disease—Combination of comprehensive diagnostic and organ-specific criteria. Mod. Rheumatol. 2017, 27, 381–391. [Google Scholar] [CrossRef]
- Ohara, H.; Okazaki, K.; Tsubouchi, H.; Inui, K.; Kawa, S.; Kamisawa, T.; Tazuma, S.; Uchida, K.; Hirano, K.; Yoshida, H.; et al. Clinical diagnostic criteria of IgG4-related sclerosing cholangitis. J. Hepato-Biliary-Pancreat. Sci. 2012, 19, 536–542. [Google Scholar] [CrossRef]
- Kawano, M.; Saeki, T.; Nakashima, H.; Nishi, S.; Yamaguchi, Y.; Hisano, S.; Yamanaka, N.; Inoue, D.; Yamamoto, M.; Takahashi, H.; et al. Proposal for diagnostic criteria for IgG4-related kidney disease. Clin. Exp. Nephrol. 2011, 15, 615–626. [Google Scholar] [CrossRef]
- Salvadori, M.; Tsalouchos, A. Immunoglobulin G4-related kidney diseases: An updated review. World J. Nephrol. 2018, 6, 29–40. [Google Scholar] [CrossRef]
- Lee, H.E.; Zhang, L. Immunoglobulin G4 related hepatobiliary disease. Semin. Diagn. Pathol. 2019, 36, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Ghazale, A.H.; Smyrk, T.C.; Mandrekar, J.N.; Chari, S.T. Possible association between IgG4-associated systemic disease with or without autoimmune pancreatitis and non-Hodgkin lymphoma. Pancreas 2009, 38, 523–526. [Google Scholar] [CrossRef]
- Carruthers, M.N.; Topazian, M.D.; Khosroshahi, A.; Witzig, T.E.; Wallace, Z.S.; Hart, P.A.; Deshpande, V.; Smyrk, T.C.; Chari, S.; Stone, J.H. Rituximab for IgG4-related disease: A prospective, open-label trial. Ann. Rheum. Dis. 2015, 74, 1171–1177. [Google Scholar] [CrossRef]
- Han, G.J.; Hu, H.; Mao, D.; Bai, X.; She, D.Y.; Zhao, S.F.; Wen, Z.L.; Gao, J. IgG4-related lung disease: Analysis of 8 cases and literature review. Chin. J. Tuberc. Respir. Dis. 2017, 12, 193–198. [Google Scholar]
- Ebbo, M.; Grados, A.; Samson, M.; Groh, M.; Loundou, A.; Rigolet, A.; Terrier, B.; Guillaud, C.; Carra-Dallière, C.; Renou, F.; et al. Long-term efficacy and safety of rituximab in IgG4-related disease: Data from a French nationwide study of thirty-three patients. PLoS ONE 2017, 12, e0183844. [Google Scholar] [CrossRef] [PubMed]
- Al-Mujaini, A.; Al-Khabori, M.; Shenoy, K.; Wali, U. Immunoglobulin G4-Related Disease: An Update. Oman Med. J. 2018, 33, 97–103. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1. Clinically: characteristic diffuse/localized swelling or masses in single or multiple organs |
| 2. Hematologically: an elevated serum IgG4 level (>135 mg/dL) |
| 3. Histologically: |
| (i) marked lymphoplasmacytic infiltration and fibrosis; |
| (ii) infiltration of IgG4+ plasma cells with a ratio of IgG4/IgG cells of more than 40% and more than 10 IgG4+ cells/ high power field |
| 1 + 2 + 3 as “definite” |
| 1 + 3 as “probable” |
| 1 + 2 as “possible” |
| 1. Presence of some kidney damage, as manifested by abnormal urinalysis or urine marker(s) or decreased kidney function with either elevated serum IgG level, hypocomplementemia, or elevated serum IgE level |
| 2. Abnormal renal radiologic findings: |
| a. Multiple low-density lesions on enhanced computed tomography |
| b. Diffuse kidney enlargement |
| c. Hypovascular solitary mass in the kidney |
| d. Hypertrophic lesion of renal pelvic wall without irregularity of the renal pelvic surface |
| 3. Elevated serum IgG4 level (IgG4 ≥ 135 mg/dL) |
| 4. Histologic findings in the kidney |
| a. Dense lymphoplasmacytic infiltration with infiltrating IgG4-positive plasma cells [10/high power field (HPF) and/or IgG4/IgG-positive plasma cells (40%) |
| b. Characteristic fibrosis surrounding nests of lymphocytes and/or plasma cells |
| 5. Histologic findings in extra-renal organ(s): |
| Dense lymphoplasmacytic infiltration with infiltrating IgG4-positive plasma cells [10/HPF and/or IgG4/IgG-positive plasma cells 40% in extra-renal organ(s) |
| Definite: |
| 1 + 3 + 4 a, b |
| 2 + 3 + 4 a, b |
| 2 + 3 + 5 |
| 1 + 3 + 4 a + 5 |
| Probable: |
| 1 + 4 a, b |
| 2 + 4 a, b |
| 2 + 5 |
| 3 + 4 a, b |
| Possible: |
| 1 + 3 |
| 2 + 3 |
| 1 + 4 a |
| 2 + 4 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nour, E.; Hammami, A.; Missaoui, N.; Bdioui, A.; Dahmani, W.; Ben Ameur, W.; Braham, A.; Ajmi, S.; Ben Slama, A.; Ksiaa, M.; et al. Multi-Organ Involvement of Immunoglobulin G4-Related Disease. Gastroenterol. Insights 2021, 12, 350-357. https://0-doi-org.brum.beds.ac.uk/10.3390/gastroent12030033
Nour E, Hammami A, Missaoui N, Bdioui A, Dahmani W, Ben Ameur W, Braham A, Ajmi S, Ben Slama A, Ksiaa M, et al. Multi-Organ Involvement of Immunoglobulin G4-Related Disease. Gastroenterology Insights. 2021; 12(3):350-357. https://0-doi-org.brum.beds.ac.uk/10.3390/gastroent12030033
Chicago/Turabian StyleNour, Elleuch, Aya Hammami, Nabiha Missaoui, Ahlem Bdioui, Wafa Dahmani, Wafa Ben Ameur, Ahlem Braham, Salem Ajmi, Aida Ben Slama, Mehdi Ksiaa, and et al. 2021. "Multi-Organ Involvement of Immunoglobulin G4-Related Disease" Gastroenterology Insights 12, no. 3: 350-357. https://0-doi-org.brum.beds.ac.uk/10.3390/gastroent12030033