Natural Occurrence in Venomous Arthropods of Antimicrobial Peptides Active against Protozoan Parasites

 , , and
, , and
Abstract
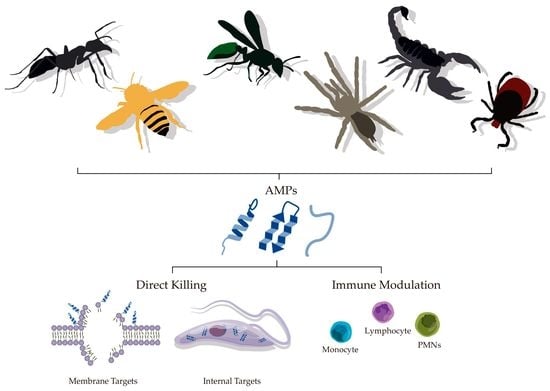
:
1. Introduction
2. AMPs
3. Differences between Plasma Membranes of Protozoan and Mammalian Cells
4. Mode of Action of Antiprotozoal AMPs
4.1. Direct Killing
4.2. Immune Modulatory Effects
5. Protozoonosis
5.1. Chagas Disease
Anti-Chagas diseaseAMPs
5.2. Human African Trypanosomiasis
Anti-Human African Trypanosomiasis AMPs
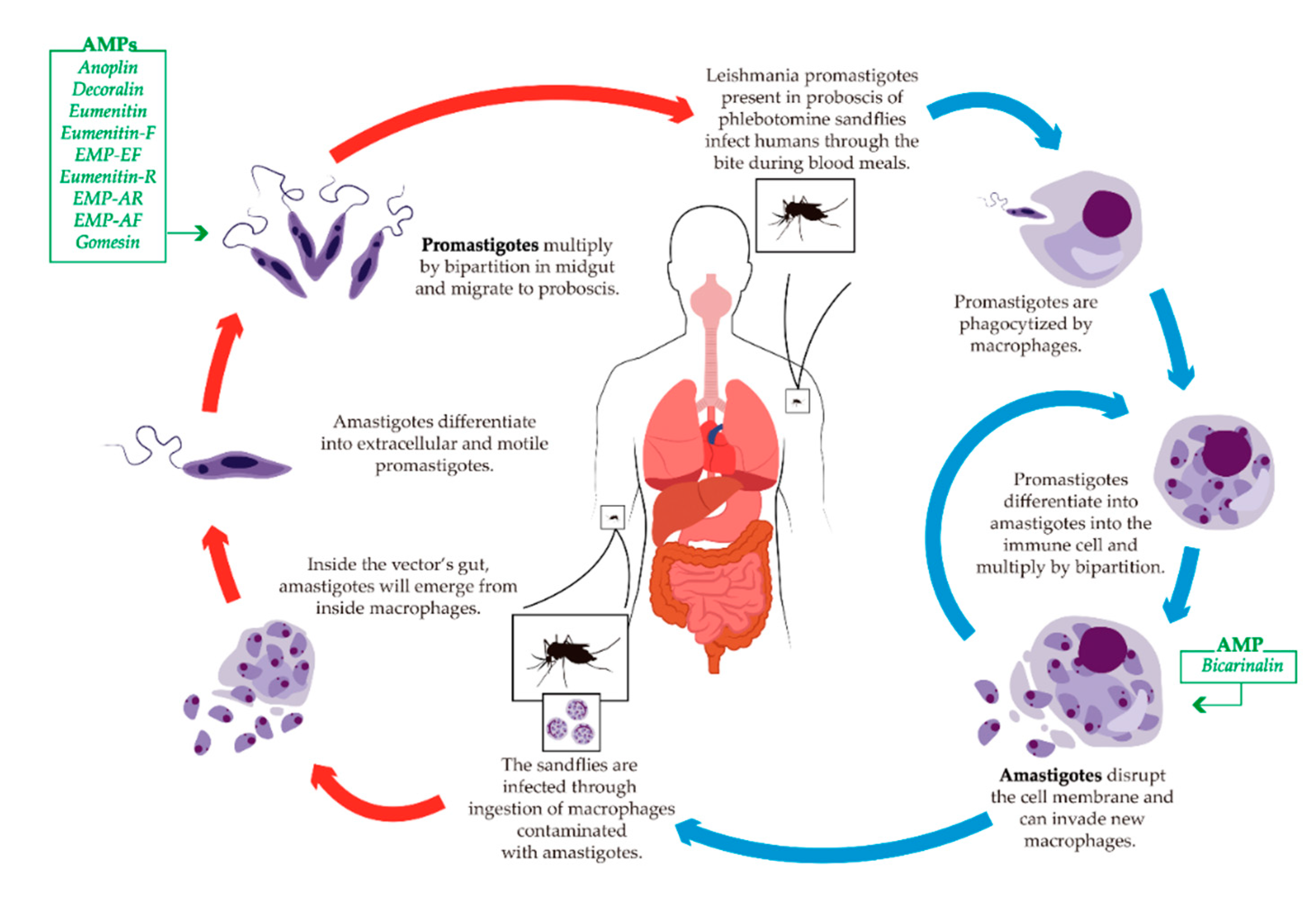
5.3. Leishmaniasis
Antileishmanial AMPs
5.4. Malaria
Antimalarial AMPs
5.5. Toxoplasmosis
Anti-Toxoplasma AMPs
6. Future Prospects
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Junior, V.H.; De Amorim, P.C.H.; Junior, W.T.H.; Cardoso, J.L.C. Venomous and poisonous arthropods: Identification, clinical manifestations of envenomation, and treatments used in human injuries. Rev. Soc. Bras. Med. Trop. 2015, 48, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Giribet, G.; Edgecombe, G.D. Reevaluating the arthropod tree of life. Annu. Rev. Èntomol. 2012, 57, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, E.F.; Mourão, C.B.F.; Moreira, K.G.; Camargos, T.S.; Mortari, M.R. Arthropod venoms: A vast arsenal of insecticidal neuropeptides. Biopolymers 2012, 98, 385–405. [Google Scholar] [CrossRef] [PubMed]
- Daly, N.L.; Wilson, D. Structural diversity of arthropod venom toxins. Toxicon 2018, 152, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.-Q. Animal biodiversity: An outline of higher-level classification and survey of taxonomic richness (COVER). Zootaxa 2011, 3148, 1–2. [Google Scholar] [CrossRef]
- Stork, N.E.; Mc Broom, J.; Gely, C.; Hamilton, A.J. New approaches narrow global species estimates for beetles, insects, and terrestrial arthropods. Proc. Natl. Acad. Sci. USA 2015, 112, 7519–7523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyun, S.-I.; Soh, H.Y.; Posavi, M.; Munro, J.B.; Hughes, D.S.; Murali, S.C.; Qu, J.; Dugan, S.; Lee, S.L.; Chao, H.; et al. Evolutionary History of Chemosensory-Related Gene Families across the Arthropoda. Mol. Boil. Evol. 2017, 34, 1838–1862. [Google Scholar] [CrossRef] [Green Version]
- Cloudsley-Thompson, J.L. Adaptations of Arthropoda to arid environments. Annu. Rev. Èntomol. 1975, 20, 261–283. [Google Scholar] [CrossRef]
- Sømme, L. Adaptations Of Terrestrial Arthropods To The Alpine Environment. Boil. Rev. 1989, 64, 367–407. [Google Scholar] [CrossRef]
- Glenner, H.; Thomsen, P.F.; Hebsgaard, M.B.; Sørensen, M.V.; Willerslev, E. The origin of insects. Science 2006, 314, 1883–1884. [Google Scholar] [CrossRef]
- Kelley, J.L.; Peyton, J.T.; Fiston-Lavier, A.-S.; Teets, N.M.; Yee, M.-C.; Johnston, J.S.; Bustamante, C.D.; Lee, R.E.; Denlinger, D.L. Compact genome of the Antarctic midge is likely an adaptation to an extreme environment. Nat. Commun. 2014, 5, 4611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcussi, S.; Arantes, E.C.; Soares, A.M. Escorpiões: Biologia, Envenenamento e Mecanismos de Ação de Suas Toxinas, 1ª edição; FUNPEC Editora: Ribeirão Preto, SP, Brasil, 2011. [Google Scholar]
- Senji Laxme, R.R.; Suranse, V.; Sunagar, K. Arthropod venoms: Biochemistry, ecology and evolution. Toxicon 2019, 158, 84–103. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.A.; Robinson, S.D.; Yeates, D.K.; Jin, J.; Baumann, K.; Dobson, J.; Fry, B.G.; King, G.F. Entomo-venomics: The evolution, biology and biochemistry of insect venoms. Toxicon 2018, 154, 15–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casewell, N.R.; Wüster, W.; Vonk, F.J.; Harrison, R.A.; Fry, B.G. Complex cocktails: The evolutionary novelty of venoms. Trends Ecol. Evol. 2013, 28, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Suranse, V.; Srikanthan, A.; Sunagar, K. Animal Venoms: Origin, diversity and evolution. In eLS; John Wiley & Sons, Ltd.: Chichester, UK, 2018; pp. 1–20. [Google Scholar]
- Schmidt, J.O. Biochemistry of Insect Venoms. Annu. Rev. Entomol. 1982, 27, 339–368. [Google Scholar] [CrossRef] [PubMed]
- Beard, R.L. Insect Toxins and Venoms. Annu. Rev. Entomol. 1963, 8, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Herzig, V. Arthropod assassins: Crawling biochemists with diverse toxin pharmacopeias. Toxicon 2019, 158, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Walker, A.A.; Hernández-Vargas, M.J.; Corzo, G.; Fry, B.G.; King, G.F. Giant fish-killing water bug reveals ancient and dynamic venom evolution in Heteroptera. Cell. Mol. Life Sci. 2018, 75, 3215–3229. [Google Scholar] [CrossRef]
- Rong, M.; Yang, S.; Wen, B.; Mo, G.; Kang, D.; Liu, J.; Lin, Z.; Jiang, W.; Li, B.; Du, C.; et al. Peptidomics combined with cDNA library unravel the diversity of centipede venom. J. Proteom. 2015, 114, 28–37. [Google Scholar] [CrossRef]
- Hakim, M.A.; Yang, S.; Lai, R. Centipede Venoms and Their Components: Resources for Potential Therapeutic Applications. Toxins 2015, 7, 4832–4851. [Google Scholar] [CrossRef] [Green Version]
- Amorim, F.G.; Longhim, H.T.; Cologna, C.T.; Degueldre, M.; De Pauw, E.; Quinton, L.; Arantes, E.C. Proteome of fraction from Tityus serrulatus venom reveals new enzymes and toxins. J. Venom. Anim. Toxins Incl. Trop. Dis. 2019, 25, 25. [Google Scholar] [CrossRef]
- Escoubas, P. Structure and pharmacology of spider venom neurotoxins. Biochimie 2000, 82, 893–907. [Google Scholar] [CrossRef]
- García-Arredondo, A.; Rodríguez-Rios, L.; Díaz-Peña, L.F.; Vega-Ángeles, R. Pharmacological characterization of venoms from three theraphosid spiders: Poecilotheria regalis, Ceratogyrus darlingi and Brachypelma epicureanum. J. Venom. Anim. Toxins Incl. Trop. Dis. 2015, 21, 555. [Google Scholar] [CrossRef]
- Von Reumont, B.M.; Blanke, A.; Richter, S.; Alvarez, F.; Bleidorn, C.; Jenner, R.A. The first venomous crustacean revealed by transcriptomics and functional morphology: Remipede venom glands express a unique toxin cocktail dominated by enzymes and a neurotoxin. Mol. Biol. Evol. 2014, 31, 48–58. [Google Scholar] [CrossRef]
- Stork, N.E. How many species of insects and other terrestrial arthropods are there on Earth? Annu. Rev. Entomol. 2018, 63, 31–45. [Google Scholar] [CrossRef]
- Bonato, L.; Chagas, A., Jr.; Edgecombe, G.D.; Lewis, J.G.E.; Minelli, A.; Pereira, L.A.; Shelley, R.M.; Stoev, P.; Zapparoli, M. ChiloBase 2.0—A World Catalogue of Centipedes (Chilopoda). Available online: http://chilobase.biologia.unipd.it/ (accessed on 12 July 2019).
- Rein, J.O. The Scorpion Files. Available online: https://www.ntnu.no/ub/scorpion-files/ (accessed on 12 July 2019).
- Natural History Museum Bern World Spider Catalog. Version 20.0. Available online: https://wsc.nmbe.ch/ (accessed on 12 July 2019).
- Cabezas-Cruz, A.; Valdés, J.J. Are ticks venomous animals? Front. Zool. 2014, 11, 47. [Google Scholar] [CrossRef]
- Wermeling, D.; Drass, M.; Ellis, D.; Mayo, M.; McGuire, D.; O’Connell, D.; Hale, V.; Chao, S. Pharmacokinetics and Pharmacodynamics of Intrathecal Ziconotide in Chronic Pain Patients. J. Clin. Pharmacol. 2003, 43, 624–636. [Google Scholar] [CrossRef]
- Ferreira, S.H.; Greene, L.J.; Alabaster, V.A.; Bakhle, Y.S.; Vane, J.R. Activity of various fractions of bradykinin potentiating factor against angiotensin I converting enzyme. Nature 1970, 225, 33. [Google Scholar] [CrossRef]
- Furman, B.L. The development of Byetta (exenatide) from the venom of the Gila monster as an anti-diabetic agent. Toxicon 2012, 59, 464–471. [Google Scholar] [CrossRef]
- Hultmark, D. Drosophila immunity: Paths and patterns. Curr. Opin. Immunol. 2003, 15, 12–19. [Google Scholar] [CrossRef]
- Barra, D.; Simmaco, M. Amphibian skin: A promising resource for antimicrobial peptides. Trends Biotechnol. 1995, 13, 205–209. [Google Scholar] [CrossRef]
- Reddy, K.; Yedery, R.; Aranha, C. Antimicrobial peptides: Premises and promises. Int. J. Antimicrob. Agents 2004, 24, 536–547. [Google Scholar] [CrossRef]
- Brogden, K.A.; Ackermann, M.; McCray, P.B., Jr.; Tack, B.F. Antimicrobial peptides in animals and their role in host defences. Int. J. Antimicrob. Agents 2003, 22, 465–478. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.L.; Hancock, R.E. Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol. 2006, 18, 24–30. [Google Scholar] [CrossRef]
- Gentilucci, L.; Tolomelli, A.; Squassabia, F. Peptides and peptidomimetics in medicine, surgery and biotechnology. Curr. Med. Chem. 2006, 13, 2449–2466. [Google Scholar] [CrossRef]
- Bessalle, R.; Haas, H.; Goria, A.; Shalit, I.; Fridkin, M. Augmentation of the antibacterial activity of magainin by positive-charge chain extension. Antimicrob. Agents Chemother. 1992, 36, 313–317. [Google Scholar] [CrossRef] [Green Version]
- Finger, S.; Kerth, A.; Dathe, M.; Blume, A. The efficacy of trivalent cyclic hexapeptides to induce lipid clustering in PG/PE membranes correlates with their antimicrobial activity. Biochim. Biophys. Acta-Biomembr. 2015, 1848, 2998–3006. [Google Scholar] [CrossRef] [Green Version]
- Patterson-Delafield, J.; Szklarek, D.; Martinez, R.J.; Lehrer, R.I. Microbicidal cationic proteins of rabbit alveolar macrophages: Amino acid composition and functional attributes. Infect. Immun. 1981, 31, 723–731. [Google Scholar]
- Mygind, P.H.; Fischer, R.L.; Schnorr, K.M.; Hansen, M.T.; Sönksen, C.P.; Ludvigsen, S.; Raventós, D.; Buskov, S.; Christensen, B.; De Maria, L.; et al. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 2005, 437, 975–980. [Google Scholar] [CrossRef]
- Dai, C.; Ma, Y.; Zhao, Z.; Zhao, R.; Wang, Q.; Wu, Y.; Cao, Z.; Li, W. Mucroporin, the first cationic host defense peptide from the venom of Lychas mucronatus. Antimicrob. Agents Chemother. 2008, 52, 3967–3972. [Google Scholar] [CrossRef]
- Zhao, Z.; Ma, Y.; Dai, C.; Zhao, R.; Li, S.; Wu, Y.; Cao, Z.; Li, W. Imcroporin, a new cationic antimicrobial peptide from the venom of the scorpion Isometrus maculates. Antimicrob. Agents Chemother. 2009, 53, 3472–3477. [Google Scholar] [CrossRef]
- Zanjani, N.T.; Miranda-Saksena, M.; Cunningham, A.L.; Dehghani, F. Antimicrobial peptides of marine crustaceans: The potential and challenges of developing therapeutic agents. Curr. Med. Chem. 2018, 25, 2245–2259. [Google Scholar] [CrossRef]
- Nie, Y.; Zeng, X.-C.; Yang, Y.; Luo, F.; Luo, X.; Wu, S.; Zhang, L.; Zhou, J. A novel class of antimicrobial peptides from the scorpion Heterometrus spinifer. Peptides 2012, 38, 389–394. [Google Scholar] [CrossRef]
- Harrison, P.L.; Abdel-Rahman, M.A.; Miller, K.; Strong, P.N. Antimicrobial peptides from scorpion venoms. Toxicon 2014, 88, 115–137. [Google Scholar] [CrossRef]
- Hernández-Aponte, C.A.; Silva-Sánchez, J.; Quintero-Hernández, V.; Rodríguez-Romero, A.; Balderas, C.; Possani, L.D.; Gurrola, G.B. Vejovine, a new antibiotic from the scorpion venom of Vaejovis mexicanus. Toxicon 2011, 57, 84–92. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, H.; Lü, H.; Li, G.; Huang, Q. LAMP: A Database Linking Antimicrobial Peptides. PLoS ONE 2013, 8, e66557. [Google Scholar] [CrossRef]
- Di Luca, M.; Maccari, G.; Maisetta, G.; Batoni, G. BaAMPs: The database of biofilm-active antimicrobial peptides. Biofouling 2015, 31, 193–199. [Google Scholar] [CrossRef]
- Fan, L.; Sun, J.; Zhou, M.; Zhou, J.; Lao, X.; Zheng, H.; Xu, H. DRAMP: A comprehensive data repository of antimicrobial peptides. Sci. Rep. 2016, 6, 24482. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Yucesoy, D.T.; Hnilova, M.; Boone, K.; Arnold, P.M.; Snead, M.L.; Tamerler, C. Chimeric peptides as implant functionalization agents for titanium alloy implants with antimicrobial properties. JOM 2015, 67, 754–766. [Google Scholar] [CrossRef]
- Wisdom, C.; Van Oosten, S.K.; Boone, K.W.; Khvostenko, D.; Arnold, P.M.; Snead, M.L.; Tamerler, C. Controlling the Biomimetic Implant Interface: Modulating Antimicrobial Activity by Spacer Design. J. Mol. Eng. Mater. 2016, 4, 1640005. [Google Scholar] [CrossRef]
- Tajbakhsh, M.; Karimi, A.; Tohidpour, A.; Abbasi, N.; Fallah, F.; Akhavan, M.M. The antimicrobial potential of a new derivative of cathelicidin from Bungarus fasciatus against methicillin-resistant Staphylococcus aureus. J. Microbiol. 2018, 56, 128–137. [Google Scholar] [CrossRef]
- Takahashi, D.; Shukla, S.K.; Prakash, O.; Zhang, G. Structural determinants of host defense peptides for antimicrobial activity and target cell selectivity. Biochimie 2010, 92, 1236–1241. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, J.; Chen, Y. Alpha-helical cationic antimicrobial peptides: Relationships of structure and function. Protein Cell 2010, 1, 143–152. [Google Scholar] [CrossRef]
- Scocchi, M.; Wang, S.; Zanetti, M. Structural organization of the bovine cathelicidin gene family and identification of a novel member 1. FEBS Lett. 1997, 417, 311–315. [Google Scholar] [CrossRef]
- Lewies, A.; Wentzel, J.F.; Jacobs, G.; Du Plessis, L.H. The Potential Use of natural and structural analogues of antimicrobial peptides in the fight against neglected tropical diseases. Molecules 2015, 20, 15392–15433. [Google Scholar] [CrossRef]
- Ciumac, D.; Gong, H.; Hu, X.; Lu, J.R. Membrane targeting cationic antimicrobial peptides. J. Colloid Interface Sci. 2019, 537, 163–185. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial peptides: diversity, mechanism of action and strategies to improve the activity and biocompatibility in vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef]
- Doherty, T.; Waring, A.J.; Hong, M. Peptide–lipid interactions of the β-hairpin antimicrobial peptide tachyplesin and its linear derivatives from solid-state NMR. Biochim. Biophys. Acta-Biomembr. 2006, 1758, 1285–1291. [Google Scholar] [CrossRef]
- Sitaram, N.; Subbalakshmi, C.; Nagaraj, R. Indolicidin, a 13-residue basic antimicrobial peptide rich in tryptophan and proline, interacts with Ca2+-calmodulin. Biochem. Biophys. Res. Commun. 2003, 309, 879–884. [Google Scholar] [CrossRef]
- Shi, J.; Ross, C.R.; Leto, T.L.; Blecha, F. PR-39, a proline-rich antibacterial peptide that inhibits phagocyte NADPH oxidase activity by binding to Src homology 3 domains of p47 phox. Proc. Natl. Acad. Sci. USA 1996, 93, 6014–6018. [Google Scholar] [CrossRef]
- Van Dijk, I.A.; Nazmi, K.; Bolscher, J.G.M.; Veerman, E.C.I.; Stap, J. Histatin-1, a histidine-rich peptide in human saliva, promotes cell-substrate and cell-cell adhesion. FASEB J. 2015, 29, 3124–3132. [Google Scholar] [CrossRef]
- Stepensky, D. Pharmacokinetics of Toxin-Derived Peptide Drugs. Toxins 2018, 10, 483. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar] [CrossRef]
- Marr, A.K.; McGwire, B.S.; McMaster, W.R. Modes of action of Leishmanicidal antimicrobial peptides. Future Microbiol. 2012, 7, 1047–1059. [Google Scholar] [CrossRef]
- Robbel, L.; Marahiel, M.A. Daptomycin, a bacterial lipopeptide synthesized by a nonribosomal machinery. J. Boil. Chem. 2010, 285, 27501–27508. [Google Scholar] [CrossRef]
- Pogliano, J.; Pogliano, N.; Silverman, J.A. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J. Bacteriol. 2012, 194, 4494–4504. [Google Scholar] [CrossRef]
- Mosaheb, M.U.W.F.Z.; Khan, N.A.; Siddiqui, R. Cockroaches, locusts, and envenomating arthropods: A promising source of antimicrobials. Iran. J. Basic Med. Sci. 2018, 21, 873–877. [Google Scholar]
- Huang, J.; Feigenson, G.W. A microscopic interaction model of maximum solubility of cholesterol in lipid bilayers. Biophys. J. 1999, 76, 2142–2157. [Google Scholar] [CrossRef]
- Ali, M.R.; Cheng, K.H.; Huang, J. Ceramide drives cholesterol out of the ordered lipid bilayer phase into the crystal phase in 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine/cholesterol/ceramide ternary mixtures. Biochemistry 2006, 45, 12629–12638. [Google Scholar] [CrossRef]
- Matsuzaki, K. Why and how are peptide–lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta-Biomembr. 1999, 1462, 1–10. [Google Scholar] [CrossRef]
- Powers, J.-P.S.; Hancock, R.E. The relationship between peptide structure and antibacterial activity. Peptides 2003, 24, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Anderson, P.; Garcia-Salcedo, J.A.; Caro, M.; Gonzalez-Rey, E. Neuropeptides kill African trypanosomes by targeting intracellular compartments and inducing autophagic-like cell death. Cell Death Differ. 2009, 16, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Luque-Ortega, J.R.; Hof, W.V.; Veerman, E.C.I.; Saugar, J.M.; Rivas, L. Human antimicrobial peptide histatin 5 is a cell-penetrating peptide targeting mitochondrial ATP synthesis in Leishmania. FASEB J. 2008, 22, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.A.J.; Hart, G.W.; Kinoshita, T. Essentials of Glycobiology, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2015. [Google Scholar]
- Barreto-Bergter, E. Structures of glycolipids found in trypanosomatids: contribution to parasite functions. Open Parasitol. J. 2010, 4, 84–97. [Google Scholar] [CrossRef]
- Souto-Padrón, T. The surface charge of trypanosomatids. An. Acad. Bras. Cienc. 2002, 74, 649–675. [Google Scholar] [CrossRef] [Green Version]
- Torrent, M.; Pulido, D.; Rivas, L.; Andreu, D. Antimicrobial peptide action on parasites. Curr. Drug Targets 2012, 13, 1138–1147. [Google Scholar] [CrossRef]
- McConville, M.J.; Mullin, K.A.; Ilgoutz, S.C.; Teasdale, R.D. Secretory pathway of trypanosomatid parasites. Microbiol. Mol. Boil. Rev. 2002, 66, 122–154. [Google Scholar] [CrossRef]
- Kulkarni, M.M.; McMaster, W.R.; Kamysz, E.; Kamysz, W.; Engman, D.M.; McGwire, B.S. The major surface-metalloprotease of the parasitic protozoan, Leishmania, protects against antimicrobial peptide-induced apoptotic killing. Mol. Microbiol. 2006, 62, 1484–1497. [Google Scholar] [CrossRef]
- Homans, S.W.; Mehlert, A.; Turco, S.J. Solution structure of the lipophosphoglycan of Leishmania donovani. Biochemistry 1992, 31, 654–661. [Google Scholar] [CrossRef]
- Razzazan, A.; Saberi, M.R.; Jaafari, M.R. Hypothesis Insights from the analysis of a predicted model of gp63 in Leishmania donovani. Bioinformation 2008, 3, 114. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Maisetta, G.; Di Luca, M.; Gaddi, L.M.H.; Esin, S.; Florio, W.; Brancatisano, F.L.; Barra, D.; Campa, M.; Batoni, G. Comparative analysis of the bactericidal activities of amphibian peptide analogues against multidrug-resistant nosocomial bacterial strains. Antimicrob. Agents Chemother. 2008, 52, 85. [Google Scholar] [CrossRef]
- Mangoni, M.L.; Marcellini HG, L.; Simmaco, M. Biological characterization and modes of action of temporins and bombinins H, multiple forms of short and mildly cationic anti-microbial peptides from amphibian skin. J. Pept. Sci. 2007, 13, 603–613. [Google Scholar] [CrossRef]
- Späth, G.F.; Epstein, L.; Leader, B.; Singer, S.M.; Avila, H.A.; Turco, S.J.; Beverley, S.M. Lipophosphoglycan is a virulence factor distinct from related glycoconjugates in the protozoan parasite Leishmania major. Proc. Natl. Acad. Sci. USA 2000, 97, 9258–9263. [Google Scholar] [CrossRef]
- Pimenta, P.F.; De Souza, W. Leishmania mexicana amazonensis: Surface charge of amastigote and promastigote forms. Exp. Parasitol. 1983, 56, 194–206. [Google Scholar] [CrossRef]
- Burleigh, B.A.; Andrews, N.W. The Mechanisms of Trypanosoma cruzi invasion of mammalian cells. Annu. Rev. Microbiol. 1995, 49, 175–200. [Google Scholar] [CrossRef]
- De Lederkremer, R.M.; Colli, W. Galactofuranose-containing glycoconjugates in trypanosomatids. Glycobiology 1995, 5, 547–552. [Google Scholar] [CrossRef]
- Nardy, A.F.F.R.; Freire-De-Lima, C.G.; Pérez, A.R.; Morrot, A. Role of Trypanosoma cruzi trans-sialidase on the escape from host immune surveillance. Front. Microbiol. 2016, 7, 784. [Google Scholar] [CrossRef]
- Acosta-Serrano, A. The mucin-like glycoprotein super-family of Trypanosoma cruzi: Structure and biological roles. Mol. Biochem. Parasitol. 2001, 114, 143–150. [Google Scholar] [CrossRef]
- Yao, C. Major surface protease of trypanosomatids: One size fits all? Infect. Immun. 2010, 78, 22–31. [Google Scholar] [CrossRef]
- Mehlert, A.; Bond, C.S.; Ferguson, M.A. The glycoforms of a Trypanosoma brucei variant surface glycoprotein and molecular modeling of a glycosylated surface coat. Glycobiology 2002, 12, 607–612. [Google Scholar] [CrossRef]
- Pays, E.; Vanhamme, L.; Perez-Morga, D. Antigenic variation in Trypanosoma brucei: Facts, challenges and mysteries. Curr. Opin. Microbiol. 2004, 7, 369–374. [Google Scholar] [CrossRef]
- Pays, E. Expression and function of surface proteins in Trypanosoma brucei. Mol. Biochem. Parasitol. 1998, 91, 3–36. [Google Scholar] [CrossRef]
- Hsiao, L.L.; Howard, R.J.; Aikawa, M.; Taraschi, T.F. Modification of host cell membrane lipid composition by the intra-erythrocytic human malaria parasite Plasmodium falciparum. Biochem. J. 1991, 274, 121–132. [Google Scholar] [CrossRef]
- Gelhaus, C.; Jacobs, T.; Andrä, J.; Leippe, M. The Antimicrobial Peptide NK-2, the Core Region of Mammalian NK-Lysin, Kills Intraerythrocytic Plasmodium falciparum. Antimicrob. Agents Chemother. 2008, 52, 1713–1720. [Google Scholar] [CrossRef]
- Vale, N.; Aguiar, L.; Gomes, P.; Aguiar, L. Antimicrobial peptides: A new class of antimalarial drugs? Front. Pharmacol. 2014, 5, 275. [Google Scholar] [CrossRef]
- Weiss, L.M.; Kim, K. Toxoplasma Gondii: The Model Apicomplexan. Perspectives and Methods, 2nd ed.; Elsevier: Amsterdam, The Netherlands; Academic Press: Cambridge, MA, USA, 2013; ISBN 9780123964816. [Google Scholar]
- Tsai, Y.-H.; Liu, X.; Seeberger, P.H. Chemical Biology of Glycosylphosphatidylinositol Anchors. Angew. Chem. Int. Ed. 2012, 51, 11438–11456. [Google Scholar] [CrossRef]
- Harrington, J.M. Antimicrobial peptide killing of African trypanosomes. Parasite Immunol. 2011, 33, 461–469. [Google Scholar] [CrossRef] [Green Version]
- McGwire, B.S.; Kulkarni, M.M. Interactions of antimicrobial peptides with Leishmania and trypanosomes and their functional role in host parasitism. Exp. Parasitol. 2010, 126, 397–405. [Google Scholar] [CrossRef]
- Lacerda, A.F.; Pelegrini, P.B.; De Oliveira, D.M.; Vasconcelos, É.A.; Grossi-De-Sa, M.F. Anti-parasitic Peptides from Arthropods and their Application in Drug Therapy. Front. Microbiol. 2016, 7, 232. [Google Scholar] [CrossRef]
- Yeaman, M.R.; Yount, N.Y.; Hauger, R.L.; Grigoriadis, D.E.; Dallman, M.F.; Plotsky, P.M.; Vale, W.W.; Dautzenberg, F.M. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Li, X.; Wang, Z. APD2: The updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2008, 37, D933–D937. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Genet. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Lee, M.-T.; Sun, T.-L.; Hung, W.-C.; Huang, H.W. Process of inducing pores in membranes by melittin. Proc. Natl. Acad. Sci. USA 2013, 110, 14243–14248. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Prieto, L.; Lazaridis, T. Modeling peptide binding to anionic membrane pores. J. Comput. Chem. 2013, 34, 1463–1475. [Google Scholar] [CrossRef]
- Pokorny, A.; Almeida, P.F.F. Kinetics of Dye Efflux and Lipid Flip-Flop Induced by δ-lysin in phosphatidylcholine vesicles and the mechanism of graded release by amphipathic, α-helical peptides. Biochemistry 2004, 43, 8846–8857. [Google Scholar] [CrossRef]
- Lee, T.-H.; N Hall, K.; Aguilar, M.-I. Antimicrobial peptide structure and mechanism of action: A focus on the role of membrane structure. Curr. Top. Med. Chem. 2015, 16, 25–39. [Google Scholar] [CrossRef]
- Mattila, J.-P.; Sabatini, K.; Kinnunen, P.K. Oxidized phospholipids as potential molecular targets for antimicrobial peptides. Biochim. Biophys. Acta-Biomembr. 2008, 1778, 2041–2050. [Google Scholar] [CrossRef] [Green Version]
- Rokitskaya, T.I.; Kolodkin, N.I.; Kotova, E.A.; Antonenko, Y.N. Indolicidin action on membrane permeability: Carrier mechanism versus pore formation. Biochim. Biophys. Acta-Biomembr. 2011, 1808, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Rogers, M.J. Inhibition of Plasmodium falciparum protein synthesis. Targeting the plastid-like organelle with thiostrepton. J. Boil. Chem. 1997, 272, 2046–2049. [Google Scholar]
- Clough, B.; Strath, M.; Preiser, P.; Denny, P.; Wilson, I.R.; Wilson, I. Thiostrepton binds to malarial plastid rRNA. FEBS Lett. 1997, 406, 123–125. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.; Mao, Y.; Wang, J.; Liu, M.; Qiao, Y.; Zheng, L.; Su, Y.; Ke, Q.; Zheng, W. Molecular mechanisms of an antimicrobial peptide piscidin (Lc-pis) in a parasitic protozoan, Cryptocaryon irritans. BMC Genom. 2018, 19, 192. [Google Scholar] [CrossRef]
- Lin, Q.; Katakura, K.; Suzuki, M. Inhibition of mitochondrial and plastid activity of Plasmodium falciparum by minocycline. FEBS Lett. 2002, 515, 71–74. [Google Scholar] [CrossRef]
- Goodman, C.D.; Su, V.; McFadden, G.I. The effects of anti-bacterials on the malaria parasite Plasmodium falciparum. Mol. Biochem. Parasitol. 2007, 152, 181–191. [Google Scholar] [CrossRef]
- Aminake, M.N.; Schoof, S.; Sologub, L.; Leubner, M.; Kirschner, M.; Arndt, H.-D.; Pradel, G. Thiostrepton and Derivatives Exhibit Antimalarial and gametocytocidal activity by dually targeting parasite proteasome and apicoplast. Antimicrob. Agents Chemother. 2011, 55, 1338–1348. [Google Scholar] [CrossRef]
- Vinhote, J.F.C.; Lima, D.B.; Mello, C.P.; de Souza, B.M.; Havt, A.; Palma, M.S.; dos Santos, R.P.; de Albuquerque, E.L.; Freire, V.N.; Martins, A.M.C. Trypanocidal activity of mastoparan from Polybia paulista wasp venom by interaction with TcGAPDH. Toxicon 2017, 137, 168–172. [Google Scholar] [CrossRef]
- Kulkarni, M.M.; McMaster, W.R.; Kamysz, W.; McGwire, B.S. Antimicrobial peptide-induced apoptotic death of Leishmania results from calcium-de pendent, caspase-independent mitochondrial toxicity. J. Biol. Chem. 2009, 284, 15496–15504. [Google Scholar] [CrossRef]
- Moreno, S.N.J.; Docampo, R. Calcium regulation in protozoan parasites. Curr. Opin. Microbiol. 2003, 6, 359–364. [Google Scholar] [CrossRef]
- Gupta, S.; Raychaudhury, B.; Banerjee, S.; Das, B.; Datta, S.C. An intracellular calcium store is present in Leishmania donovani glycosomes. Exp. Parasitol. 2006, 113, 161–167. [Google Scholar] [CrossRef]
- Da Costa, J.P.; Cova, M.; Ferreira, R.; Vitorino, R. Antimicrobial peptides: An alternative for innovative medicines? Appl. Microbiol. Biotechnol. 2015, 99, 2023–2040. [Google Scholar] [CrossRef]
- Fura, J.; Sarkar, S.; Pidgeon, S.; Pires, M. Combatting bacterial pathogens with immunomodulation and infection tolerance strategies. Curr. Top. Med. Chem. 2016, 16, 1. [Google Scholar] [CrossRef]
- Niyonsaba, F.; Hirata, M.; Ogawa, H.; Nagaoka, I. Epithelial cell-derived antibacterial peptides human beta-defensins and cathelicidin: Multifunctional activities on mast cells. Curr. Drug Targets. Inflamm. Allergy 2003, 2, 224–231. [Google Scholar] [CrossRef]
- Oppenheim, J.J.; Biragyn, A.; Kwak, L.W.; Yang, D. Roles of antimicrobial peptides such as defensins in innate and adaptive immunity. Ann. Rheum. Dis. 2003, 62, ii17–ii21. [Google Scholar] [CrossRef]
- Yang, R.; Zhang, Z.; Pei, X.; Han, X.; Wang, J.; Wang, L.; Long, Z.; Shen, X.; Li, Y. Immunomodulatory effects of marine oligopeptide preparation from Chum Salmon (Oncorhynchus keta) in mice. Food Chem. 2009, 113, 464–470. [Google Scholar] [CrossRef]
- Lahov, E.; Regelson, W. Antibacterial and immunostimulating casein-derived substances from milk: Casecidin, isracidin peptides. Food Chem. Toxicol. 1996, 34, 131–145. [Google Scholar] [CrossRef]
- Hilchie, A.L.; Wuerth, K.; Hancock, R.E.W. Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat. Methods 2013, 9, 761–768. [Google Scholar] [CrossRef]
- Nijnik, A.; Hancock, R. Host defence peptides: Antimicrobial and immunomodulatory activity and potential applications for tackling antibiotic-resistant infections. Emerg. Heal. Threat. J. 2009, 2, 7078. [Google Scholar] [CrossRef]
- Afacan, N.J.; Yeung, A.T.; Pena, O.M.; Hancock, R.E. Therapeutic potential of host defense peptides in antibiotic-resistant infections. Curr. Pharm. Des. 2012, 18, 807–819. [Google Scholar] [CrossRef]
- Lai, Y.; Gallo, R.L. AMPed Up immunity: How antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009, 30, 131–141. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Nijnik, A.; Philpott, D.J. Modulating immunity as a therapy for bacterial infections. Nat. Rev. Genet. 2012, 10, 243–254. [Google Scholar] [CrossRef]
- Bowdish, D.; Davidson, D.; Hancock, R. A Re-evaluation of the role of host defence peptides in mammalian immunity. Curr. Protein Pept. Sci. 2005, 6, 35–51. [Google Scholar] [CrossRef]
- Karaś, M.A.; Turska-Szewczuk, A.; Janczarek, M.; Szuster-Ciesielska, A. Glycoconjugates of Gram-negative bacteria and parasitic protozoa—Are they similar in orchestrating the innate immune response? Innate Immun. 2019, 25, 73–96. [Google Scholar] [CrossRef]
- Achtman, A.H.; Pilat, S.; Law, C.W.; Lynn, D.J.; Janot, L.; Mayer, M.L.; Ma, S.; Kindrachuk, J.; Finlay, B.B.; Brinkman, F.S.L. Effective adjunctive therapy by an innate defense regulatory peptide in a preclinical model of severe malaria. Sci. Transl. Med. 2012, 4, 135ra64. [Google Scholar] [CrossRef]
- Zhao, H.; Kinnunen, P.K.J. Modulation of the activity of secretory phospholipase A2 by antimicrobial peptides. Antimicrob. Agents Chemother. 2003, 47, 965–971. [Google Scholar] [CrossRef]
- Boutrin, M.-C.; Foster, H.; Pentreath, V.; Foster, H. The effects of bee (Apis mellifera) venom phospholipase A2 on Trypanosoma brucei brucei and enterobacteria. Exp. Parasitol. 2008, 119, 246–251. [Google Scholar] [CrossRef]
- Grabner, A.N.; Alfonso, J.; Kayano, A.M.; Moreira-Dill, L.S.; Santos, A.P.D.A.D.; Caldeira, C.A.; Sobrinho, J.C.; Gómez, A.; Grabner, F.P.; Cardoso, F.F.; et al. BmajPLA 2-II, a basic Lys49-phospholipase A2 homologue from Bothrops marajoensis snake venom with parasiticidal potential. Int. J. Boil. Macromol. 2017, 102, 571–581. [Google Scholar] [CrossRef]
- Barros, G.A.C.; Pereira, A.V.; Barros, L.C.; Lourenco, A.L., Jr.; Calvi, S.A.; Santos, L.D.; Barraviera, B.; Ferreira, R.S. In vitro activity of phospholipase A2 and of peptides from Crotalus durissus terrificus venom against amastigote and promastigote forms of Leishmania (L.) infantum chagasi. J. Venom. Anim. Toxins Incl. Trop. Dis. 2015, 21, 537. [Google Scholar] [CrossRef]
- Nicholls, E.F.; Madera, L.; Hancock, R.E.W. Immunomodulators as adjuvants for vaccines and antimicrobial therapy. Ann. N. Y. Acad. Sci. 2010, 1213, 46–61. [Google Scholar] [CrossRef]
- Steverding, D. The history of Chagas disease. Parasit. Vectors 2014, 7, 317. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention; Global Health, D. of P.D. and M. CDC—American trypanosomiasis. Available online: https://www.cdc.gov/parasites (accessed on 25 June 2019).
- World Health Organization. WHO Chagas disease (American trypanosomiasis). Available online: https://www.who.int/chagas/en/ (accessed on 25 June 2019).
- Antinori, S.; Galimberti, L.; Bianco, R.; Grande, R.; Galli, M.; Corbellino, M. Chagas disease in Europe: A review for the internist in the globalized world. Eur. J. Intern. Med. 2017, 43, 6–15. [Google Scholar] [CrossRef]
- Pérez-Molina, J.A.; Molina, I. Chagas disease. Lancet 2018, 391, 82–94. [Google Scholar] [CrossRef]
- Bermudez, J.; Davies, C.; Simonazzi, A.; Real, J.P.; Palma, S. Current drug therapy and pharmaceutical challenges for Chagas disease. Acta Trop. 2016, 156, 1–16. [Google Scholar] [CrossRef]
- Palmeiro-Roldán, R.; Fonseca-Berzal, C.; Gómez-Barrio, A.; Arán, V.J.; Escario, J.A.; Torrado-Duran, S.; Torrado-Santiago, S. Development of novel benznidazole formulations: Physicochemical characterization and in vivo evaluation on parasitemia reduction in Chagas disease. Int. J. Pharm. 2014, 472, 110–117. [Google Scholar] [CrossRef] [Green Version]
- Mejia, A.M.; Hall, B.S.; Taylor, M.C.; Gómez-Palacio, A.; Wilkinson, S.R.; Triana-Chávez, O.; Kelly, J.M. Benznidazole-Resistance in Trypanosoma cruzi is a readily acquired trait that can arise independently in a single population. J. Infect. Dis. 2012, 206, 220–228. [Google Scholar] [CrossRef]
- Adade, C.M.; Oliveira, I.R.; Pais, J.A.; Souto-Padrón, T. Melittin peptide kills Trypanosoma cruzi parasites by inducing different cell death pathways. Toxicon 2013, 69, 227–239. [Google Scholar] [CrossRef]
- Adade, C.M.; Chagas, G.S.F.; Souto-Padrón, T. Apis mellifera venom induces different cell death pathways in Trypanosoma cruzi. Parasitology 2012, 139, 1444–1461. [Google Scholar] [CrossRef]
- Ruben, L.; Akins, C.D.; Haghighat, N.G.; Xue, L. Calcium influx in Trypanosoma brucei can be induced by amphiphilic peptides and amines. Mol. Biochem. Parasitol. 1996, 81, 191–200. [Google Scholar] [CrossRef]
- Chicharro, C.; Granata, C.; Lozano, R.; Andreu, D.; Rivas, L. N-terminal fatty acid substitution increases the leishmanicidal activity of CA(1-7)M(2-9), a cecropin-melittin hybrid peptide. Antimicrob. Agents Chemother. 2001, 45, 2441–2449. [Google Scholar] [CrossRef]
- Khalili, S.; Mohebali, M.; Ebrahimzadeh, E.; Shayan, E.; Mohammadi-Yeganeh, S.; Moghaddam, M.M.; Elikaee, S.; Akhoundi, B.; Sharifi-Yazdi, M.K. Antimicrobial activity of an antimicrobial peptide against amastigote forms of Leishmania major. Veter-Res. Forum Int. Q. J. 2018, 9, 323–328. [Google Scholar]
- Fieck, A.; Hurwitz, I.; Kang, A.S.; Durvasula, R. Trypanosoma cruzi: Synergistic cytotoxicity of multiple amphipathic anti-microbial peptides to T. cruzi and potential bacterial hosts. Exp. Parasitol. 2010, 125, 342–347. [Google Scholar] [CrossRef]
- Lima, D.B.; Mello, C.P.; Bandeira, I.C.J.; De Menezes, R.R.P.P.B.; Sampaio, T.L.; Falcão, C.B.; Morlighem, J.-R.L.; Rádis-Baptista, G.; Martins, A.M.C. The dinoponeratoxin peptides from the giant ant Dinoponera quadriceps display in vitro antitrypanosomal activity. Boil. Chem. 2018, 399, 187–196. [Google Scholar] [CrossRef]
- Parente, A.M.S.; Daniele-Silva, A.; Furtado, A.A.; Melo, M.A.; Lacerda, A.F.; Queiroz, M.; Moreno, C.; Santos, E.; Rocha, H.A.O.; Barbosa, E.G.; et al. Analogs of the scorpion venom peptide Stigmurin: Structural assessment, toxicity, and increased antimicrobial activity. Toxins 2018, 10, 161. [Google Scholar] [CrossRef]
- Kuhn-Nentwig, L.; Willems, J.; Seebeck, T.; Shalaby, T.; Kaiser, M.; Nentwig, W. Cupiennin 1a exhibits a remarkably broad, non-stereospecific cytolytic activity on bacteria, protozoan parasites, insects, and human cancer cells. Amino Acids 2011, 40, 69–76. [Google Scholar] [CrossRef]
- Mashaghi, A.; Bezrukavnikov, S.; Minde, D.P.; Wentink, A.S.; Kityk, R.; Zachmann-Brand, B.; Mayer, M.P.; Kramer, G.; Bukau, B.; Tans, S.J. Alternative modes of client binding enable functional plasticity of Hsp70. Nature 2016, 539, 448–451. [Google Scholar] [CrossRef]
- Kragol, G.; Lovas, S.; Váradi, G.; Condie, B.A.; Hoffmann, R.; Otvos, L. The antibacterial peptide pyrrhocoricin inhibits the atpase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry 2001, 40, 3016–3026. [Google Scholar] [CrossRef]
- Nwaka, S.; Hudson, A. Innovative lead discovery strategies for tropical diseases. Nat. Rev. Drug Discov. 2006, 5, 941–955. [Google Scholar] [CrossRef]
- Amorim-Carmo, B.; Daniele-Silva, A.; Parente, A.M.S.; Furtado, A.A.; Carvalho, E.; Oliveira, J.W.F.; Santos, E.C.G.; Silva, M.S.; Silva, S.R.B.; Silva-Júnior, A.A.; et al. Potent and broad-spectrum antimicrobial activity of analogs from the scorpion peptide stigmurin. Int. J. Mol. Sci. 2019, 20, 623. [Google Scholar] [CrossRef]
- World Health Organization. WHO Trypanosomiasis, Human African (Sleeping Sickness). Available online: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed on 8 July 2019).
- Pays, E.; Vanhollebeke, B.; Uzureau, P.; Lecordier, L.; Perez-Morga, D. The molecular arms race between African trypanosomes and humans. Nat. Rev. Genet. 2014, 12, 575–584. [Google Scholar] [CrossRef]
- Tiberti, N.; Sanchez, J.C. Sleeping Sickness in the ‘Omics Era. Proteom.-Clin. Appl. 2018, 12, 1–10. [Google Scholar] [CrossRef]
- Maxmen, A. Pill treats sleeping sickness Scientists seek approval from regulators for this relatively quick and easy therapy. Nature 2017, 550, 441. [Google Scholar] [CrossRef]
- Pink, R.; Hudson, A.; Mouriès, M.-A.; Bendig, M. Opportunities and Challenges in Antiparasitic Drug Discovery. Nat. Rev. Drug Discov. 2005, 4, 727–740. [Google Scholar] [CrossRef]
- World Health Organization. WHO Leishmaniasis. Available online: https://www.who.int/en/news-room/fact-sheets/detail/leishmaniasis (accessed on 23 July 2019).
- Myler, P.J.; Fasel, N. Leishmania: After the Genome; Caister Academic: Poole, UK, 2008; ISBN 9781904455288. [Google Scholar]
- McGwire, B.S.; Satoskar, A.R. Leishmaniasis: Clinical syndromes and treatment. QJM 2014, 107, 7–14. [Google Scholar] [CrossRef]
- Alvar, J.; Velez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; Boer, M.D. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef]
- Kaye, P.; Scott, P. Leishmaniasis: Complexity at the host–pathogen interface. Nat. Rev. Genet. 2011, 9, 604–615. [Google Scholar] [CrossRef]
- Zahedifard, F.; Rafati, S. Prospects for antimicrobial peptide-based immunotherapy approaches in Leishmania control. Expert Rev. Anti-Infect. Ther. 2018, 16, 461–469. [Google Scholar] [CrossRef]
- Ali, M.; Hofland, H.; Zijlstra, E.; El-Hassan, A.; El-Toum, I.; Satti, M.; Ghalib, H. The treatment of kala-azar in the Sudan with sodium stibogluconate: A randomized trial of three dosage regimens. Trans. R. Soc. Trop. Med. Hyg. 1993, 87, 307–309. [Google Scholar]
- Baiocco, P.; Colotti, G.; Franceschini, S.; Ilari, A. Molecular basis of antimony treatment in leishmaniasis. J. Med. Chem. 2009, 52, 2603–2612. [Google Scholar] [CrossRef]
- Ameen, M. Cutaneous and mucocutaneous leishmaniasis: Emerging therapies and progress in disease management. Expert Opin. Pharmacother. 2010, 11, 557–569. [Google Scholar] [CrossRef]
- Rojas, R.; Valderrama, L.; Valderrama, M.; Varona, M.X.; Ouellette, M.; Saravia, N.G. Resistance to Antimony and Treatment Failure in Human Leishmania (Viannia) Infection. J. Infect. Dis. 2006, 193, 1375–1383. [Google Scholar] [CrossRef]
- Croft, S.L.; Sundar, S.; Fairlamb, A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [Google Scholar] [CrossRef]
- De Menezes, J.P.B.; Guedes, C.E.S.; Petersen, A.L.D.O.A.; Fraga, D.B.M.; Veras, P.S.T. Advances in development of new treatment for leishmaniasis. Biomed Res. Int. 2015, 2015, 815023. [Google Scholar] [CrossRef]
- Berman, J. Clinical status of agents being developed for leishmaniasis. Expert Opin. Investig. Drugs 2005, 14, 1337–1346. [Google Scholar] [CrossRef]
- Sundar, S.; Olliaro, P.L. Miltefosine in the treatment of leishmaniasis: Clinical evidence for informed clinical risk management. Ther. Clin. Risk Manag. 2007, 3, 733–740. [Google Scholar]
- Chattopadhyay, A.; Jafurulla, M. A novel mechanism for an old drug: Amphotericin B in the treatment of visceral leishmaniasis. Biochem. Biophys. Res. Commun. 2011, 416, 7–12. [Google Scholar] [CrossRef]
- Sundar, S.; Rai, M.; Chakravarty, J.; Agarwal, D.; Agrawal, N.; Vaillant, M.; Olliaro, P.; Murray, H.W. New treatment approach in indian visceral leishmaniasis: Single-dose liposomal amphotericin b followed by short-course oral miltefosine. Clin. Infect. Dis. 2008, 47, 1000–1006. [Google Scholar] [CrossRef]
- Silva, P.I.; Daffre, S.; Bulet, P. Isolation and characterization of gomesin, an 18-residue cysteine-rich defense peptide from the spider Acanthoscurria gomesiana hemocytes with sequence similarities to horseshoe crab antimicrobial peptides of the tachyplesin family. J. Biol. Chem. 2000, 275, 33464–33470. [Google Scholar] [CrossRef]
- Schaeffer, M.; De Miranda, A.; Mottram, J.C.; Coombs, G.H. Differentiation of Leishmania major is impaired by over-expression of pyroglutamyl peptidase I. Mol. Biochem. Parasitol. 2006, 150, 318–329. [Google Scholar] [CrossRef]
- Konno, K.; Rangel, M.; Oliveira, J.S.; dos Santos Cabrera, M.P.; Fontana, R.; Hirata, I.Y.; Hide, I.; Nakata, Y.; Mori, K.; Kawano, M.; et al. Decoralin, a novel linear cationic α-helical peptide from the venom of the solitary eumenine wasp Oreumenes decoratus. Peptides 2007, 28, 2320–2327. [Google Scholar] [CrossRef]
- Konno, K.; Hisada, M.; Naoki, H.; Itagaki, Y.; Fontana, R.; Rangel, M.; Oliveira, J.S.; Cabrera, M.P.D.S.; Neto, J.R.; Hide, I.; et al. Eumenitin, a novel antimicrobial peptide from the venom of the solitary eumenine wasp Eumenes rubronotatus. Peptides 2006, 27, 2624–2631. [Google Scholar] [CrossRef]
- Konno, K.; Hisada, M.; Naoki, H.; Itagaki, Y.; Kawai, N.; Miwa, A.; Yasuhara, T.; Morimoto, Y.; Nakata, Y. Structure and biological activities of eumenine mastoparan-AF (EMP-AF), a new mast cell degranulating peptide in the venom of the solitary wasp (Anterhynchium flavomarginatum micado). Toxicon 2000, 38, 1505–1515. [Google Scholar] [CrossRef]
- Rangel, M.; Cabrera, M.P.D.S.; Kazuma, K.; Ando, K.; Wang, X.; Kato, M.; Nihei, K.-I.; Hirata, I.Y.; Cross, T.J.; Garcia, A.N.; et al. Chemical and biological characterization of four new linear cationic α-helical peptides from the venoms of two solitary eumenine wasps. Toxicon 2011, 57, 1081–1092. [Google Scholar] [CrossRef]
- Pérez-Cordero, J.J.; Lozano, J.M.; Cortés, J.; Delgado, G. Leishmanicidal activity of synthetic antimicrobial peptides in an infection model with human dendritic cells. Peptides 2011, 32, 683–690. [Google Scholar] [CrossRef]
- Diaz-Achirica, P.; Ubach, J.; Guinea, A.; Andreu, D.; Rivas, L. The plasma membrane of Leishmania donovani promastigotes is the main target for CA(1–8)M(1–18), a synthetic cecropin A–melittin hybrid peptide. Biochem. J. 1998, 330, 453–460. [Google Scholar] [CrossRef]
- Téné, N.; Bonnafé, E.; Berger, F.; Rifflet, A.; Guilhaudis, L.; Ségalas-Milazzo, I.; Pipy, B.; Coste, A.; Leprince, J.; Treilhou, M. Biochemical and biophysical combined study of bicarinalin, an ant venom antimicrobial peptide. Peptides 2016, 79, 103–113. [Google Scholar] [CrossRef]
- Borges, A.; Silva, S.; Camp, H.J.O.D.; Velasco, E.; Alvarez, M.; Alfonzo, M.J.; Jorquera, A.; De Sousa, L.; Delgado, O.; De Sousa, J.L. In vitro leishmanicidal activity of Tityus discrepans scorpion venom. Parasitol. Res. 2006, 99, 167–173. [Google Scholar] [CrossRef]
- Lima, D.B.; Sousa, P.L.; Torres, A.F.C.; Rodrigues, K.A.D.F.; Mello, C.P.; De Menezes, R.R.P.P.B.; Tessarolo, L.D.; Quinet, Y.P.; De Oliveira, M.R.; Martins, A.M.C. Antiparasitic effect of Dinoponera quadriceps giant ant venom. Toxicon 2016, 120, 128–132. [Google Scholar] [CrossRef]
- Alberola, J.; Rodriguez, A.; Francino, O.; Roura, X.; Rivas, L.; Andreu, D. Safety and efficacy of antimicrobial peptides against naturally acquired leishmaniasis. Antimicrob. Agents Chemother. 2004, 48, 641–643. [Google Scholar] [CrossRef]
- White, N.J.; Pukrittayakamee, S.; Hien, T.T.; Faiz, M.A.; Mokuolu, O.A.; Dondorp, A.M. Malaria. Lancet 2014, 383, 723–735. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, X.-Q. A Malaria transmission model with temperature-dependent incubation period. Bull. Math. Boil. 2017, 72, 1155–1182. [Google Scholar] [CrossRef]
- Watson, J.; Taylor, W.R.J.; Bancone, G.; Chu, C.S.; Jittamala, P.; White, N.J. Implications of current therapeutic restrictions for primaquine and tafenoquine in the radical cure of vivax malaria. PLoS Negl. Trop. Dis. 2018, 12, e0006440. [Google Scholar] [CrossRef]
- Sawa, P.; Shekalaghe, S.A.; Drakeley, C.J.; Sutherland, C.J.; Mweresa, C.K.; Baidjoe, A.Y.; Manjurano, A.; Kavishe, R.A.; Beshir, K.B.; Yussuf, R.U.; et al. Malaria transmission after artemether-lumefantrine and dihydroartemisinin-piperaquine: A randomized trial. J. Infect. Dis. 2013, 207, 1637–1645. [Google Scholar] [CrossRef]
- Jones, J.G. Malaria chemoprophylaxis and travel immunizations. Am. Fam. Physician 2010, 82, 583–584. [Google Scholar]
- Conde, R.; Zamudio, F.Z.; Rodr, M.H. Scorpine, an anti-malaria and anti-bacterial agent purified from scorpion venom. FEBS Lett. 2000, 471, 165–168. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Xu, J.; Rodriguez, M.D.C.; Lanz-Mendoza, H.; Hernández-Rivas, R.; Du, W.; Zhu, S. Characterization of two linear cationic antimalarial peptides in the scorpion Mesobuthus eupeus. Biochimie 2010, 92, 350–359. [Google Scholar] [CrossRef]
- Vizioli, J.; Bulet, P.; Hoffmann, J.A.; Kafatos, F.C.; Müller, H.-M.; Dimopoulos, G. Gambicin: A novel immune responsive antimicrobial peptide from the malaria vector Anopheles gambiae. Proc. Natl. Acad. Sci. USA 2001, 98, 12630–12635. [Google Scholar] [CrossRef]
- Zhu, S.; Gao, B.; Aumelas, A.; Rodriguez, M.D.C.; Lanz-Mendoza, H.; Peigneur, S.; Diego-Garcia, E.; Martin-Eauclaire, M.-F.; Tytgat, J.; Possani, L.D. MeuTXKβ1, a scorpion venom-derived two-domain potassium channel toxin-like peptide with cytolytic activity. Biochim. Biophys. Acta-Proteins Proteom. 2010, 1804, 872–883. [Google Scholar] [CrossRef]
- Choi, S.-J.; Parent, R.; Guillaume, C.; Deregnaucourt, C.; Delarbre, C.; Ojcius, D.M.; Montagne, J.-J.; Célérier, M.-L.; Phelipot, A.; Amiche, M.; et al. Isolation and characterization of Psalmopeotoxin I and II: Two novel antimalarial peptides from the venom of the tarantula Psalmopoeus cambridgei. FEBS Lett. 2004, 572, 109–117. [Google Scholar] [CrossRef]
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Zuegg, J.; Blaskovich, M.A.T.; Cooper, M.A. Contribution of amphipathicity and hydrophobicity to the antimicrobial activity and cytotoxicity of β-hairpin peptides. ACS Infect. Dis. 2016, 2, 442–450. [Google Scholar] [CrossRef]
- Moreira, C.K.; Rodrigues, F.G.; Ghosh, A.; Varotti, F.D.P.; Miranda, A.; Daffre, S.; Jacobs-Lorena, M.; Moreira, L.A. Effect of the antimicrobial peptide Gomesin against different life stages of Plasmodium spp. Exp. Parasitol. 2007, 116, 346–353. [Google Scholar] [CrossRef]
- Carter, V.; Underhill, A.; Baber, I.; Sylla, L.; Baby, M.; Larget-Thiery, I.; Zettor, A.; Bourgouin, C.; Langel, Ü.; Faye, I.; et al. Killer bee molecules: antimicrobial peptides as effector molecules to target sporogonic stages of Plasmodium. PLoS Pathog. 2013, 9, e1003790. [Google Scholar] [CrossRef] [PubMed]
- Cabezas-Cruz, A.; Tonk, M.; Bouchut, A.; Pierrot, C.; Pierce, R.J.; Kotsyfakis, M.; Rahnamaeian, M.; Vilcinskas, A.; Khalife, J.; Valdés, J.J. Antiplasmodial activity is an ancient and conserved feature of tick defensins. Front. Microbiol. 2016, 7, 2954. [Google Scholar] [CrossRef] [PubMed]
- Tonk, M.; Cabezas-Cruz, A.; Valdés, J.J.; Rego, R.O.; Rudenko, N.; Golovchenko, M.; Bell-Sakyi, L.; De La Fuente, J.; Grubhoffer, L. Identification and partial characterisation of new members of the Ixodes ricinus defensin family. Gene 2014, 540, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Asthana, N.; Yadav, S.P.; Ghosh, J.K. Dissection of antibacterial and toxic activity of melittin. J. Biol. Chem. 2004, 279, 55042–55050. [Google Scholar] [CrossRef] [PubMed]
- Konno, K.; Hisada, M.; Fontana, R.; Lorenzi, C.C.; Naoki, H.; Itagaki, Y.; Miwa, A.; Kawai, N.; Nakata, Y.; Yasuhara, T.; et al. Anoplin, a novel antimicrobial peptide from the venom of the solitary wasp Anoplius samariensis. Biochim. Biophys. Acta-Protein Struct. Mol. Enzym. 2001, 1550, 70–80. [Google Scholar] [CrossRef]
- Hirai, Y.; Yasuhara, T.; Yoshida, H.; Nakajima, T.; Fujino, M.; Kitada, C. A new mast cell degranulating peptide “mastoparan” in the venom of Vespula lewisii. Chem. Pharm. Bull. 1979, 27, 1942–1944. [Google Scholar] [CrossRef]
- Khan, K.; Khan, W. Congenital toxoplasmosis: An overview of the neurological and ocular manifestations. Parasitol. Int. 2018, 67, 715–721. [Google Scholar] [CrossRef]
- Martins-Duarte, E.S.; De Souza, W.; Vommaro, R.C. Toxoplasma gondii: The effect of fluconazole combined with sulfadiazine and pyrimethamine against acute toxoplasmosis in murine model. Exp. Parasitol. 2013, 133, 294–299. [Google Scholar] [CrossRef]
- Tang, Y.; Hou, S.; Li, X.; Wu, M.; Ma, B.; Wang, Z.; Jiang, J.; Deng, M.; Duan, Z.; Tang, X.; et al. Anti-parasitic effect on Toxoplasma gondii induced by a spider peptide lycosin-I. Exp. Parasitol. 2019, 198, 17–25. [Google Scholar] [CrossRef]
- Tanaka, T.; Maeda, H.; Galay, R.L.; Boldbattar, D.; Umemiya-Shirafuji, R.; Suzuki, H.; Xuan, X.; Tsuji, N.; Fujisaki, K. tick longicin implicated in the arthropod transmission of Toxoplasma Gondii. J. Vet. Sci. Technol. 2012, 3, 3633–3640. [Google Scholar]
- Tsuji, N.; Battsetseg, B.; Boldbaatar, D.; Miyoshi, T.; Xuan, X.; Oliver, J.H.; Fujisaki, K. Babesial vector tick defensin against Babesia sp. Parasites. Infect. Immun. 2007, 75, 3633–3640. [Google Scholar] [CrossRef] [PubMed]
- Fratini, F.; Cilia, G.; Turchi, B.; Felicioli, A. Insects, arachnids and centipedes venom: A powerful weapon against bacteria. A literature review. Toxicon 2017, 130, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Primon-Barros, M.; Macedo, A.J. Animal venom peptides: potential for new antimicrobial agents. Curr. Top. Med. Chem. 2017, 17, 1119–1156. [Google Scholar] [CrossRef]
- Rajamuthiah, R.; Jayamani, E.; Conery, A.L.; Fuchs, B.B.; Kim, W.; Johnston, T.; Vilcinskas, A.; Ausubel, F.M.; Mylonakis, E. A defensin from the model beetle Tribolium castaneum acts synergistically with telavancin and daptomycin against multidrug resistant Staphylococcus aureus. PLoS ONE 2015, 10, e0128576. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.P.; Yoshimatsu, K.; Suzuki, T.; Kato, T.; Park, E.Y. Expression and purification of cyto-insectotoxin (Cit1a) using silkworm larvae targeting for an antimicrobial therapeutic agent. Appl. Microbiol. Biotechnol. 2014, 98, 6973–6982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bommarius, B.; Jenssen, H.; Elliott, M.; Kindrachuk, J.; Pasupuleti, M.; Gieren, H.; Jaeger, K.-E.; Hancock, R.W.; Kalman, D. Cost-effective expression and purification of antimicrobial and host defense peptides in Escherichia coli. Peptides 2010, 31, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. Recombinant production of antimicrobial peptides in Escherichia coli: A review. Protein Expr. Purif. 2011, 80, 260–267. [Google Scholar] [CrossRef]
- Kulkarni, M.M.; Karafova, A.; Kamysz, W.; McGwire, B.S. Design of protease-resistant pexiganan enhances antileishmanial activity. Parasitol. Res. 2014, 113, 1971–1976. [Google Scholar] [CrossRef]
- Lipsky, B.A.; Holroyd, K.J.; Zasloff, M. Topical versus systemic antimicrobial therapy for treating mildly infected diabetic foot ulcers: A randomized, controlled, double-blinded, multicenter trial of pexiganan cream. Clin. Infect. Dis. 2008, 47, 1537–1545. [Google Scholar] [CrossRef]
- Fox, J.L. Antimicrobial peptides stage a comeback. Nat. Biotechnol. 2013, 31, 379–382. [Google Scholar] [CrossRef]
- World Health Organization. WHO Malaria. Available online: https://www.who.int/malaria/en/ (accessed on 28 July 2019).
- Wanted: A reward for antibiotic development. Nat. Biotechnol. 2018, 36, 555. [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | AMP | Parasite Stage | Inhibition Activity a | Reference |
|---|---|---|---|---|
| Insect | ||||
| Apis mellifera | Melittin | Epimastigote Trypomastigote Amastigote | IC50 = 2.44 μg/mL IC50 = 0.14 μg/mL IC50 = 0.22 μg/mL | [155] |
| A. mellifera | Apiadecin 14 | Epimastigote | LD100 = 199 μM | [160] |
| Polybia paulista | Mastoparan | Epimastigote Trypomastigote Amastigote | IC50 = 61.4 μM IC50 = 5.31 μM b | [124] |
| Dinoponera quadriceps | M-PONTX-Dq3a | Epimastigote Trypomastigote Amastigote | IC50 = 4.7 μM IC50 = 0.32 μM b | [163] |
| D. quadriceps | M-PONTX-Dq3b | Epimastigote Trypomastigote | IC50 = 48.8 μM IC50 = 7.4 μM | [163] |
| D. quadriceps | M-PONTX-Dq3c | Trypomastigote | IC50 = 34.8 μM | [163] |
| D. quadriceps | M-PONTX-Dq4e | Epimastigote Trypomastigote Amastigote | IC50 = 23.5 μM IC50 = 4.7 μM b | [163] |
| Scorpion | ||||
| Tityus stigmurus | Stigmurin | Epimastigote Trypomastigote | GI = 90% (25μM) GI = 100% (25μM) | [164] |
| Spider | ||||
| Cupiennius salei | Cupiennin 1a | Amastigote | IC50 = 0.92 μM | [165] |
| Source | AMP | Activity against | Parasite Stage | Inhibition Activity a | Reference |
|---|---|---|---|---|---|
| Insect | |||||
| Apis mellifera | Melittin | L. major L. panamensis | Promastigote | EC50 = 74.01 μg/mL EC50 ≥ 100 μg/mL | [195] |
| Anoplius samariensis | Anoplin | L. major | Promastigote | IC50 ≥ 87 μM | [191] |
| Oreumenes decoratus | Decoralin | L. major | Promastigote | IC50 = 72 μM | [191] |
| Eumenes rubronotatus | Eumenitin | L. major | Promastigote | IC50 = 35 μM | [191] |
| Eumenes fraterculus | Eumenitin-F | L. major | Promastigote | IC50 = 52 μM | [194] |
| E. fraterculus | Eumenine mastoparan-EF (EMP-EF) | L. major | Promastigote | IC50 = 40 μM | [194] |
| E. rubrofemoratus | eumenitin-R | L. major | Promastigote | IC50 ≥ 62 μM | [194] |
| E. rubrofemoratus | Eumenine mastoparan-ER (EMP-AR) | L. major | Promastigote | IC50 = 20 μM | [194] |
| Anterhynchium flavomarginatum micado | Eumenine mastoparan-AF (EMP-AF) | L. major | Promastigote | IC50 = 35 μM | [194] |
| Tetramorium bicarinatum | Bicarinalin | L. infantum | Amastigote | IC50 = 1.5 μM | [197] |
| Spider | |||||
| Acanthoscurria gomesiana | Gomesin * | L. amazonensis L. major | Promastigote | IC50 = ~5.0 μM IC50 = ~2.5 μM | [189,190] |
| Source | AMP | Activity against | Parasite Stage | Inhibition Activity | Reference |
|---|---|---|---|---|---|
| Insect | |||||
| Apis mellifera | Melittin | P. berghei P. falciparum | Ookinete | GI = 100% (50 µM) GI = 60% (50 µM) | [213] |
| Anoplius samariensis | Anoplin | P. berghei | Ookinete | GI = 100% (100 µM) | [213] |
| Vespula lewisii | Mastoparan X | P. berghei P. falciparum | Ookinete | GI = 100% (100 µM) | [213] |
| Scorpion | |||||
| Pandinus imperator | Scorpine | P. berghei | Gametocyte Ookinete | ED50 = 10 µM ED50 = 0.7 µM | [206] |
| Mesobuthus eupeus | Meucin-24 | P. berghei P. falciparum | Ookinete Trophozoite | GI = 40% (20 µM) GI = 100% (10 µM) | [207] |
| M. eupeus | Meucin-25 | P. berghei P. falciparum | Ookinete Trophozoite | GI = 50% (20 µM) GI = 100% (10 µM) | [207] |
| M. eupeus | MeuTXKβ1 | P. berghei | Ookinete | GI = 89–98.8% (10-20 µM) | [209] |
| Spider | |||||
| Psalmopoeus cambridgei | Psalmopeotoxin I (PcFK1) | P. falciparum | Trophozoite | IC50c = 1.59 µM | [210] |
| P. cambridgei | Psalmopeotoxin II (PcFK2) | P. falciparum | Trophozoite | IC50 = 1.15 µM | [210] |
| Acanthoscurria gomesiana | Gomesin * | P. berghei P. falciparum | Trophozoite Ookinete Oocysts | IC50 = 46.8 µM GI = 100% (50 µM) GI = 86% (100 µM) | [212] |
| A. gomesiana | Gomesin * | P. falciparum | Oocysts | GI = 100% (100 µM) | [212] |
| Cupiennius salei | Cupiennin 1a | P. falciparum | Trophozoite | IC50 = 0.032 µM | [165] |
| Tick | |||||
| Ixodes ricinus | DefMT2 * | P. falciparum | Trophozoite | GI = 70% (50 µM) | [214] |
| I. ricinus | DefMT3 * | P. falciparum | Trophozoite | GI = 50% (50 µM) | [214] |
| I. ricinus | DefMT5 * | P. falciparum | Trophozoite | GI = 100% (50 µM) | [214] |
| I. ricinus | DefMT7 * | P. falciparum | Trophozoite | GI = 30% (50 µM) | [214] |
| Source | AMP | Activity Against | Parasite Stage | Inhibition Activity a | Reference |
|---|---|---|---|---|---|
| Spider | |||||
| Lycosa singoriensis | Lycosin-I | T. gondii | Tachyzoite | IC50 b = 28 μM IC50 c = 10.08 μM | [221] |
| Tick | |||||
| Haemaphysalis longicornis | Longicin * | T. gondii | Tachyzoite | - d | [222] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sabiá Júnior, E.F.; Menezes, L.F.S.; de Araújo, I.F.S.; Schwartz, E.F. Natural Occurrence in Venomous Arthropods of Antimicrobial Peptides Active against Protozoan Parasites. Toxins 2019, 11, 563. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11100563
Sabiá Júnior EF, Menezes LFS, de Araújo IFS, Schwartz EF. Natural Occurrence in Venomous Arthropods of Antimicrobial Peptides Active against Protozoan Parasites. Toxins. 2019; 11(10):563. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11100563
Chicago/Turabian StyleSabiá Júnior, Elias Ferreira, Luis Felipe Santos Menezes, Israel Flor Silva de Araújo, and Elisabeth Ferroni Schwartz. 2019. "Natural Occurrence in Venomous Arthropods of Antimicrobial Peptides Active against Protozoan Parasites" Toxins 11, no. 10: 563. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11100563