



Genome-Wide Association Study Based on Plant Height and Drought-Tolerance Indices Reveals Two Candidate Drought-Tolerance Genes in Sweet Sorghum

Abstract
:1. Introduction
2. Materials and Methods
2.1. Test Materials and Experimental Design
2.2. Measurements and Data Analysis
3. Results
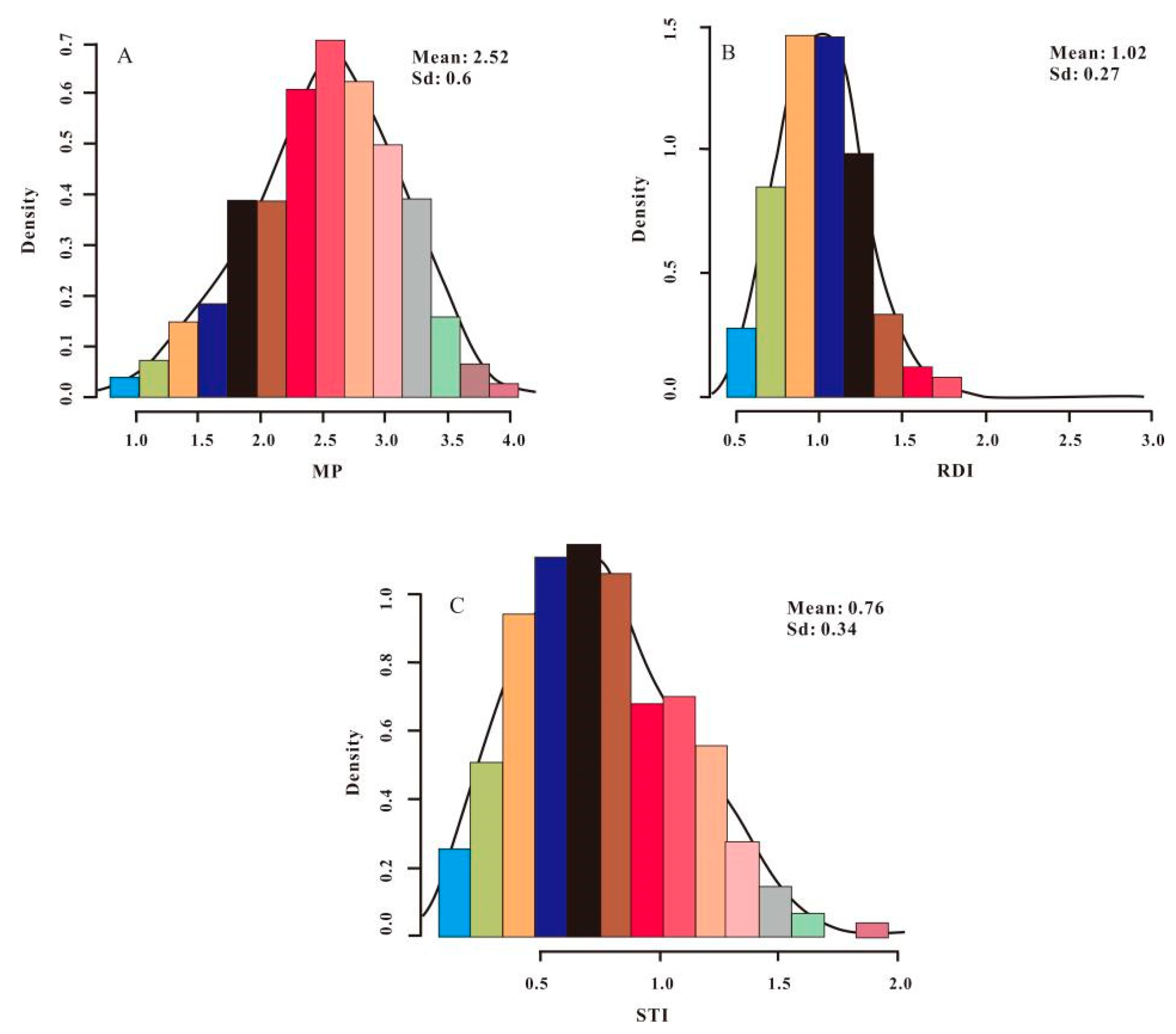
3.1. Distribution of PH and Drought-Tolerance Indices in Sweet Sorghum
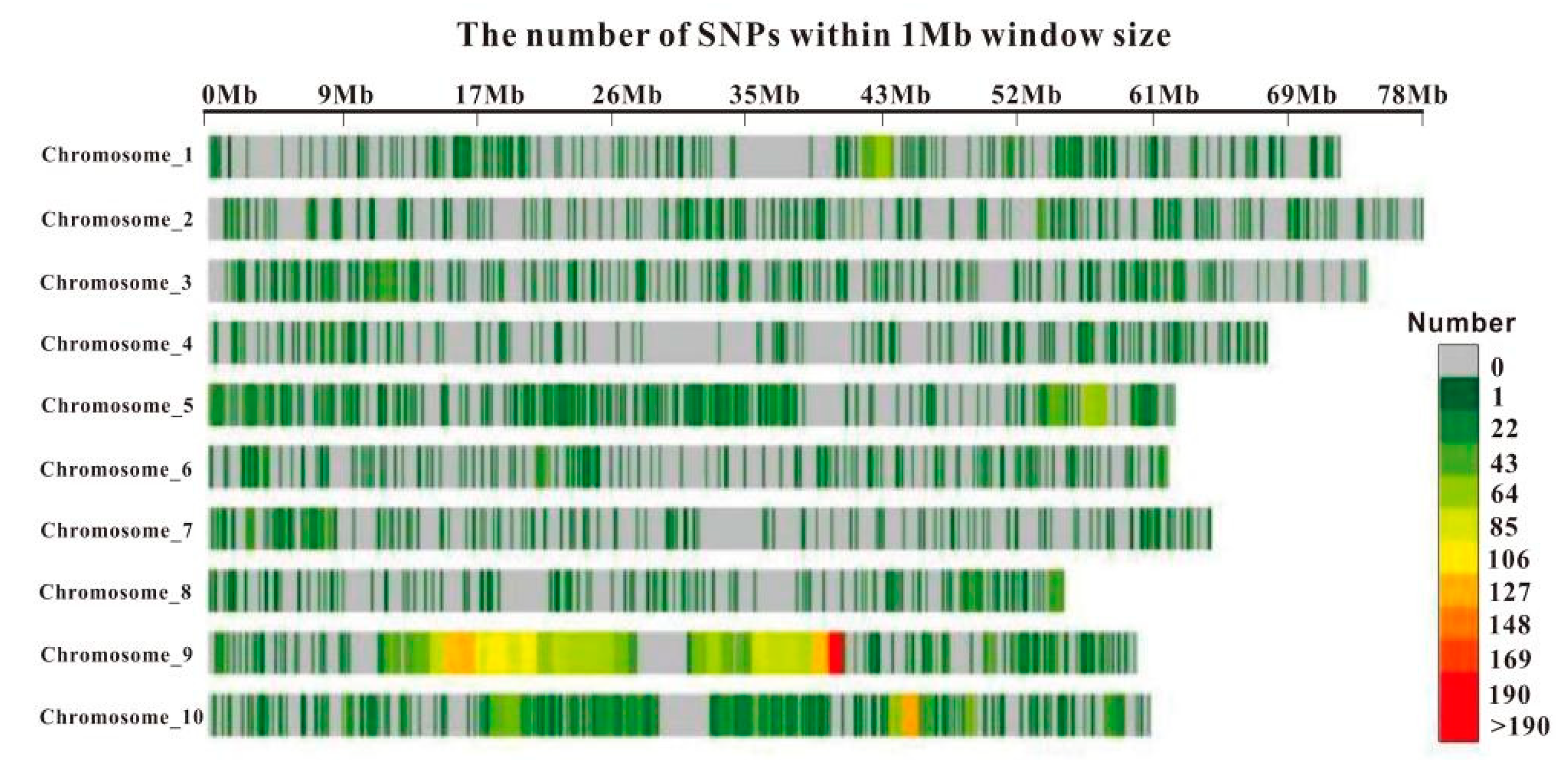
3.2. SNP Marker Density Distribution
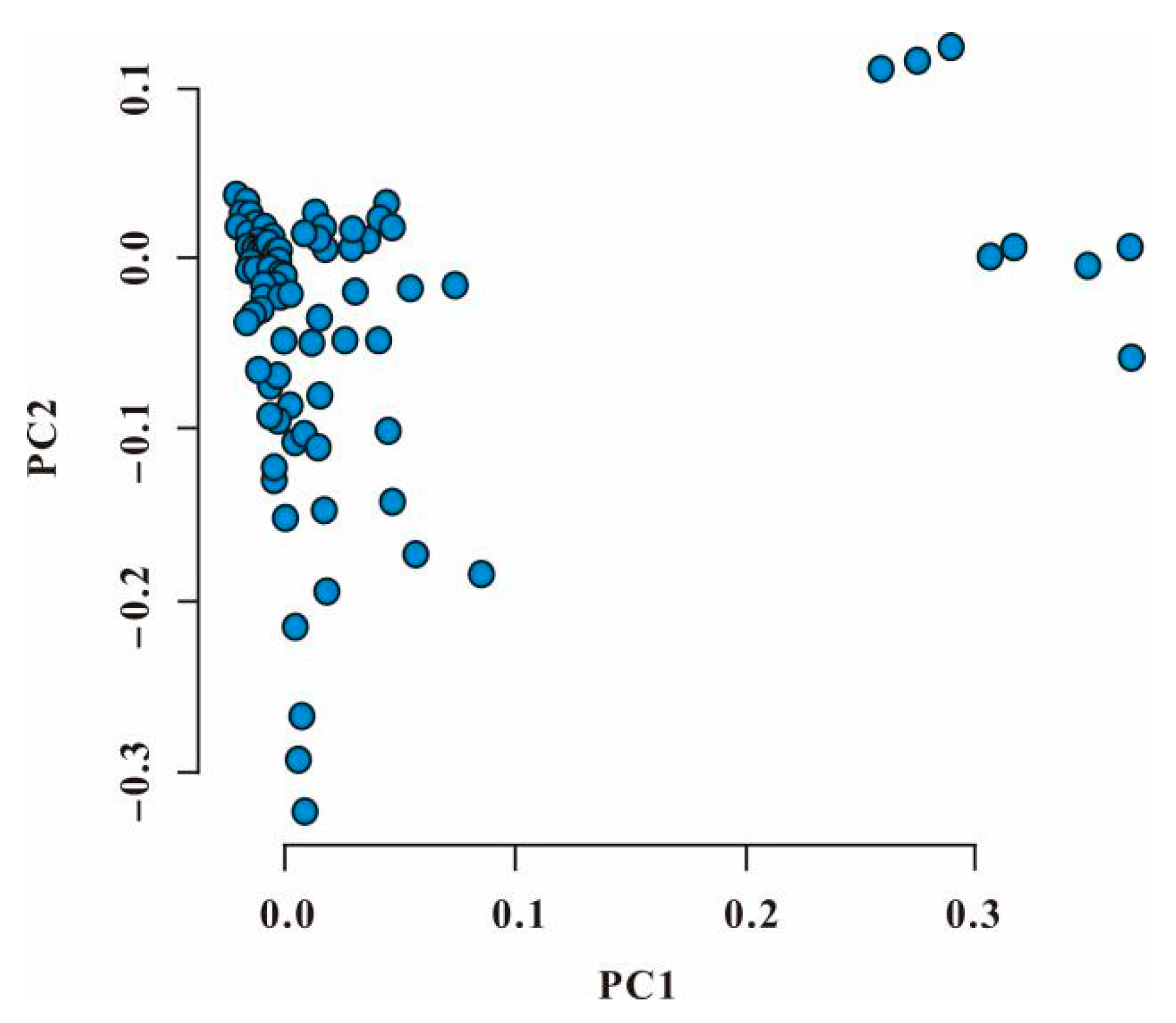
3.3. Principal Component Analysis of Genotype Data
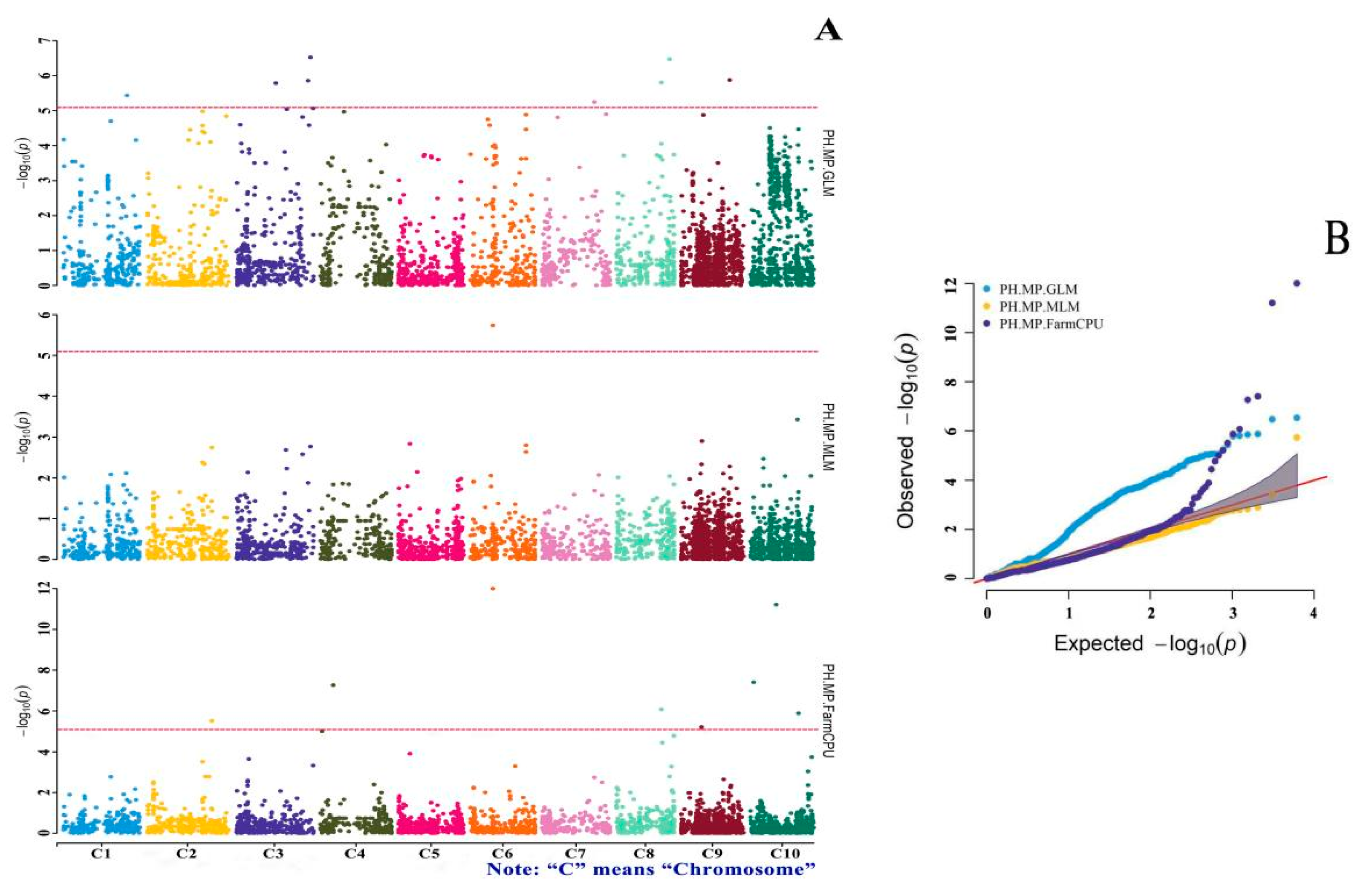
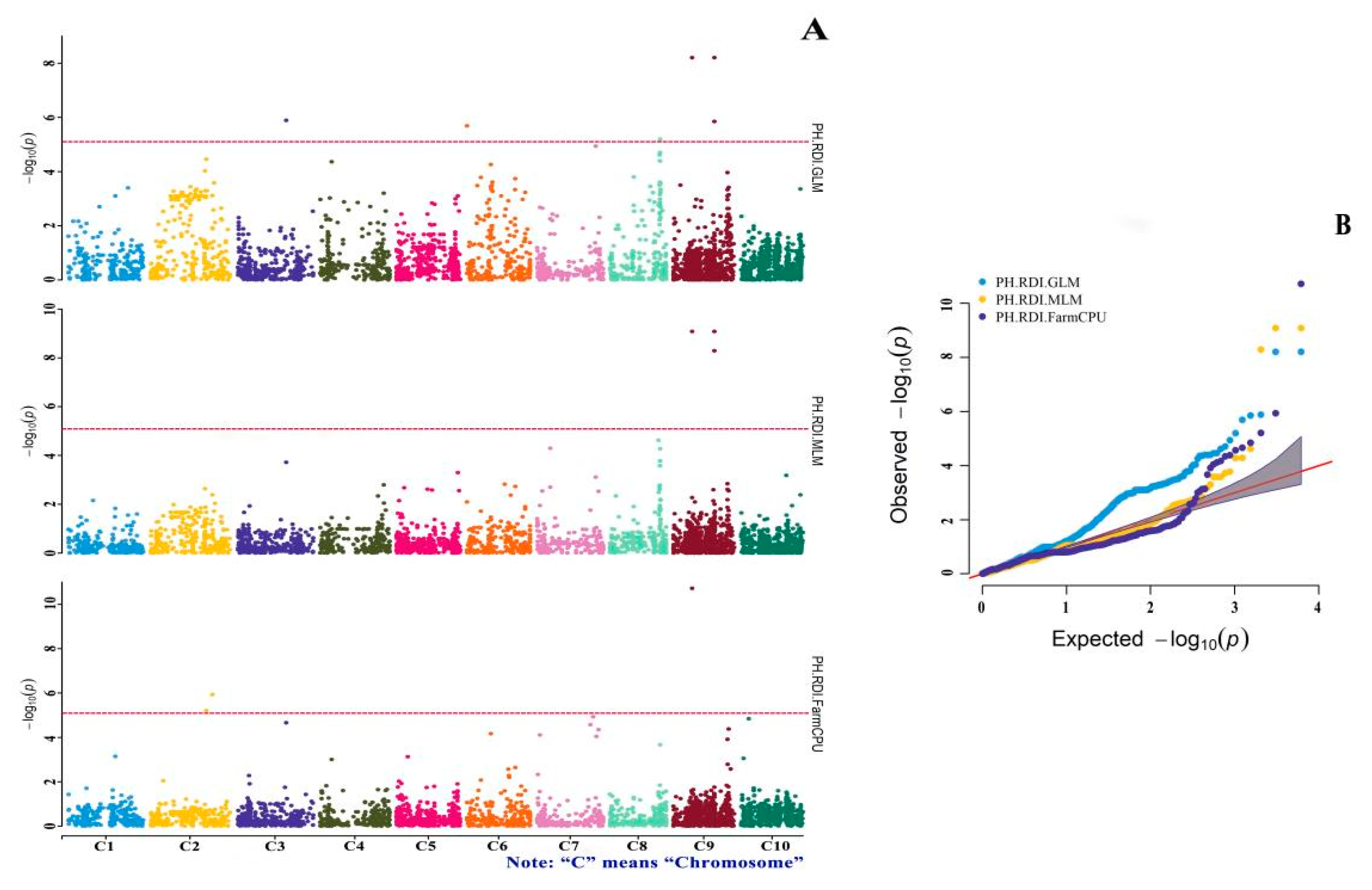
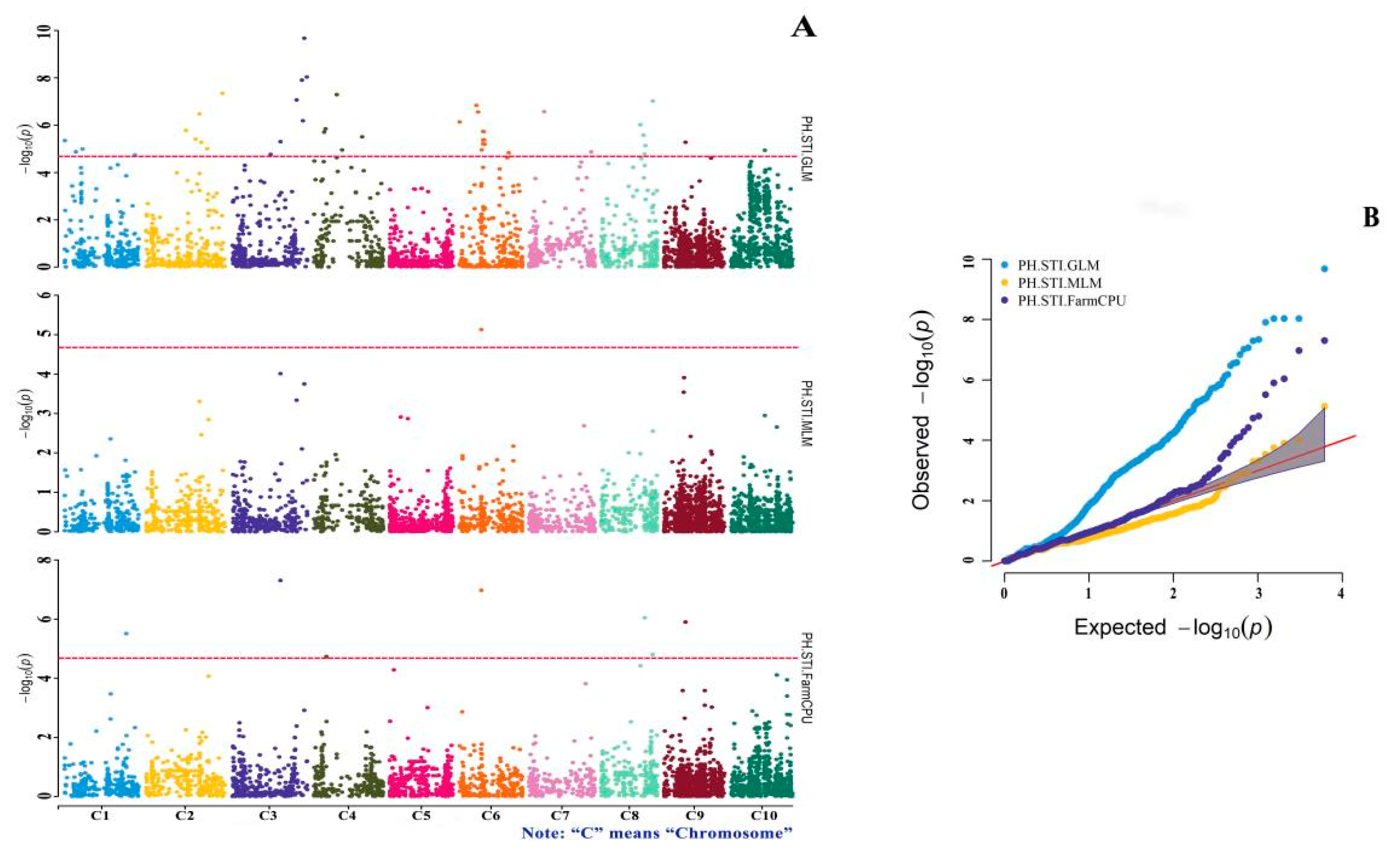
3.4. GWAS of Drought-Tolerance Indices
3.5. Gene Function Annotation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lei, Y.J.; Zhang, Y.F.; Zhang, M.H.; Liang, X.M.; Shao, X.J. Calculation and prediction of water resource ecological footprint in Xinjiang. Agric. Res. Arid. Areas 2017, 35, 142–150. [Google Scholar]
- Hu, M.F.; Tian, C.Y.; Zhao, Z.Y.; Wang, L.X. Salinization causes and research progress of technologies improving saline-alkali soil in Xinjiang. J. Northwest A&F Univ. 2012, 40, 111–117. [Google Scholar]
- Mohamed, M.F.M.; Emam, M.M.; Salama, K.H.A.; Morsy, A.A. Sorghum under saline conditions: Responses, tolerance mechanisms, and management strategies. Planta 2021, 254, 24. [Google Scholar]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef]
- Ma, X.; Feng, F.; Wei, H.; Mei, H.; Xu, K.; Chen, S.; Li, T.; Liang, X.; Liu, H.; Luo, L. Genome-Wide Association Study for Plant Height and Grain Yield in Rice under Contrasting Moisture Regimes. Front. Plant Sci. 2016, 7, 1801. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Yang, W.; Chang, Y.; Ma, X.; Tu, H.; Xiong, F.; Jiang, N.; Feng, H.; Huang, C.; Yang, P.; et al. Genome-Wide Association Studies of Image Traits Reveal Genetic Architecture of Drought Resistance in Rice. Mol. Plant. 2018, 11, 789–805. [Google Scholar] [CrossRef] [Green Version]
- Pantalião, G.F.; Narciso, M.; Guimarães, C.; Castro, A.; Colombari, J.M.; Breseghello, F.; Rodrigues, L.; Vianello, R.P.; Borba, T.O.; Brondani, C. Genome-wide association study (GWAS) for grain yield in rice cultivated under water deficit. Genetica 2016, 144, 651–664. [Google Scholar] [CrossRef]
- Hoang, G.T.; Van Dinh, L.; Nguyen, T.T.; Ta, N.K.; Gathignol, F.; Mai, C.D.; Jouannic, S.; Tran, K.D.; Khuat, T.H.; Do, V.N.; et al. Genome-wide Association Study of a Panel of Vietnamese Rice Landraces Reveals New QTLs for Tolerance to Water Deficit During the Vegetative Phase. Rice 2019, 12, 4. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, S.; Dreisigacker, S.; Lopes, M.; Chavez, P.; Reynolds, M.P. Genome-wide association study for grain yield and related traits in an elite spring wheat population grown in temperate irrigated environments. Theor. Appl. Genet. 2015, 128, 353–363. [Google Scholar] [CrossRef]
- Mei, F.; Chen, B.; Du, L.; Li, S.; Zhu, D.; Chen, N.; Zhang, Y.; Li, F.; Wang, Z.; Cheng, X.; et al. A gain-of-function allele of a DREB transcription factor gene ameliorates drought tolerance in wheat. Plant Cell. 2022, 34, 4472–4494. [Google Scholar] [CrossRef]
- Zheng, X.; Qiao, L.; Liu, Y.; Wei, N.; Zhao, J.; Wu, B.; Yang, B.; Wang, J.; Zheng, J. Genome-Wide Association Study of Grain Number in Common Wheat from Shanxi Under Different Water Regimes. Front. Plant Sci. 2022, 12, 806295. [Google Scholar] [CrossRef] [PubMed]
- Mathew, I.; Shimelis, H.; Shayanowako, A.; Laing, M.; Chaplot, V. Genome-wide association study of drought tolerance and biomass allocation in wheat. PLoS ONE 2019, 14, e0225383. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Wang, Z.P.; Liang, X.L.; Weng, J.F.; Lv, X.L.; Zhang, D.G.; Yang, J.; Yong, H.J.; Li, M.S.; Li, H.F.; et al. Identification of loci contributing to maize drought tolerance in a genome-wide association study. Euphytica 2016, 210, 165–179. [Google Scholar] [CrossRef]
- Wu, X.; Feng, H.; Wu, D.; Yan, S.; Zhang, P.; Wang, W.; Zhang, J.; Ye, J.; Dai, G.; Fan, Y.; et al. Using high-throughput multiple optical phenotyping to decipher the genetic architecture of maize drought tolerance. Genome Biol. 2021, 22, 185. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Liu, S.; Ferjani, A.; Li, J.; Yan, J.; Yang, X.; Qin, F. Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat. Genet. 2016, 48, 1233–1241. [Google Scholar] [CrossRef]
- Sun, M.; Li, Y.; Zheng, J.; Wu, D.; Li, C.; Li, Z.; Zang, Z.; Zhang, Y.; Fang, Q.; Li, W.; et al. A Nuclear Factor Y-B Transcription Factor, GmNFYB17, Regulates Resistance to Drought Stress in Soybean. Int. J. Mol. Sci. 2022, 23, 7242. [Google Scholar] [CrossRef]
- Chen, L.; Fang, Y.; Li, X.; Zeng, K.; Chen, H.; Zhang, H.; Yang, H.; Cao, D.; Hao, Q.; Yuan, S.; et al. Identification of soybean drought-tolerant genotypes and loci correlated with agronomic traits contributes new candidate genes for breeding. Plant. Mol. Biol. 2020, 102, 109–122. [Google Scholar] [CrossRef]
- Spindel, J.E.; Dahlberg, J.; Colgan, M.; Hollingsworth, J.; Sievert, J.; Staggenborg, S.H.; Hutmacher, R.; Jansson, C.; Vogel, J.P. Association mapping by aerial drone reveals 213 genetic associations for Sorghum bicolor biomass traits under drought. BMC Genom. 2018, 19, 679. [Google Scholar] [CrossRef] [Green Version]
- Maina, F.; Harou, A.; Hamidou, F.; Morris, G.P. Genome-wide association studies identify putative pleiotropic locus mediating drought tolerance in sorghum. Plant Direct. 2022, 6, 413. [Google Scholar] [CrossRef]
- Hou, S.; Zhu, G.; Li, Y.; Li, W.; Fu, J.; Niu, E.; Li, L.; Zhang, D.; Guo, W. Genome-Wide Association Studies Reveal Genetic Variation and Candidate Genes of Drought Stress Related Traits in Cotton (Gossypium hirsutum L.). Front. Plant. Sci. 2018, 9, 1276. [Google Scholar] [CrossRef]
- Guan, S.Y.; Jiao, P.; Jiang, Z.Z.; Qi, Z.; Xia, H.F.; Qu, J.; Ma, Y.Y. Research Progress of MYB Transcription Factors in Plant Abiotic Stress. J. Jilin Agric. Univ. 2019, 41, 253–260. [Google Scholar]
- Xu, L.; Wang, Y.C.; He, X.L. Huang, Y.H.; Xu, Z.L.; Shao, H.B.; Zhang, D.Y. Isolation, Expression and Binding Function Analysis of the Transcription Factor GmMYB52 in Soybean. Acta Agron. Sin. 2017, 43, 1458–1467. [Google Scholar] [CrossRef]
- Wang, C.; Lu, G.; Hao, Y.; Guo, H.; Guo, Y.; Zhao, J.; Cheng, H. ABP9, a maize bZIP transcription factor, enhances tolerance to salt and drought in transgenic cotton. Planta 2017, 246, 453–469. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Yang, J.; Ferreira, T.; Morris, A.P.; Medland, S.E.; Genetic Investigation of ANthropometric Traits (GIANT) Consortium; DIAbetes Genetics Replication and Meta-Analysis (DIAGRAM) Consortium; Madden, P.A.; Heath, A.C.; Martin, N.G.; Montgomery, G.W.; et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 2012, 44, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Wang, L.; Jiang, N.; Jia, T.; Luo, Z. A robust and efficient statistical method for genetic association studies using case and control samples from multiple cohorts. BMC Genom. 2013, 14, 88. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Buckler, E.S.; Casstevens, T.M.; Bradbury, P.J. Software engineering the mixed model for genome-wide association studies on large samples. Brief Bioinform. 2009, 10, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Korte, A.; Vilhjálmsson, B.J.; Segura, V.; Platt, A.; Long, Q.; Nordborg, M. A mixed-model approach for genome-wide association studies of correlated traits in structured populations. Nat. Genet. 2012, 44, 1066–1071. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zaitlen, N.A.; Goddard, M.E.; Visscher, P.M.; Price, A.L. Advantages and pitfalls in the application of mixed-model association methods. Nat. Genet. 2014, 46, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Ersoz, E.; Lai, C.Q.; Todhunter, R.J.; Tiwari, H.K.; Gore, M.A.; Bradbury, P.J.; Yu, J.; Arnett, D.K.; Ordovas, J.M.; et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 2010, 42, 355–360. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Liu, X.; Bradbury, P.; Yu, J.; Zhang, Y.M.; Todhunter, R.J.; Buckler, E.S.; Zhang, Z. Enrichment of statistical power for genome-wide association studies. BMC Biol. 2014, 12, 73. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Dai, Y.H.; Guan, Y.; Liu, M.Q.; Zhang, Q.K.; He, X.H. Dynamic Monitoring and Evaluation of Ecological Environment Quality in Alar Reclamation Area from 1990 to 2020. Bull. Soil Water Conserv. 2022, 42, 122–128. [Google Scholar]
- Amiri, R.; Bahraminejad, S.; Sasani, S.; Ghobadi, M. Genetic evaluation of 80 irrigated bread wheat genotypes for drought tolerance indices. Bulg. J. Agric. Sci. 2014, 20, 101–111. [Google Scholar]
- Li, C.; Sun, B.; Li, Y.; Liu, C.; Wu, X.; Zhang, D.; Shi, Y.; Song, Y.; Buckler, E.S.; Zhang, Z.; et al. Numerous genetic loci identified for drought tolerance in the maize nested association mapping populations. BMC Genom. 2016, 17, 894. [Google Scholar] [CrossRef]
- Khan, S.U.; Zheng, Y.; Chachar, Z.; Zhang, X.; Zhou, G.; Zong, N.; Leng, P.; Zhao, J. Dissection of Maize Drought Tolerance at the Flowering Stage Using Genome-Wide Association Studies. Genes 2022, 13, 564. [Google Scholar] [CrossRef]
- Khayatnezhad, M.; Zaefizadeh, M.; Gholamin, R.; Jamaati-E-Somarin, S. Study of Genetic Diversity and Path Analysis for Yield in Durum Wheat Genotypes under Water and Dry Conditions. World Appl. Sci. J. 2010, 9, 655–665. [Google Scholar]
- Saeidi, M.; Abdoli, M.; Azhand, M.; Khas-amiri, M. Evaluation of drought resistance of barley (Hordeum vulgare L.) cultivars using agronomic characteristics and drought tolerance indices. Albanian J. Agric. Sci. 2013, 12, 545–554. [Google Scholar]
- Aditya, J.; Bhartiya, A.; Pal, R.S.; Kant, L.; Pattanayak, A. Identification of drought tolerant, high yielding rice genotypes for rainfed upland ecosystem of uttarakhand hills through different drought tolerance indices. J. Environ. Biol. 2022, 43, 306–316. [Google Scholar] [CrossRef]
- Mueen, A.K.; Hafiz Muhammad, F.U.; Iqbal, M.; Rehman, A.; Chattha, W.S. Evaluation of high-yielding wheat (Triticum aestivum L.) varieties under water limitation. Plant. Genet. Resour. 2021, 19, 245–251. [Google Scholar]
- Ballesta, P.; Mora, F.; Del, P.A. Association mapping of drought tolerance indices in wheat: QTL-rich regions on chromosome 4A. Sci. Agric. 2020, 77, 153. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Pressoir, G.; Briggs, W.H.; Vroh, B.I.; Yamasaki, M.; Doebley, J.F.; McMullen, M.D.; Gaut, B.S.; Nielsen, D.M.; Holland, J.B.; et al. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 2006, 38, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.P.; Chen, P.; Hong, W.J.; Zhao, X.Y.; Liu, X.M. Research Progress of MYB Transcription Factor Family in Arabidopsis thaliana. Life Sci. Res. 2016, 20, 555–560. [Google Scholar]
- Xiong, H.; Li, J.; Liu, P.; Duan, J.; Zhao, Y.; Guo, X.; Li, Y.; Zhang, H.; Ali, J.; Li, Z. Overexpression of OsMYB48-1, a novel MYB-related transcription factor, enhances drought and salinity tolerance in rice. PLoS ONE 2014, 9, e92913. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.; Liu, C.; Li, Z.; Ran, Q.; Xie, G.; Wang, B.; Fang, S.; Chu, J.; Zhang, J. ZmbZIP4 Contributes to Stress Resistance in Maize by Regulating ABA Synthesis and Root Development. Plant Physiol. 2018, 178, 753–770. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chromosome | Length (bp) | No. of SNPs | Average Density (SNPs/Mb) |
|---|---|---|---|
| 1 | 72,621,628 | 392 | 5.4 |
| 2 | 77,923,599 | 340 | 4.4 |
| 3 | 74,347,826 | 379 | 5.1 |
| 4 | 67,928,809 | 243 | 3.6 |
| 5 | 61,993,318 | 615 | 10.0 |
| 6 | 61,563,909 | 284 | 4.6 |
| 7 | 64,298,007 | 266 | 4.1 |
| 8 | 54,875,046 | 271 | 4.9 |
| 9 | 59,493,343 | 2401 | 40.4 |
| 10 | 60,355,397 | 995 | 16.5 |
| Marker | Variant | Effect | p-Value | Methods |
|---|---|---|---|---|
| SNP-9-46359555 | T/C | 0.259 | 1.34 × 10−6 | MP.GLM |
| SNP-9-32010157 | T/C | −0.800 | 5.43 × 10−6 | MP.FarmCPU |
| SNP-8-54701961 | C/T | −0.280 | 5.27 × 10−6 | MP.FarmCPU |
| SNP-8-50726311 | C/G | −0.180 | 3.37 × 10−7 | MP.GLM |
| SNP-8-42803746 | G/A | −0.100 | 1.22 × 10−7 | MP.FarmCPU |
| SNP-8-42803746 | G/A | −0.140 | 1.57 × 10−6 | MP.GLM |
| SNP-7-49761327 | G/C | −0.503 | 5.68 × 10−6 | MP.GLM |
| SNP-6-21397577 | T/C | −0.776 | 1.73 × 10−12 | MP.FarmCPU |
| SNP-6-21397577 | T/C | −0.745 | 1.84 × 10−6 | MP.MLM |
| SNP-4-1724051 | C/T | −0.099 | 3.23 × 10−6 | MP.FarmCPU |
| SNP-4-12346536 | A/T | −0.293 | 2.50 × 10−9 | MP.FarmCPU |
| SNP-3-71564436 | A/C | −0.336 | 2.98 × 10−7 | MP.GLM |
| SNP-3-69226243 | A/G | −0.287 | 1.40 × 10−6 | MP.GLM |
| SNP-3-38408072 | C/A | −0.140 | 1.63 × 10−6 | MP.GLM |
| SNP-2-62168227 | A/C | 0.137 | 1.72 × 10−6 | MP.FarmCPU |
| SNP-1-60603003 | T/C | 0.316 | 3.69 × 10−6 | MP.GLM |
| SNP-10-45922678 | T/C | −0.208 | 1.57 × 10−7 | MP.FarmCPU |
| SNP-10-3166515 | C/T | 0.349 | 1.21 × 10−9 | MP.FarmCPU |
| SNP-10-24652464 | T/C | 0.561 | 5.82 × 10−12 | MP.FarmCPU |
| Marker | Variant | Effect | p-Value | Methods |
|---|---|---|---|---|
| SNP-9-40211466 | A/T | −0.920 | 1.42 × 10−6 | RDI.GLM |
| SNP-9-40211466 | A/T | −1.479 | 5.15 × 10−9 | RDI.MLM |
| SNP-9-40211299 | T/A | −1.059 | 6.28 × 10−9 | RDI.GLM |
| SNP-9-40211299 | T/A | −1.504 | 8.32 × 10−10 | RDI.MLM |
| SNP-9-18625001 | G/A | −0.991 | 3.42 × 10−18 | RDI.FarmCPU |
| SNP-9-18625001 | G/A | −1.059 | 6.28 × 10−9 | RDI.GLM |
| SNP-9-18625001 | G/A | −1.504 | 8.32 × 10−10 | RDI.MLM |
| SNP-8-48976460 | T/C | −0.237 | 6.46 × 10−6 | RDI.GLM |
| SNP-7-59879988 | T/A | 0.098 | 8.80 × 10−8 | RDI.FarmCPU |
| SNP-7-57772159 | T/A | −0.059 | 3.74 × 10−7 | RDI.FarmCPU |
| SNP-7-51913862 | T/C | 0.069 | 5.20 × 10−8 | RDI.FarmCPU |
| SNP-7-2876065 | A/G | −0.102 | 5.05 × 10−7 | RDI.FarmCPU |
| SNP-6-23330464 | C/G | −0.064 | 5.51 × 10−7 | RDI.FarmCPU |
| SNP-6-153673 | A/G | −0.091 | 2.07 × 10−6 | RDI.GLM |
| SNP-3-48129228 | T/G | 0.183 | 3.09 × 10−8 | RDI.FarmCPU |
| SNP-3-48129228 | T/G | 0.270 | 1.30 × 10−6 | RDI.GLM |
| SNP-2-60765047 | T/C | −0.064 | 5.09 × 10−10 | RDI.FarmCPU |
| SNP-2-54990996 | T/A | −0.076 | 3.20 × 10−9 | RDI.FarmCPU |
| SNP-10-7591972 | C/T | −0.125 | 1.82 × 10−7 | RDI.FarmCPU |
| Marker | Variant | Effect | p-Value | Methods |
|---|---|---|---|---|
| SNP-1-60603003 | T/C | 0.122 | 3.11 × 10−6 | STI.FarmCPU |
| SNP-9-21304761 | G/C | 0.169 | 1.26 × 10−6 | STI.FarmCPU |
| SNP-9-21304761 | G/C | 0.210 | 5.34 × 10−6 | STI.GLM |
| SNP-8-50726311 | C/G | −0.112 | 9.58 × 10−8 | STI.GLM |
| SNP-8-43496503 | A/G | −0.136 | 7.33 × 10−6 | STI.GLM |
| SNP-8-42803746 | G/A | −0.068 | 9.16 × 10−7 | STI.FarmCPU |
| SNP-8-41546525 | T/C | −0.161 | 2.65 × 10−6 | STI.GLM |
| SNP-8-38378608 | G/T | −0.157 | 9.75 × 10−7 | STI.GLM |
| SNP-7-14894170 | T/C | −0.198 | 2.70 × 10−7 | STI.GLM |
| SNP-6-24638786 | T/C | −0.116 | 6.40 × 10−6 | STI.GLM |
| SNP-6-24155091 | C/G | −0.119 | 4.23 × 10−6 | STI.GLM |
| SNP-6-23766693 | G/C | −0.120 | 1.90 × 10−6 | STI.GLM |
| SNP-6-23020118 | T/C | −0.114 | 6.21 × 10−6 | STI.GLM |
| SNP-6-22967016 | C/T | −0.114 | 4.32 × 10−6 | STI.GLM |
| SNP-6-22966971 | A/G | −0.120 | 1.86 × 10−6 | STI.GLM |
| SNP-6-21397577 | T/C | −0.398 | 1.07 × 10−7 | STI.FarmCPU |
| SNP-6-21397577 | T/C | −0.426 | 7.49 × 10−6 | STI.MLM |
| SNP-6-18184340 | T/C | −0.138 | 2.78 × 10−7 | STI.GLM |
| SNP-6-16486758 | T/C | −0.147 | 1.48 × 10−7 | STI.GLM |
| SNP-6-153673 | A/G | −0.108 | 7.35 × 10−7 | STI.GLM |
| SNP-4-47497728 | T/C | −0.178 | 3.13 × 10−6 | STI.GLM |
| SNP-4-22602002 | A/G | −0.249 | 5.12 × 10−8 | STI.GLM |
| SNP-4-11561108 | T/G | −0.164 | 1.44 × 10−6 | STI.GLM |
| SNP-4-11561107 | G/C | −0.164 | 1.44 × 10−6 | STI.GLM |
| SNP-4-10100565 | G/C | −0.206 | 1.98 × 10−6 | STI.GLM |
| SNP-3-73977961 | C/A | −0.145 | 9.30 × 10−9 | STI.GLM |
| SNP-3-73977959 | T/A | −0.145 | 9.30 × 10−9 | STI.GLM |
| SNP-3-73977947 | T/A | −0.145 | 9.30 × 10−9 | STI.GLM |
| SNP-3-71564436 | A/C | −0.246 | 2.12 × 10−10 | STI.GLM |
| SNP-3-70162204 | A/G | −0.165 | 6.51 × 10−7 | STI.GLM |
| SNP-3-69226243 | A/G | −0.201 | 1.24 × 10−8 | STI.GLM |
| SNP-3-63949295 | G/C | −0.213 | 8.63 × 10−8 | STI.GLM |
| SNP-3-48129228 | T/G | 0.240 | 4.95 × 10−8 | STI.FarmCPU |
| SNP-3-48129228 | T/G | 0.293 | 4.99 × 10−6 | STI.GLM |
| SNP-2-75743471 | G/T | −0.159 | 4.57 × 10−8 | STI.GLM |
| SNP-2-54990998 | T/C | −0.120 | 5.33 × 10−6 | STI.GLM |
| SNP-2-54990996 | T/A | −0.120 | 5.33 × 10−6 | STI.GLM |
| SNP-2-53243721 | C/T | 0.486 | 3.35 × 10−7 | STI.GLM |
| SNP-2-49409474 | C/G | −0.176 | 3.93 × 10−6 | STI.GLM |
| SNP-2-39852357 | A/G | −0.205 | 1.67 × 10−6 | STI.GLM |
| SNP-1-159169 | G/T | −0.182 | 4.54 × 10−6 | STI.GLM |
| SNP | Genes | Gene Function Annotation |
|---|---|---|
| SNP-1-159169 | Sb01g000300.1 | transcriptional corepressor Leunig-homolog-like [Sorghum bicolor], |
| Zea mays LOC100285229 (pco116270) | ||
| SNP-1-60603003 | Sb01g037050.1 | TF TGA2.2 [Sorghum bicolor], |
| Zea mays putative bZIP TF | ||
| (LOC100274089) | ||
| SNP-1-60603003 | Sb01g037050.2 | TF TGA2.2 [Sorghum bicolor], |
| Zea mays putative bZIP TF | ||
| (LOC100274089) | ||
| SNP-1-60603003 | Sb01g037050.3 | TF TGA2.2 [Sorghum bicolor], |
| Setaria italica TF HBP-1b(c1)-like (LOC101767047), | ||
| transcript variant X1, mRNA | ||
| SNP-1-60603003 | Sb01g037050.4 | TF TGA2.2 [Sorghum bicolor], |
| Zea mays putative bZIP TF | ||
| (LOC100274089) | ||
| SNP-8-50726311 | Sb08g019720.1 | TF LUX-like [Oryza brachyantha], |
| Sorghum bicolor hypothetical protein, mRNA, | ||
| MYB family TF EFM, Arabidopsis thaliana |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xin, Y.; Gao, L.; Hu, W.; Gao, Q.; Yang, B.; Zhou, J.; Xu, C. Genome-Wide Association Study Based on Plant Height and Drought-Tolerance Indices Reveals Two Candidate Drought-Tolerance Genes in Sweet Sorghum. Sustainability 2022, 14, 14339. https://0-doi-org.brum.beds.ac.uk/10.3390/su142114339
Xin Y, Gao L, Hu W, Gao Q, Yang B, Zhou J, Xu C. Genome-Wide Association Study Based on Plant Height and Drought-Tolerance Indices Reveals Two Candidate Drought-Tolerance Genes in Sweet Sorghum. Sustainability. 2022; 14(21):14339. https://0-doi-org.brum.beds.ac.uk/10.3390/su142114339
Chicago/Turabian StyleXin, Yue, Lina Gao, Wenming Hu, Qi Gao, Bin Yang, Jianguo Zhou, and Cuilian Xu. 2022. "Genome-Wide Association Study Based on Plant Height and Drought-Tolerance Indices Reveals Two Candidate Drought-Tolerance Genes in Sweet Sorghum" Sustainability 14, no. 21: 14339. https://0-doi-org.brum.beds.ac.uk/10.3390/su142114339