Environmental Variations in Mycobacterium ulcerans Transcriptome: Absence of Mycolactone Expression in Suboptimal Environments


Abstract
:1. Introduction
2. Results
2.1. Differences in Gene Expression in Experimental Environments Relatively to Control Condition
2.2. Plasmid Genes Involved in Mycolactone Production
2.3. Utilization of Chitin by M. ulcerans and Chitinases Expression
2.4. Biological Components Impacted by the Changes in Gene Expression
2.5. Additional Information Driven from MU/MT Comparisons
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Strain and Experiment Protocols
5.2. RNA Extraction
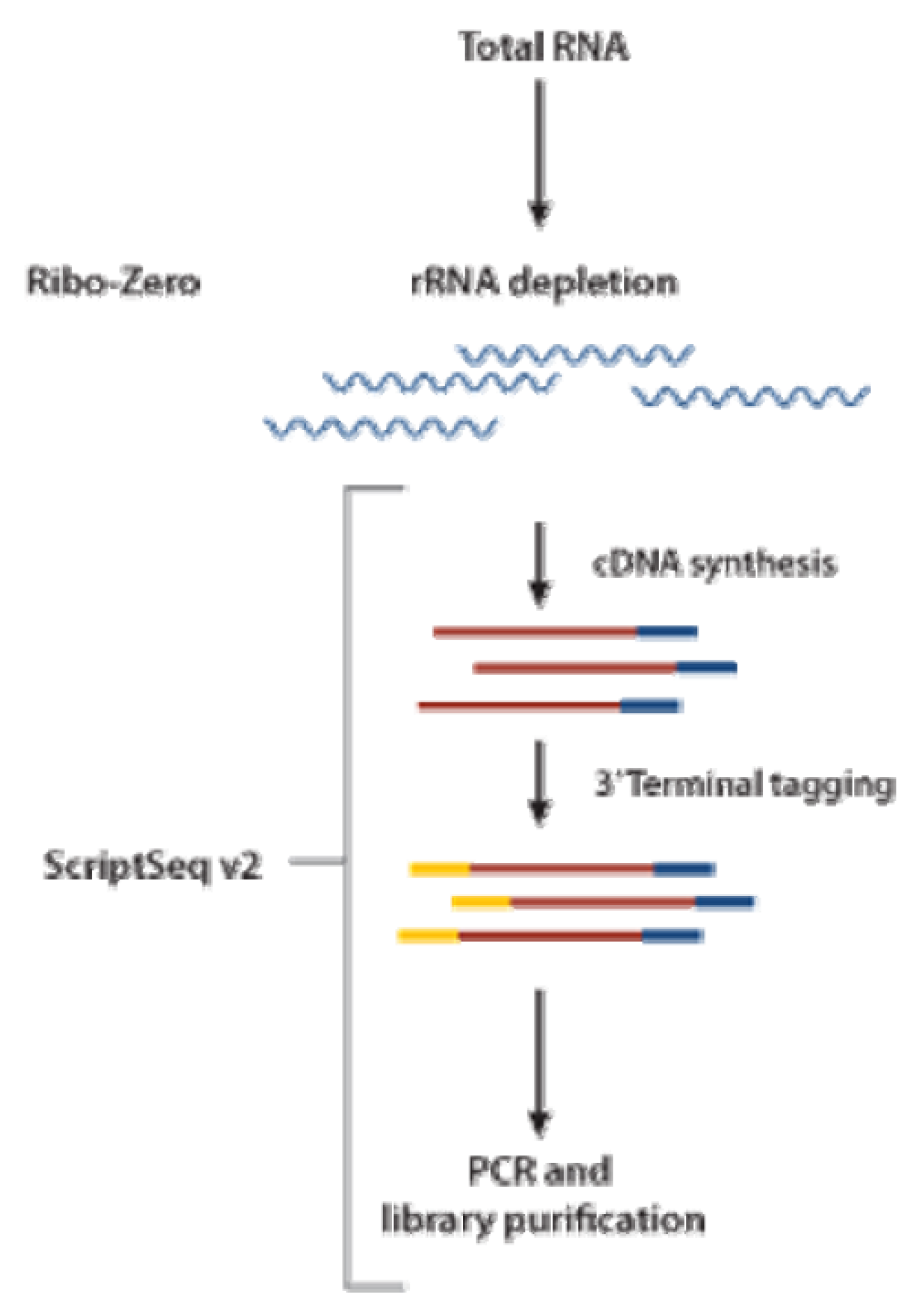
5.3. RNA Sequencing
5.4. Statistics
5.5. Identification of the Cellular Component, Molecular Function and/or Processes Affected
5.6. Use of M. tuberculosis Genome to Complete the Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Detailed RNA Sequencing Protocol
RNA Quality Control
Total RNA Amplification

Library Generation

ANNEX—Sequencing vocabulary

References
- Asiedu, K.; Scherpbier, R.; Raviglione, M. Infection à Mycobacterium Ulcère de Buruli; World Health Organization: Geneva, Switzerland, 2000. [Google Scholar]
- Zingue, D.; Bouam, A.; Roger, B.D.T.; Drancourt, M. Buruli Ulcer, a Prototype for Ecosystem-Related Infection, Caused by Mycobacterium ulcerans. Clin. Microbiol. Rev. 2018, 31, 1–52. [Google Scholar] [CrossRef] [PubMed]
- Merritt, R.W.; Walker, E.D.; Small, P.L.C.; Wallace, J.R.; Johnson, P.D.R.; Benbow, M.E.; Boayke, D.A. Ecology and transmission of Buruli ulcer disease: A systematic review. PLoS Negl. Trop. Dis. 2010, 4, e911. [Google Scholar] [CrossRef] [PubMed]
- Roche, B.; Benbow, M.E.; Merritt, R.; Kimbirauskas, R.; McIntosh, M.; Small, P.L.C.; Williamson, H.; Guégan, J.-F. Identifying the Achilles’ heel of multi-host pathogens: The concept of keystone “host” species illustrated by Mycobacterium ulcerans transmission. Environ. Res. Lett. 2013, 8, 045009. [Google Scholar] [CrossRef] [PubMed]
- Garchitorena, A.; Guégan, J.-F.; Léger, L.; Eyangoh, S.; Marsollier, L.; Roche, B. Mycobacterium ulcerans dynamics in aquatic ecosystems are driven by a complex interplay of abiotic and biotic factors. eLife 2015, 4, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Stinear, T.P.; Seemann, T.; Pidot, S.; Frigui, W.; Reysset, G.; Garnier, T.; Meurice, G.; Simon, D.; Bouchier, C.; Tichit, M.; et al. Reductive evolution and niche adaptation inferred from the genome of Mycobacterium ulcerans, the causative agent of Buruli ulcer. Genome Res. 2007, 17, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Demangel, C.; Stinear, T.; Cole, S. Buruli ulcer: Reductive evolution enhances pathogenicity of Mycobacterium ulcerans. Nat. Rev. Microbiol. 2009, 6, 799–799. [Google Scholar] [CrossRef] [PubMed]
- Stinear, T.P.; Mve-Obiang, A.; Small, P.L.C.; Frigui, W.; Pryor, M.J.; Brosch, R.; Jenkin, G.A.; Jonhson, P.D.; Davies, J.K.; Lee, R.E.; et al. Giant plasmid-encoded polyketide synthases produce the macrolide toxin of Mycobacterium ulcerans. Proc. Natl. Acad. Sci. USA 2004, 101, 1345–1349. [Google Scholar] [CrossRef] [PubMed]
- Röltgen, K.; Pluschke, G. Mycobacterium ulcerans Disease (Buruli Ulcer): Potential Reservoirs and Vectors. Curr. Clin. Microbiol. Rep. 2015, 2, 35–43. [Google Scholar] [CrossRef]
- Doig, K.D.; Holt, K.E.; Fyfe, J.A.; Lavender, C.J.; Eddyani, M.; Portaels, F.; Yeboah-Manu, D.; Pluschke, G.; Seeman, T.; Stinear, T.P. On the origin of Mycobacterium ulcerans, the causative agent of Buruli ulcer. BMC Genom. 2012, 13, 258. [Google Scholar] [CrossRef] [PubMed]
- Portaels, F.; Meyers, W.M.; Ablordey, A.; Castro, A.G.; Chemlal, K.; de Rijk, P.; Elsen, P.; Fissette, K.; Fraga, A.G.; Lee, R.; et al. First cultivation and characterization of Mycobacterium ulcerans from the environment. PLoS Negl. Trop. Dis. 2008, 2, e178. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, J.A.M.; Lavender, C.J.; Johnson, P.D.R.; Globan, M.; Sievers, A.; Azuolas, J.; Stinear, T.P. Development and application of two multiplex real-time PCR assays for the detection of Mycobacterium ulcerans in clinical and environmental samples. Appl. Environ. Microbiol. 2007, 73, 4733–4740. [Google Scholar] [CrossRef] [PubMed]
- Williamson, H.R.; Benbow, M.E.; Campbell, L.P.; Johnson, C.R.; Sopoh, G.; Barogui, Y.; Merrit, M.W.; Small, P.L.C. Detection of Mycobacterium ulcerans in the environment predicts prevalence of Buruli ulcer in Benin. PLoS Negl. Trop. Dis. 2012, 6, e1506. [Google Scholar] [CrossRef] [PubMed]
- Combe, M.; Velvin, C.J.; Morris, A.; Garchitorena, A.; Carolan, K.; Sanhueza, D.; Roche, B.; Couppié, P.; Guégan, J.-F. Global and local environmental changes as drivers of Buruli ulcer emergence. Emerg. Microbes Infect. 2017, 6, e22. [Google Scholar]
- Deshayes, C.; Angala, S.K.; Marion, E.; Brandli, I.; Babonneau, J.; Preisser, L.; Eyangoh, S.; Delneste, Y.; Legras, P.; De Chastellier, C.; et al. Regulation of Mycolactone, the Mycobacterium ulcerans Toxin, Depends on Nutrient Source. PLoS Negl. Trop. Dis. 2013, 7, e2502. [Google Scholar] [CrossRef]
- Sanhueza, D.; Chevillon, C.; Colwell, R.; Marion, E.; Marsollier, L. Chitin promotes Mycobacterium ulcerans growth. FEMS Microbiol. Ecol. 2016, 92, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sanhueza, D.; Chevillon, C.; Bouzinbi, N.; Godreuil, S.; Guegan, J. Chitin Increases Mycobacterium ulcerans Growth in Acidic Environments. Microbes Environ. 2018, 33, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Zingue, D.; Bouam, A.; Militello, M.; Drancourt, M. High-Throughput Carbon Substrate Profiling of Mycobacterium ulcerans Suggests Potential Environmental Reservoirs. PLoS Negl. Trop. Dis. 2017, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Croucher, N.J.; Thomson, N.R. Studying bacterial transcriptomes using RNA-seq. Curr. Opin. Microbiol. 2010, 13, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Tortoli, E.; Fedrizzi, T.; Meehan, C.J.; Trovato, A.; Grottola, A.; Giacobazzi, E.; Sepini, G.F.; Tagliazucchi, S.; Fabio, A.; Bettua, C.; et al. The new phylogeny of the genus Mycobacterium: The old and the news. Infect. Genet. Evol. 2017, 56, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Primm, T.P.; Lucero, C.A.; Falkinham, J.O. Health impacts of environmental mycobacteria. Clin. Microbiol. Rev. 2004, 17, 98–106. [Google Scholar] [CrossRef] [PubMed]
- George, K.M. Mycolactone: A Polyketide Toxin from Mycobacterium ulcerans Required for Virulence. Science 1999, 283, 854–857. [Google Scholar] [CrossRef] [PubMed]
- George, K.M.; Barker, L.P.; Welty, D.M.; Small, P.L. Partial purification and characterization of biological effects of a lipid toxin produced by Mycobacterium ulcerans. Infect. Immun. 1998, 66, 587–593. [Google Scholar] [PubMed]
- Marsollier, L.; Aubry, J.; Coutanceau, E.; Saint André, J.P.; Small, P.L.; Milon, G.; Legras, P.; Guadagnini, S.; Carbonelle, B.; Cole, S.T. Colonization of the salivary glands of Naucoris cimicoides by Mycobacterium ulcerans requires host plasmatocytes and a macrolide toxin, mycolactone. Cell Microbiol. 2005, 7, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Hammoudi, N.; Cassagne, C.; Armstrong, N.; Ranque, S.; Henrissat, B.; Drancourt, M.; Bouam, A. Mycobacterium ulcerans mycolactones-fungi crosstalking. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Aronson, A.I. MicroReview The two faces of Bacillus thuringiensis: Insecticidal proteins and post-exponential survival. Mol. Microbiol. 1993, 7, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Melo, J.V.; De Jones, G.W.; Berry, C.; Vasconcelos, R.H.T.; Fontes de Oliveira, C.M.; Furtado, A.F.; Peixoto, C.A.; Silva-Filha, M.H.N.L. Cytopathological Effects of Bacillus sphaericus Cry48Aa/Cry49Aa Toxin on Binary Toxin-Susceptible and -Resistant Culex quinquefasciatus Larvae. Appl. Environ. Microbiol. 2009, 75, 4782–4789. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, A.; Trauth, S.; Ziesack, M.; Nagler, K.; Bergeest, J.P.; Rohr, K.; Becker, N.; Hôfer, T.; Bischofs, I.B. Phenotypic memory in Bacillus subtilis links quality tradeoff. Nat. Commun. 2018, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Nakanaga, K.; Ogura, Y.; Toyoda, A.; Yoshida, M.; Fukano, H.; Fujiwara, N.; Miyamoto, Y.; Nakata, N.; Kazumi, Y.; Maeda, S.; et al. Naturally occurring a loss of a giant plasmid from Mycobacterium ulcerans subsp. shinshuense makes it non-pathogenic. Sci. Rep. 2018, 8, e8218. [Google Scholar]
- De Gentile, P.L.; Mahaza, C.; Rolland, F.; Carbonnelle, B.; Verret, J.L.; Chabasse, D. Cutaneous ulcer from Mycobacterium ulcerans. A propos of 1 case in French Guyana. Bull. Soc. Pathol. Exot. 1992, 85, 212–214. [Google Scholar] [PubMed]
- Robbe-Saule, M.; Babonneau, J.; Sismeiro, O.; Marsollier, L.; Marion, E. An Optimized Method for Extracting Bacterial RNA from Mouse Skin Tissue Colonized by Mycobacterium ulcerans. Front. Microbiol. 2017, 8, 512. [Google Scholar] [CrossRef] [PubMed]
- De Meeûs, T.; Guégan, J.F. Teriokhin. MultiTest V.1.2, a program to binomially combine independent tests and performance comparison with other related methods on proportional data. BMC Bioinform. 2009, 10, 443. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Sherman, B.T.; Tan, Q.; Collins, J.R.; Alvord, W.G.; Roayaei, J.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. The DAVID Gene Functional Classification Tool: A novel biological module-centric algorithm to functionally a nalyze large gene lists. Genome Biol. 2007, 8, R183. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, T.; Zhang, R.; Brash, D.E.; Bystrykh, L.V. Enhancing the detection of barcoded reads in high throughput DNA sequencing data by controlling the false discovery rate. BMC Bioinf. 2014, 15, 264. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatments | Total Number of Affected Genes | With GI N (%) | At Least One GO Retrieved | |
|---|---|---|---|---|
| Comparisons: | ||||
| ‘Calcium vs. Control’ | 73 | 65 | (89) | YES |
| ‘Chitin vs. Control’ | 115 | 79 | (69) | YES |
| ‘Calcium + Chitin vs. Control’ | 30 | 27 | (90) | YES |
| ‘Starch vs. Control’ | 14 | 10 | (71) | NO |
| Genes in common between: | ||||
| ‘Calcium vs. Control’ & ‘Chitin vs. Control’ | 22 | 21 | (95) | YES |
| ‘Chitin vs. Control’ & ‘Calcium + Chitin vs. Control’ | 4 | 3 | (75) | NO |
| ‘Calcium vs. control’ & ‘Calcium + Chitin vs. Control’ | 15 | 12 | (80) | NO |
| All three comparisons | 4 | 3 | (75) | NO |
| Treatments | GO Type | GO ID | GO Term | p-Value | Fold Enrichment | Genes (N) | Deregulated Genes (N) | Gene List (Expression Change) |
|---|---|---|---|---|---|---|---|---|
| A: Calcium vs. control | Biological Process | 0043933 | Macromolecular complex subunit organization | 0.0039 | 11.481 | 27 | 4 (1 upregulated, 3 downregulated) | MUL_0626(2.23)//MUL_1353(−2.62)//MUL_2075(−1.56)//MUL_3967(−1.73) |
| Biological Process | 0034621 | Cellular macromolecular complex subunit organization | 0.0074 | 21.136 | 11 | 3 (1 upregulated, 2 downregulated) | MUL_0626(2.23)//MUL_2075(−1.56)//MUL_3967(−1.73) | |
| Biological Process | 0006412 | Translation | 0.0288 | 3.954 | 98 | 5 (5 downregulated) | MUL_0073(−2.58)//MUL_0741(−2.74)//MUL_0865(−2.21)//MUL_2075(−1.56)//MUL_4181(−1.81) | |
| Biological Process | 0009165 | Nucleotide biosynthetic process | 0.0537 | 4.366 | 71 | 4 (2 upregulated, 2 downregulated) | MUL_1364(2.58)//MUL_1784(1.83)//MUL_2370(−2.25)//MUL_3967(−1.73) | |
| Molecular Function | 0008080 | N-acetyltransferase activity | 0.0038 | 11.796 | 23 | 4 (1 upregulated, 3 downregulated) | MUL_1771(−2.59)//MUL_3389(−2.51)//MUL_4691(1.81)//MUL_4933(−3.07) | |
| Molecular Function | 0016410 | N-acyltransferase activity | 0.0048 | 10.852 | 25 | 4 (1 upregulated, 3 downregulated) | MUL_1771(−2.59)//MUL_3389(−2.51)//MUL_4691(1.81)//MUL_4933(−3.07) | |
| B: Chitin vs. control | Biological Process | 0006260 | DNA replication | 0.0068 | 9.472 | 30 | 4 (3 upregulated, downregulated) | MUL_0072(2.47)//MUL_0517(−3.27)//MUL_1222(2.61)//MUL_3525(2.44) |
| Molecular Function | 0004089 | Carbonate dehydratase activity | 0.0332 | 57.853 | 2 | 2 (1 upregulated, 1 downregulated) | MUL_2767 (−2.8)//MUL_3373(1.89) | |
| Molecular Function | 0008270 | Zinc ion binding | 0.0440 | 3.571 | 81 | 5 (2 upregulated, 3 downregulated) | MUL_0517(-3.27)//MUL_2210(-1.51)//MUL_2342(1.73)//MUL_2767(−2.8)//MUL_3373(1.89) | |
| C: Chitin + calcium vs. control | Biological Process | 0009116 | Nucleoside metabolic process | 0.0135 | 15.223 | 24 | 3 (1 upregulated, 2 downregulated) | MUL_3335(2.22)//MUL_4200(−1.58)//MUL_4862(−3.14) |
| Biological Process | 0043094 | Cellular metabolic compound salvage | 0.0302 | 60.862 | 4 | 2 (2 downregulated) | MUL_4200(−1.58)//MUL_4862(−3.14) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanhueza, D.; Guégan, J.-F.; Jordan, H.; Chevillon, C. Environmental Variations in Mycobacterium ulcerans Transcriptome: Absence of Mycolactone Expression in Suboptimal Environments. Toxins 2019, 11, 146. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030146
Sanhueza D, Guégan J-F, Jordan H, Chevillon C. Environmental Variations in Mycobacterium ulcerans Transcriptome: Absence of Mycolactone Expression in Suboptimal Environments. Toxins. 2019; 11(3):146. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030146
Chicago/Turabian StyleSanhueza, Daniel, Jean-François Guégan, Heather Jordan, and Christine Chevillon. 2019. "Environmental Variations in Mycobacterium ulcerans Transcriptome: Absence of Mycolactone Expression in Suboptimal Environments" Toxins 11, no. 3: 146. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11030146