Influence of Tree Vegetation on Soil Microbial Communities in Temperate Forests and Their Potential as a Proactive Indicator of Vegetation Shift Due to Climate Change

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Areas and Soil Sampling
2.2. Soil Properties Analysis
2.3. DNA Library Preparation and Amplicon Sequencing
2.4. Sequence Processing and OTU Clustering
2.5. Bioinformatics and Statistical Analysis
3. Results
3.1. Environmental Properties
3.2. Illumina Sequencing Results and Diversity Indices
3.3. Microbial Community Compositions
3.4. Biomarkers and Their Correlations with Environmental Factors
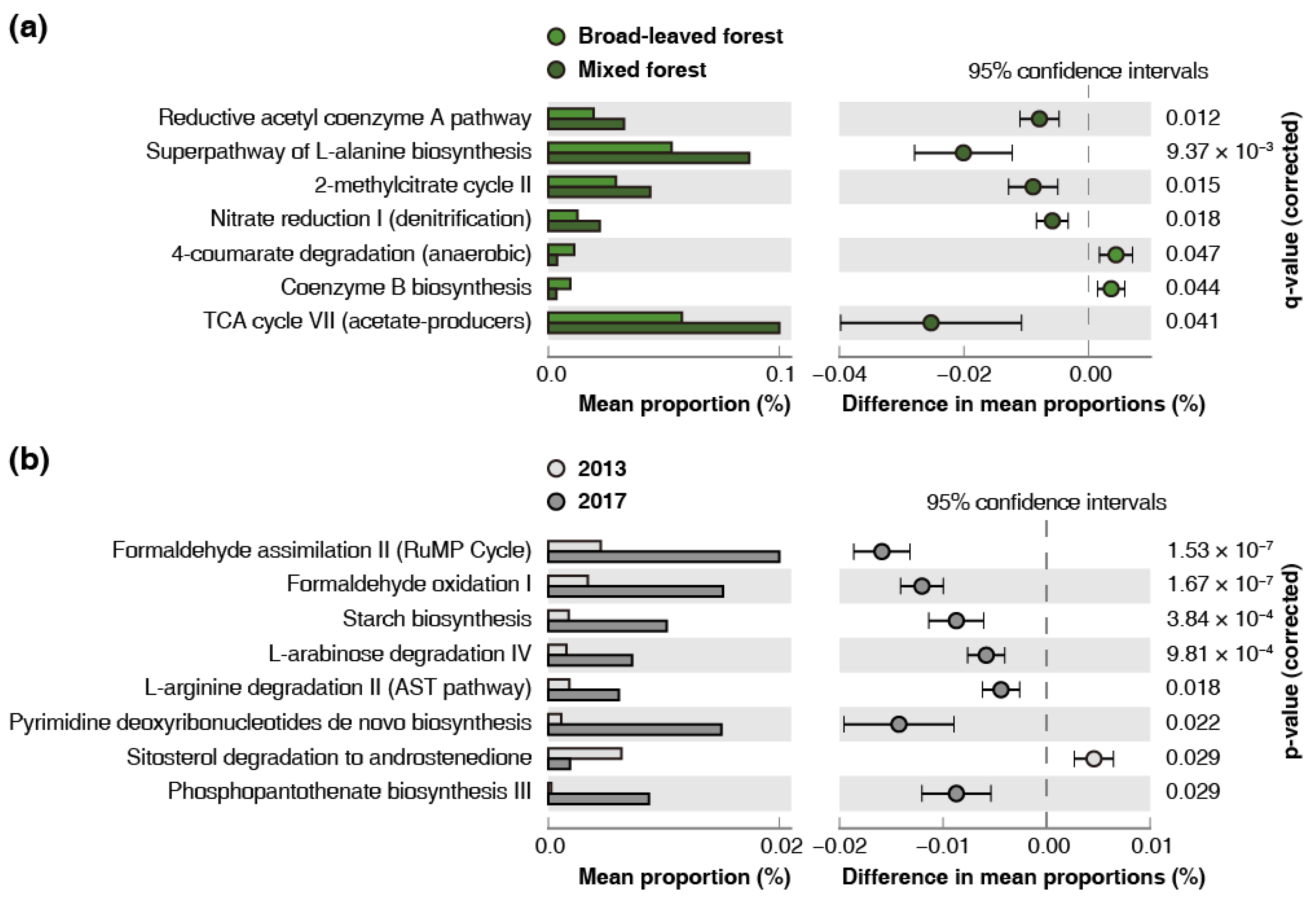
3.5. Genome-Based Prediction of Functional Abundance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, C.-H.; Oh, J.-G.; Kang, E.-O.; Yun, C.-S.; Lim, J.-K. Community Distribution on Mountain Forest Vegetation of the Gyebangsan Area in the Odaesan National Park, Korea. Korean J. Ecol. Environ. 2014, 47, 135–145. [Google Scholar] [CrossRef]
- Lee, H.; Oh, S.-Y.; Lee, Y.M.; Jang, Y.; Jang, S.; Kim, C.; Lim, Y.W.; Kim, J. Successional Variation in the Soil Microbial Community in Odaesan National Park, Korea. Sustainability 2020, 12, 4795. [Google Scholar] [CrossRef]
- Lladó, S.; López-Mondéjar, R.; Baldrian, P. Forest Soil Bacteria: Diversity, Involvement in Ecosystem Processes, and Response to Global Change. Microbiol. Mol. Biol. Rev. 2017, 81, e00063-16. [Google Scholar] [CrossRef] [Green Version]
- Nkongolo, K.K.; Narendrula-Kotha, R. Advances in monitoring soil microbial community dynamic and function. J. Appl. Genet. 2020, 61, 249–263. [Google Scholar] [CrossRef]
- Clark, J.S.; Campbell, J.H.; Grizzle, H.; Acosta-Martìnez, V.; Zak, J.C. Soil Microbial Community Response to Drought and Precipitation Variability in the Chihuahuan Desert. Microb. Ecol. 2008, 57, 248–260. [Google Scholar] [CrossRef]
- Office of Odaesan; Korea National Park Service. Odaesan National Park Resource Monitoring (4th Year); Korea National Park Service: Pyeongchang-gun, Korea, 2008. [Google Scholar]
- Imaji, A.; Seiwa, K. Carbon allocation to defense, storage, and growth in seedlings of two temperate broad-leaved tree species. Oecologia 2009, 162, 273–281. [Google Scholar] [CrossRef]
- Lee, D.-H. Above-and below-ground biomass of Abies holophylla under different stand conditions. Life Sci. J. 2013, 10, 751–758. [Google Scholar]
- Johnson, E.A.; Miyanishi, K. Disturbance and succession. In Plant Disturbance Ecology—The Process and the Response; Academic Press: Cambridge, MA, USA, 2007; pp. 1–14. [Google Scholar]
- Keith, D.A.; Rodoreda, S.; Bedward, M. Decadal change in wetland-woodland boundaries during the late 20th century reflects climatic trends. Glob. Chang. Biol. 2009, 16, 2300–2306. [Google Scholar] [CrossRef]
- De Vos, B.; Lettens, S.; Muys, B.; Deckers, J.A. Walkley? Black analysis of forest soil organic carbon: Recovery, limitations and uncertainty. Soil Use Manag. 2007, 23, 221–229. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2′s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- UNITE Community. UNITE QIIME Release for Fungi; 2018-11-18 Edition; UNITE Community: London, UK, 2019. [Google Scholar]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2: An improved and customizable approach for metagenome inference. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’hara, R.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Package ‘vegan’. Community ecology package, version. J. Stat. Softw. 2013, 2, 1–295. [Google Scholar]
- Roberts, D.W. Labdsv: Ordination and Multivariate Analysis for Ecology, R Package Version 2.0-1. 2019. Available online: https://cran.r-project.org/web/packages/labdsv/labdsv.pdf (accessed on 3 June 2020).
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.-K.; Lee, C.-W. Distribution Status and Age Structure of Abies holophylla Population in Sudo-Am Temple Forest. Korean J. Ecol. Environ. 2014, 47, 160–166. [Google Scholar] [CrossRef]
- Cruz-Martínez, K.; Suttle, K.B.; Brodie, E.L.; Power, M.E.; Andersen, G.L.; Banfield, J.F. Despite strong seasonal responses, soil microbial consortia are more resilient to long-term changes in rainfall than overlying grassland. ISME J. 2009, 3, 738–744. [Google Scholar] [CrossRef]
- Deltedesco, E.; Keiblinger, K.M.; Piepho, H.-P.; Antonielli, L.; Pötsch, E.M.; Zechmeister-Boltenstern, S.; Gorfer, M. Soil microbial community structure and function mainly respond to indirect effects in a multifactorial climate manipulation experiment. Soil Biol. Biochem. 2020, 142, 107704. [Google Scholar] [CrossRef]
- Barnard, R.L.; Osborne, C.A.; Firestone, M.K. Changing precipitation pattern alters soil microbial community response to wet-up under a Mediterranean-type climate. ISME J. 2014, 9, 946–957. [Google Scholar] [CrossRef] [Green Version]
- De Biasi, A.M.; Bianchi, C.N.; Morri, C. Analysis of macrobenthic communities at different taxonomic levels: An example from an estuarine environment in the Ligurian Sea (NW Mediterranean). Estuarine Coast. Shelf Sci. 2003, 58, 99–106. [Google Scholar] [CrossRef]
- Carvalho, F.M.; Souza, R.C.; Barcellos, F.G.; Hungria, M.; De Vasconcelos, A.T.R. Genomic and evolutionary comparisons of diazotrophic and pathogenic bacteria of the order Rhizobiales. BMC Microbiol. 2010, 10, 37. [Google Scholar] [CrossRef] [Green Version]
- Trappe, J.M. Fungus associates of ectotrophic mycorrhizae. Bot. Rev. 1962, 28, 538–606. [Google Scholar] [CrossRef]
- Gering, J.C.; Crist, T.O.; Veech, J.A. Additive Partitioning of Species Diversity across Multiple Spatial Scales: Implications for Regional Conservation of Biodiversity. Conserv. Biol. 2003, 17, 488–499. [Google Scholar] [CrossRef]
- De Celis, M.; Belda, I.; Ortiz-Álvarez, R.; Arregui, L.; Marquina, D.; Serrano, S.; Santos, A. Tuning up microbiome analysis to monitor WWTPs’ biological reactors functioning. Sci. Rep. 2020, 10, 4079. [Google Scholar] [CrossRef]
- Kaneko, T.; Nakamura, Y.; Sato, S.; Asamizu, E.; Kato, T.; Sasamoto, S.; Watanabe, A.; Idesawa, K.; Ishikawa, A.; Kawashima, K. Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA Res. 2000, 7, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Reich, P.B.; Oleksyn, J.; Modrzynski, J.; Mrozinski, P.; Hobbie, S.E.; Eissenstat, D.M.; Chorover, J.; Chadwick, O.A.; Hale, C.M.; Tjoelker, M.G. Linking litter calcium, earthworms and soil properties: A common garden test with 14 tree species. Ecol. Lett. 2005, 8, 811–818. [Google Scholar] [CrossRef]
- Eriksson, H.M.; Jönsson, C. Four tree species and the calcium, magnesium and potassium budgets of a Swedish forest site. N. Z. J. For. Sci. 1994, 24, 415–426. [Google Scholar]
- Ying, J.; Li, X.; Wang, N.; Lan, Z.; He, J.; Bai, Y. Contrasting effects of nitrogen forms and soil pH on ammonia oxidizing microorganisms and their responses to long-term nitrogen fertilization in a typical steppe ecosystem. Soil Biol. Biochem. 2017, 107, 10–18. [Google Scholar] [CrossRef]
- Van Der Lee, G. Anoxic microsites in Douglas fir litter. Soil Biol. Biochem. 1999, 31, 1295–1301. [Google Scholar] [CrossRef]
- Zhang, D.; Hui, D.; Luo, Y.; Zhou, G. Rates of litter decomposition in terrestrial ecosystems: Global patterns and controlling factors. J. Plant Ecol. 2008, 1, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Giblin, A.E.; Tobias, C.R.; Song, B.; Weston, N.; Banta, G.T.; Rivera-Monroy, V.H. The Importance of Dissimilatory Nitrate Reduction to Ammonium (DNRA) in the Nitrogen Cycle of Coastal Ecosystems. Oceanography 2013, 26, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Shu, D.; Liu, D.; Liu, S.; Deng, N.; An, S. Passive and active ecological restoration strategies for abandoned farmland leads to shifts in potential soil nitrogen loss by denitrification and soil denitrifying microbes. Land Degrad. Dev. 2020, 31, 1086–1098. [Google Scholar] [CrossRef]
- Han, S.-I.; Lee, Y.-R.; Kim, J.-O.; Whang, K.-S. Terrimonas rhizosphaerae sp. nov., isolated from ginseng rhizosphere soil. Int. J. Syst. Evol. Microbiol. 2017, 67, 391–395. [Google Scholar] [CrossRef]
- Madhaiyan, M.; Poonguzhali, S.; Senthilkumar, M.; Pragatheswari, D.; Lee, J.-S.; Lee, K.-C. Arachidicoccus rhizosphaerae gen. nov., sp. nov., a plant-growth-promoting bacterium in the family Chitinophagaceae isolated from rhizosphere soil. Int. J. Syst. Evol. Microbiol. 2015, 65, 578–586. [Google Scholar] [CrossRef]
- Ofek, M.; Hadar, Y.; Minz, D. Ecology of Root Colonizing Massilia (Oxalobacteraceae). PLoS ONE 2012, 7, e40117. [Google Scholar] [CrossRef]
- Hardoim, P. The Ecology of Seed Microbiota. In Seed Endophytes; Springer Science and Business Media LLC: Secaucus, NJ, USA, 2019; pp. 103–125. [Google Scholar]
- Knief, C. Diversity and Habitat Preferences of Cultivated and Uncultivated Aerobic Methanotrophic Bacteria Evaluated Based on pmoA as Molecular Marker. Front. Microbiol. 2015, 6, 1346. [Google Scholar] [CrossRef] [Green Version]
- Kalyuzhnaya, M.G.; Gomez, O.A.; Murrell, J.C. The Methane-Oxidizing Bacteria (Methanotrophs). Taxon. Genom. Ecophysiol. Hydrocarb.-Degrad. Microbes 2019, 245–278. [Google Scholar] [CrossRef]
- Müller, J.E.N.; Heggeset, T.M.B.; Wendisch, V.F.; Vorholt, J.A.; Brautaset, T. Methylotrophy in the thermophilic Bacillus methanolicus, basic insights and application for commodity production from methanol. Appl. Microbiol. Biotechnol. 2014, 99, 535–551. [Google Scholar] [CrossRef]
- Kelly, D.P.; McDonald, I.R.; Wood, A.P. The Family Methylobacteriaceae. In The Prokaryotes: Alphaproteobacteria and Betaproteobacteria; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 313–340. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Study Area | Alpha Diversity Index | ||
|---|---|---|---|---|
| Forest Vegetation Type | Location | Shannon 1 | Simpson 2 | |
| 2013 | Mixed forest | P01 | 4.787 | 0.980 |
| (Mongolian oak & Manchurian fir) | P06 | 4.965 | 0.977 | |
| P07 | 5.044 | 0.982 | ||
| P13 | 4.821 | 0.973 | ||
| P14 | 5.046 | 0.980 | ||
| P15 | 5.119 | 0.976 | ||
| mean | 4.964 | 0.978 | ||
| Broad-leaved forest | P02 | 4.832 | 0.976 | |
| (Mongolian oak) | P03 | 4.403 | 0.966 | |
| P04 | 4.429 | 0.959 | ||
| P05 | 4.912 | 0.982 | ||
| P08 | 4.988 | 0.983 | ||
| P09 | 4.195 | 0.943 | ||
| P10 | 4.963 | 0.983 | ||
| P11 | 4.564 | 0.961 | ||
| P12 | 4.405 | 0.935 | ||
| mean | 4.632 | 0.966 | ||
| 2017 | Mixed forest | P01 | 5.140 | 0.980 |
| P06 | 4.862 | 0.977 | ||
| P07 | 4.452 | 0.946 | ||
| P13 | 4.288 | 0.942 | ||
| P14 | 4.326 | 0.926 | ||
| P15 | 5.323 | 0.983 | ||
| mean | 4.732 | 0.959 | ||
| Broad-leaved forest | P02 | 4.755 | 0.969 | |
| P03 | 4.440 | 0.964 | ||
| P04 | 4.669 | 0.970 | ||
| P05 | 4.408 | 0.956 | ||
| P08 | 4.824 | 0.974 | ||
| P09 | 4.834 | 0.972 | ||
| P10 | 4.795 | 0.979 | ||
| P11 | 4.665 | 0.973 | ||
| P12 | 4.296 | 0.950 | ||
| mean | 4.632 | 0.967 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, Y.-M.; Lee, H.; Kwon, S.-L.; Yoo, Y.; Kim, D.; Han, S.-I.; Lee, A.-H.; Kim, C.; Kim, G.-H.; Kim, J.-J. Influence of Tree Vegetation on Soil Microbial Communities in Temperate Forests and Their Potential as a Proactive Indicator of Vegetation Shift Due to Climate Change. Sustainability 2020, 12, 10591. https://0-doi-org.brum.beds.ac.uk/10.3390/su122410591
Heo Y-M, Lee H, Kwon S-L, Yoo Y, Kim D, Han S-I, Lee A-H, Kim C, Kim G-H, Kim J-J. Influence of Tree Vegetation on Soil Microbial Communities in Temperate Forests and Their Potential as a Proactive Indicator of Vegetation Shift Due to Climate Change. Sustainability. 2020; 12(24):10591. https://0-doi-org.brum.beds.ac.uk/10.3390/su122410591
Chicago/Turabian StyleHeo, Young-Mok, Hanbyul Lee, Sun-Lul Kwon, Yeonjae Yoo, Dongjun Kim, Sang-Il Han, Aslan-Hwanhwi Lee, Changmu Kim, Gyu-Hyeok Kim, and Jae-Jin Kim. 2020. "Influence of Tree Vegetation on Soil Microbial Communities in Temperate Forests and Their Potential as a Proactive Indicator of Vegetation Shift Due to Climate Change" Sustainability 12, no. 24: 10591. https://0-doi-org.brum.beds.ac.uk/10.3390/su122410591