

The Microbiome and Antibiotic Resistome in Soil under Biodegradable Composite Carbon Source Amendment
 , ,
, ,
Abstract
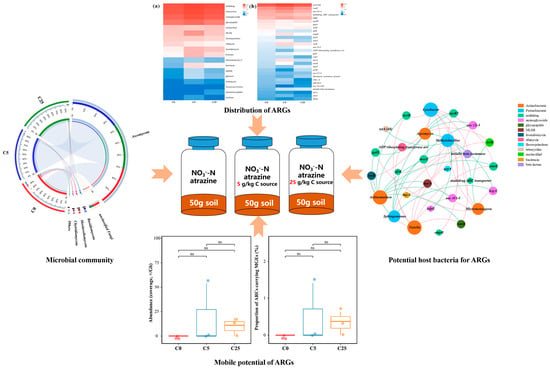
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials and Sample Collection
2.2. Microcosm Experiment
2.3. DNA Extraction and Illumina MiSeq Sequencing Analysis
2.4. Metagenomic Analysis
2.5. Network Analysis
2.6. Statistical Analyses
3. Results and Discussion
3.1. The Effect of a Composite Carbon Source on the Microbial Community
3.2. Network Analysis of the Microbial Community
3.3. Changes in Nitrogen Cycling Processes and Involved Functional Genes
3.4. The Antibiotic Resistome in the Soil Samples
3.5. Potential Host Bacteria for ARGs
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tilman, D.; Cassman, K.G.; Matson, P.A.; Naylor, R.; Polasky, S. Agricultural sustainability and intensive production practices. Nature 2002, 418, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Castaldelli, G.; Colombani, N.; Soana, E.; Vincenzi, F.; Fano, E.A.; Mastrocicco, M. Reactive nitrogen losses via denitrification assessed in saturated agricultural soils. Geoderma 2019, 337, 91–98. [Google Scholar] [CrossRef]
- Ahmed, M.; Rauf, M.; Mukhtar, Z.; Saeed, N.A. Excessive use of nitrogenous fertilizers: An unawareness causing serious threats to environment and human health. Environ. Sci. Pollut. Res. 2017, 24, 26983–26987. [Google Scholar] [CrossRef]
- Romantschuk, M.; Sarand, I.; Petänen, T.; Peltola, R.; Jonsson-Vihanne, M.; Koivula, T.; Yrjälä, K.; Haahtela, K. Means to improve the effect of in situ bioremediation of contaminated soil: An overview of novel approaches. Environ. Pollut. 2000, 107, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Vidali, M. Bioremediation. An overview. Pure Appl. Chem. 2001, 73, 1163–1172. [Google Scholar] [CrossRef]
- Tomei, M.C.; Daugulis, A.J. Ex situ bioremediation of contaminated soils: An overview of conventional and innovative technologies. Crit. Rev. Environ. Sci. Technol. 2013, 43, 2107–2139. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Lou, Y.; Pan, H.; Wang, H.; Yang, Q.; Sun, Y.; Zhuge, Y. Reinforced Bioremediation of Excessive Nitrate in Atrazine-Contaminated Soil by Biodegradable Composite Carbon Source. Polymers 2023, 15, 2765. [Google Scholar] [CrossRef]
- Wang, J.; Chu, L. Biological nitrate removal from water and wastewater by solid-phase denitrification process. Biotechnol. Adv. 2016, 34, 1103–1112. [Google Scholar] [CrossRef]
- Yang, Z.C.; Sun, H.M.; Zhou, Q.; Zhao, L.; Wu, W.Z. Nitrogen removal performance in pilot-scale solid-phase denitrification systems using novel biodegradable blends for treatment of waste water treatment plants effluent. Bioresour. Technol. 2020, 305, 122994. [Google Scholar] [CrossRef]
- Yang, Z.; Zhou, Q.; Sun, H.; Jia, L.; Zhao, L.; Wu, W. Metagenomic analyses of microbial structure and metabolic pathway in solid-phase denitrification systems for advanced nitrogen removal of wastewater treatment plant effluent: A pilot-scale study. Water Res. 2021, 196, 117067. [Google Scholar] [CrossRef]
- Fuller, S.; Gautam, A. A procedure for measuring microplastics using pressurized fluid extraction. Environ. Sci. Technol. 2016, 50, 5774–5780. [Google Scholar] [CrossRef] [Green Version]
- Hernando-Amado, S.; Coque, T.M.; Baquero, F.; Martínez, J.L. Defining and combating antibiotic resistance from One Health and Global Health perspectives. Nat. Microbiol. 2019, 4, 1432–1442. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-G.; Zhao, Y.; Zhu, D.; Gillings, M.; Penuelas, J.; Ok, Y.S.; Capon, A.; Banwart, S. Soil biota, antimicrobial resistance and planetary health. Environ. Int. 2019, 131, 105059. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, W.; Yang, X.; Wang, J.; Lin, H.; Yang, Y. Microplastics are a hotspot for antibiotic resistance genes: Progress and perspective. Sci. Total Environ. 2021, 773, 145643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, S.; Zhang, Q.; Long, Z.; Yu, Y.; Fang, H. Fungicides enhanced the abundance of antibiotic resistance genes in greenhouse soil. Environ. Pollut. 2020, 259, 113877. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhang, J.; Lu, T.; Zhang, K. Metagenomics revealed the mobility and hosts of antibiotic resistance genes in typical pesticide wastewater treatment plants. Sci. Total Environ. 2022, 817, 153033. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lou, Y.; Pan, H.; Wang, H.; Yang, Q.; Zhuge, Y.; Hu, J. Improved Denitrification Performance of Polybutylene Succinate/Corncob Composite Carbon Source by Proper Pretreatment: Performance, Functional Genes and Microbial Community Structure. Polymers 2023, 15, 801. [Google Scholar] [CrossRef]
- Fang, H.; Lian, J.; Wang, H.; Cai, L.; Yu, Y. Exploring bacterial community structure and function associated with atrazine biodegradation in repeatedly treated soils. J. Hazard. Mater. 2015, 286, 457–465. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Arango-Argoty, G.; Garner, E.; Prudent, A.; Heath, L.S.; Vikesland, P.; Zhang, L.Q. DeepARG: A deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 2018, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Yu, K.; Zhang, J.; Zhang, G.; Huang, J.; Ma, L.; Deng, C.; Li, X.; Li, B. Deciphering the mobility and bacterial hosts of antibiotic resistance genes under antibiotic selection pressure by metagenomic assembly and binning approaches. Water Res. 2020, 186, 116318. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Wang, Y.; Sun, X.; Peng, Y.; Xiao, L. Mixing effect of polylactic acid microplastic and straw residue on soil property and ecological function. Chemosphere 2020, 243, 125271. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Wang, Y.; Ye, S.; Liu, S.; Stirling, E.; Gilbert, J.A.; Faust, K.; Knight, R.; Jansson, J.K.; Cardona, C. Earth microbial co-occurrence network reveals interconnection pattern across microbiomes. Microbiome 2020, 8, 82. [Google Scholar] [CrossRef]
- Ramette, A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, K.S.; Leff, J.W.; Barberán, A.; Bates, S.T.; Betley, J.; Crowther, T.W.; Kelly, E.F.; Oldfield, E.E.; Shaw, E.A.; Steenbock, C. Biogeographic patterns in below-ground diversity in New York City’s Central Park are similar to those observed globally. Proc. R. Soc. B 2014, 281, 20141988. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Yergeau, E.; Hogues, H.; Whyte, L.G.; Greer, C.W. The functional potential of high Arctic permafrost revealed by metagenomic sequencing, qPCR and microarray analyses. ISME J. 2010, 4, 1206–1214. [Google Scholar] [CrossRef]
- Lladó, S.; Žifčáková, L.; Větrovský, T.; Eichlerová, I.; Baldrian, P. Functional screening of abundant bacteria from acidic forest soil indicates the metabolic potential of Acidobacteria subdivision 1 for polysaccharide decomposition. Biol. Fertil. Soils 2016, 52, 251–260. [Google Scholar] [CrossRef]
- Hug, L.A.; Castelle, C.J.; Wrighton, K.C.; Thomas, B.C.; Sharon, I.; Frischkorn, K.R.; Williams, K.H.; Tringe, S.G.; Banfield, J.F. Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 2013, 1, 22. [Google Scholar] [CrossRef] [Green Version]
- Orwin, K.; Dickie, I.; Holdaway, R.; Wood, J. A comparison of the ability of PLFA and 16S rRNA gene metabarcoding to resolve soil community change and predict ecosystem functions. Soil Biol. Biochem. 2018, 117, 27–35. [Google Scholar] [CrossRef]
- Wolińska, A.; Kuźniar, A.; Zielenkiewicz, U.; Izak, D.; Szafranek-Nakonieczna, A.; Banach, A.; Błaszczyk, M. Bacteroidetes as a sensitive biological indicator of agricultural soil usage revealed by a culture-independent approach. Appl. Soil Ecol. 2017, 119, 128–137. [Google Scholar] [CrossRef]
- Ganzert, L.; Bajerski, F.; Wagner, D. Bacterial community composition and diversity of five different permafrost-affected soils of Northeast Greenland. FEMS Microbiol. Ecol. 2014, 89, 426–441. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhang, B.; Chen, J.; Mao, W.; Lou, L.; Shen, C.; Lin, Q. Biofertilizer-induced response to cadmium accumulation in Oryza sativa L. grains involving exogenous organic matter and soil bacterial community structure. Ecotoxicol. Environ. Saf. 2021, 211, 111952. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, Y.; Mao, W.; Wang, C.; Yin, S. Functional potential differences between Firmicutes and Proteobacteria in response to manure amendment in a reclaimed soil. Can. J. Microbiol. 2020, 66, 689–697. [Google Scholar] [CrossRef]
- Tang, Z.; Zhang, L.; He, N.; Gong, D.; Gao, H.; Ma, Z.; Fu, L.; Zhao, M.; Wang, H.; Wang, C. Soil bacterial community as impacted by addition of rice straw and biochar. Sci. Rep. 2021, 11, 22185. [Google Scholar] [CrossRef]
- Chen, W.-M.; Moulin, L.; Bontemps, C.; Vandamme, P.; Béna, G.; Boivin-Masson, C. Legume Symbiotic Nitrogen Fixation by β-Proteobacteria Is Widespread in Nature. J. Bacteriol. 2003, 185, 7266–7272. [Google Scholar] [CrossRef] [Green Version]
- Tedersoo, L.; Bahram, M.; Põlme, S.; Kõljalg, U.; Yorou, N.S.; Wijesundera, R.; Ruiz, L.V.; Vasco-Palacios, A.M.; Thu, P.Q.; Suija, A. Global diversity and geography of soil fungi. Science 2014, 346, 1256688. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, M.; Brunner, I.; Hagedorn, F.; Bardgett, R.D.; Stierli, B.; Herzog, C.; Chen, X.; Zingg, A.; Graf-Pannatier, E.; Rigling, A. A decade of irrigation transforms the soil microbiome of a semi-arid pine forest. Mol. Ecol. 2017, 26, 1190–1206. [Google Scholar] [CrossRef] [Green Version]
- Castillo, J.M.; Nogales, R.; Romero, E. Biodegradation of 3, 4 dichloroaniline by fungal isolated from the preconditioning phase of winery wastes subjected to vermicomposting. J. Hazard. Mater. 2014, 267, 119–127. [Google Scholar] [CrossRef]
- Dia, A.; Lauga, B.; Davranche, M.; Fahy, A.; Duran, R.; Nowack, B.; Petitjean, P.; Henin, O.; Martin, S.; Marsac, R. Bacteria-mediated reduction of As (V)-doped lepidocrocite in a flooded soil sample. Chem. Geol. 2015, 406, 34–44. [Google Scholar] [CrossRef] [Green Version]
- Postma, J.; Schilder, M.T. Enhancement of soil suppressiveness against Rhizoctonia solani in sugar beet by organic amendments. Appl. Soil Ecol. 2015, 94, 72–79. [Google Scholar] [CrossRef]
- Grady, E.N.; MacDonald, J.; Liu, L.; Richman, A.; Yuan, Z.-C. Current knowledge and perspectives of Paenibacillus: A review. Microb. Cell Factories 2016, 15, 203. [Google Scholar] [CrossRef] [Green Version]
- Ramos, S.M.S.; Cruz, R.; Barbosa, R.d.N.; Machado, A.R.; Costa, A.F.d.; Motta, C.M.d.S.; Oliveira, N.T.d. Penicillium and Talaromyces communities of sugarcane soils (Saccharum officinarum L.): Ecological and phylogenetic aspects. J. Agric. Sci. 2018, 10, 335. [Google Scholar] [CrossRef] [Green Version]
- Toma, Y.; Takakai, F.; Darung, U.; Kuramochi, K.; Limin, S.H.; Dohong, S.; Hatano, R. Nitrous oxide emission derived from soil organic matter decomposition from tropical agricultural peat soil in central Kalimantan, Indonesia. Soil Sci. Plant Nutr. 2011, 57, 436–451. [Google Scholar] [CrossRef] [Green Version]
- Soni, S.K.; Soni, R. Regulation of cellulase synthesis in Chaetomium erraticum. Bioresources 2010, 5, 81–98. [Google Scholar] [CrossRef]
- Singh, S.B.; Lal, S.P.; Pant, S.; Kulshrestha, G. Degradation of atrazine by an acclimatized soil fungal isolate. J. Environ. Sci. Health B 2008, 43, 27–33. [Google Scholar] [CrossRef]
- Fan, X.; Song, F. Bioremediation of atrazine: Recent advances and promises. J. Soils Sediments 2014, 14, 1727–1737. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, W.; Li, Y.; Zhang, C.; Wang, L.; Niu, L.; Zhang, H. Microbial community shift via black carbon: Insight into biological nitrogen removal from microbial assemblage and functional patterns. Environ. Res. 2021, 192, 110266. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Peng, C.; Dai, Y.; Li, Y.; Jiao, S.; Ma, X.; Liu, X.; Wang, L. Slower antibiotics degradation and higher resistance genes enrichment in plastisphere. Water Res. 2022, 222, 118920. [Google Scholar] [CrossRef]
- Hollister, E.B.; Engledow, A.S.; Hammett, A.J.M.; Provin, T.L.; Wilkinson, H.H.; Gentry, T.J. Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J. 2010, 4, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Yang, J.; Wang, B.C.; Liu, W.; Hua, Z.S.; Jiang, H.C. Influence of salinity on the diversity and composition of carbohydrate metabolism, nitrogen and sulfur cycling genes in lake surface sediments. Front. Microbiol. 2022, 13, 1019010. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-G.; Johnson, T.A.; Su, J.-Q.; Qiao, M.; Guo, G.-X.; Stedtfeld, R.D.; Hashsham, S.A.; Tiedje, J.M. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc. Natl. Acad. Sci. USA 2013, 110, 3435–3440. [Google Scholar] [CrossRef] [PubMed]
- Akhil, D.; Lakshmi, D.; Senthil Kumar, P.; Vo, D.-V.N.; Kartik, A. Occurrence and removal of antibiotics from industrial wastewater. Environ. Chem. Lett. 2021, 19, 1477–1507. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Wang, J.; Zhu, L.; Conkle, J.L.; Yang, R. Soil types influence the characteristic of antibiotic resistance genes in greenhouse soil with long-term manure application. J. Hazard. Mater. 2020, 392, 122334. [Google Scholar] [CrossRef]
- Xu, Y.; Li, H.; Tan, L.; Li, Q.; Liu, W.; Zhang, C.; Gao, Y.; Wei, X.; Gong, Q.; Zheng, X. What role does organic fertilizer actually play in the fate of antibiotic resistome and pathogenic bacteria in planting soil? J. Environ. Manag. 2022, 317, 115382. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Jia, S.; He, X.; Zhang, X.; Ye, L. Different impacts of manure and chemical fertilizers on bacterial community structure and antibiotic resistance genes in arable soils. Chemosphere 2017, 188, 455–464. [Google Scholar] [CrossRef]
- Park, M.; Rooney, A.P.; Hecht, D.W.; Li, J.; McClane, B.A.; Nayak, R.; Paine, D.D.; Rafii, F. Phenotypic and genotypic characterization of tetracycline and minocycline resistance in Clostridium perfringens. Arch. Microbiol. 2010, 192, 803–810. [Google Scholar] [CrossRef]
- Morar, M.; Wright, G.D. The genomic enzymology of antibiotic resistance. Annu. Rev. Genet. 2010, 44, 25–51. [Google Scholar] [CrossRef]
- Imchen, M.; Kumavath, R. Metagenomic insights into the antibiotic resistome of mangrove sediments and their association to socioeconomic status. Environ. Pollut. 2021, 268, 115795. [Google Scholar] [CrossRef] [PubMed]
- Faron, M.L.; Ledeboer, N.A.; Buchan, B.W. Resistance mechanisms, epidemiology, and approaches to screening for vancomycin-resistant Enterococcus in the health care setting. J. Clin. Microbiol. 2016, 54, 2436–2447. [Google Scholar] [CrossRef] [Green Version]
- Bank, M.S.; Ok, Y.S.; Swarzenski, P.W. Microplastic’s role in antibiotic resistance. Science 2020, 369, 1315. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-M.; Lu, P.-Z. Distribution of antibiotic resistance genes in soil amended using Azolla imbricata and its driving mechanisms. Sci. Total Environ. 2019, 692, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Wang, F.; Liu, H.; Liu, H.; Pu, S.; Lin, F.; Geng, H.; Ma, S.; Zhang, Y.; Tian, Z. Deciphering the toxic effects of metals in gold mining area: Microbial community tolerance mechanism and change of antibiotic resistance genes. Environ. Res. 2020, 189, 109869. [Google Scholar] [CrossRef] [PubMed]
- Radu, E.; Woegerbauer, M.; Rab, G.; Oismüller, M.; Strauss, P.; Hufnagl, P.; Gottsberger, R.A.; Krampe, J.; Weyermair, K.; Kreuzinger, N. Resilience of agricultural soils to antibiotic resistance genes introduced by agricultural management practices. Sci. Total Environ. 2021, 756, 143699. [Google Scholar] [CrossRef]
- Song, B.; Shang, S.; Cai, F.M.; Liu, Z.; Fang, J.; Adams, J.M.; Razavi, B.S. Microbial resistance in rhizosphere hotspots under biodegradable and conventional microplastic amendment: Community and functional sensitivity. Soil Biol. Biochem. 2023, 180, 108989. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Lou, Y.; Yan, X.; Pan, H.; Wang, H.; Yang, Q.; Sun, Y.; Zhuge, Y. The Microbiome and Antibiotic Resistome in Soil under Biodegradable Composite Carbon Source Amendment. J. Xenobiot. 2023, 13, 424-438. https://0-doi-org.brum.beds.ac.uk/10.3390/jox13030027
Yang Z, Lou Y, Yan X, Pan H, Wang H, Yang Q, Sun Y, Zhuge Y. The Microbiome and Antibiotic Resistome in Soil under Biodegradable Composite Carbon Source Amendment. Journal of Xenobiotics. 2023; 13(3):424-438. https://0-doi-org.brum.beds.ac.uk/10.3390/jox13030027
Chicago/Turabian StyleYang, Zhongchen, Yanhong Lou, Xianghui Yan, Hong Pan, Hui Wang, Quangang Yang, Yajie Sun, and Yuping Zhuge. 2023. "The Microbiome and Antibiotic Resistome in Soil under Biodegradable Composite Carbon Source Amendment" Journal of Xenobiotics 13, no. 3: 424-438. https://0-doi-org.brum.beds.ac.uk/10.3390/jox13030027