New Insights into the Roles of Monocytes/Macrophages in Cardiovascular Calcification Associated with Chronic Kidney Disease

Abstract
:1. Introduction
2. Mechanistic Insights into the Calcification of Cardiovascular Cells
3. Monocytes/Macrophages and CVC
3.1. Monocytes/Macrophages in a Physiological Setting
3.2. Roles of Monocytes/Macrophages in CVC
3.2.1. The Procalcific Actions of Monocytes/Macrophages
3.2.2. Anticalcific Actions of Monocytes/Macrophages
3.2.3. Monocytes/Macrophages as Precursors of Osteoclast-Like Cells
4. The Impact of CKD on Macrophage Functions: Consequences for CVC
4.1. Uraemic Toxicity and Monocyte/Macrophage Functions
4.1.1. Influence of CKD on Monocyte Subtypes
4.1.2. Influence of UTs on Monocyte/Macrophage-Driven CVC
4.2. Impact of CKD Treatments on Macrophage Functions
5. Conclusions
Funding
Conflicts of Interest
References
- Hénaut, L.; Mentaverri, R.; Liabeuf, S.; Bargnoux, A.S.; Delanaye, P.; Cavalier, É.; Cristol, J.P.; Massy, Z.; Kamel, S.; Néphrologie, G.; et al. Pathophysiological mechanisms of vascular calcification. Ann. Biol. Clin. 2015, 73, 271–287. [Google Scholar] [CrossRef] [PubMed]
- Goodman, W.G.; Goldin, J.; Kuizon, B.D.; Yoon, C.; Gales, B.; Sider, D.; Wang, Y.; Chung, J.; Emerick, A.; Greaser, L.; et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N. Engl. J. Med. 2000, 342, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Russo, D.; Corrao, S.; Battaglia, Y.; Andreucci, M.; Caiazza, A.; Carlomagno, A.; Lamberti, M.; Pezone, N.; Pota, A.; Russo, L.; et al. Progression of coronary artery calcification and cardiac events in patients with chronic renal disease not receiving dialysis. Kidney Int. 2011, 80, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Blacher, J.; Guerin, A.P.; Pannier, B.; Marchais, S.J.; London, G.M. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension 2001, 38, 938–942. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.J. Calciphylaxis: Diagnosis, Pathogenesis, and Treatment. Adv. Skin Wound Care 2019, 32, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Giachelli, C.M. The emerging role of phosphate in vascular calcification. Kidney Int. 2009, 75, 890–897. [Google Scholar] [CrossRef] [Green Version]
- Vavilis, G.; Bäck, M.; Occhino, G.; Trevisan, M.; Bellocco, R.; Evans, M.; Lindholm, B.; Szummer, K.; Carrero, J.J. Kidney Dysfunction and the Risk of Developing Aortic Stenosis. J. Am. Coll. Cardiol. 2019, 73, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Rajamannan, N.M.; Evans, F.J.; Aikawa, E.; Grande-Allen, K.J.; Demer, L.L.; Heistad, D.D.; Simmons, C.A.; Masters, K.S.; Mathieu, P.; O’Brien, K.D.; et al. Calcific aortic valve disease: Not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation 2011, 124, 1783–1791. [Google Scholar] [CrossRef]
- Otto, C.M. Valvular aortic stenosis: Disease severity and timing of intervention. J. Am. Coll. Cardiol. 2006, 47, 2141–2151. [Google Scholar] [CrossRef]
- Byon, C.H.; Sun, Y.; Chen, J.; Yuan, K.; Mao, X.; Heath, J.M.; Anderson, P.G.; Tintut, Y.; Demer, L.L.; Wang, D.; et al. Runx2-upregulated receptor activator of nuclear factor κB ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1387–1396. [Google Scholar] [CrossRef]
- Deuell, K.A.; Callegari, A.; Giachelli, C.M.; Rosenfeld, M.E.; Scatena, M. RANKL enhances macrophage paracrine pro-calcific activity in high phosphate-treated smooth muscle cells: Dependence on IL-6 and TNF-α. J. Vasc. Res. 2012, 49, 510–521. [Google Scholar] [CrossRef]
- Shin, V.; Zebboudj, A.F.; Boström, K. Endothelial cells modulate osteogenesis in calcifying vascular cells. J. Vasc. Res. 2004, 41, 193–201. [Google Scholar] [CrossRef]
- New, S.E.; Aikawa, E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ. Res. 2011, 108, 1381–1391. [Google Scholar] [CrossRef]
- New, S.E.; Goettsch, C.; Aikawa, M.; Marchini, J.F.; Shibasaki, M.; Yabusaki, K.; Libby, P.; Shanahan, C.M.; Croce, K.; Aikawa, E. Macrophage-derived matrix vesicles: An alternative novel mechanism for microcalcification in atherosclerotic plaques. Circ. Res. 2013, 113, 72–77. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef]
- Tintut, Y.; Patel, J.; Territo, M.; Saini, T.; Parhami, F.; Demer, L.L. Monocyte/macrophage regulation of vascular calcification in vitro. Circulation 2002, 105, 650–655. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef]
- Valledor, A.F.; Comalada, M.; Santamaría-Babi, L.F.; Lloberas, J.; Celada, A. Macrophage proinflammatory activation and deactivation: A question of balance. Adv. Immunol. 2010, 108, 1–20. [Google Scholar]
- Smith, E.R.; Hanssen, E.; McMahon, L.P.; Holt, S.G. Fetuin-A-containing calciprotein particles reduce mineral stress in the macrophage. PLoS ONE 2013, 8, e60904. [Google Scholar] [CrossRef]
- Pazár, B.; Ea, H.K.; Narayan, S.; Kolly, L.; Bagnoud, N.; Chobaz, V.; Roger, T.; Lioté, F.; So, A.; Busso, N. Basic calcium phosphate crystals induce monocyte/macrophage IL-1β secretion through the NLRP3 inflammasome in vitro. J. Immunol. 2011, 186, 2495–2502. [Google Scholar] [CrossRef]
- Viegas, C.; Araújo, N.; Marreiros, C.; Simes, D. The interplay between mineral metabolism, vascular calcification and inflammation in Chronic Kidney Disease (CKD): Challenging old concepts with new facts. Aging (Albany NY) 2019, 11, 4274–4299. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Millan, A.; Sorribas, V. Role of calcium-phosphate deposition in vascular smooth muscle cell calcification. Am. J. Physiol. Cell Physiol. 2011, 300, C210–C220. [Google Scholar] [CrossRef] [Green Version]
- Sage, A.P.; Lu, J.; Tintut, Y.; Demer, L.L. Hyperphosphatemia-induced nanocrystals upregulate the expression of bone morphogenetic protein-2 and osteopontin genes in mouse smooth muscle cells in vitro. Kidney Int. 2011, 79, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Kapustin, A.N.; Chatrou, M.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.; Alvarez-Hernandez, D.; et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef]
- Coscas, R.; Bensussan, M.; Jacob, M.P.; Louedec, L.; Massy, Z.; Sadoine, J.; Daudon, M.; Chaussain, C.; Bazin, D.; Michel, J.B. Free DNA precipitates calcium phosphate apatite crystals in the arterial wall in vivo. Atherosclerosis 2017, 259, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Stansfield, B.K.; Ingram, D.A. Clinical significance of monocyte heterogeneity. Clin. Transl. Med. 2015, 4, 5. [Google Scholar] [CrossRef]
- Tsou, C.L.; Peters, W.; Si, Y.; Slaymaker, S.; Aslanian, A.M.; Weisberg, S.P.; Mack, M.; Charo, I.F. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Investig. 2007, 117, 902–909. [Google Scholar] [CrossRef] [Green Version]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.; Liu, Y.J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef]
- Mukherjee, R.; Kanti Barman, P.; Kumar Thatoi, P.; Tripathy, R.; Das, B.K.; Ravindran, B. Non-Classical monocytes display inflammatory features: Validation in Sepsis and Systemic Lupus Erythematous. Sci. Rep. 2015, 5, 13886. [Google Scholar] [CrossRef] [Green Version]
- Nockher, W.A.; Scherberich, J.E. Expanded CD14+ CD16+ monocyte subpopulation in patients with acute and chronic infections undergoing hemodialysis. Infect. Immun. 1998, 66, 2782–2790. [Google Scholar]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.F.; Wang, H. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef]
- Rogacev, K.S.; Cremers, B.; Zawada, A.M.; Seiler, S.; Binder, N.; Ege, P.; Große-Dunker, G.; Heisel, I.; Hornof, F.; Jeken, J.; et al. CD14++CD16+ monocytes independently predict cardiovascular events: A cohort study of 951 patients referred for elective coronary angiography. J. Am. Coll. Cardiol. 2012, 60, 1512–1520. [Google Scholar] [CrossRef]
- Hristov, M.; Weber, C. Differential role of monocyte subsets in atherosclerosis. Thromb. Haemost. 2011, 106, 757–762. [Google Scholar] [CrossRef] [Green Version]
- Hewing, B.; Au, S.C.; Ludwig, A.; Ellerbroek, R.; van Dijck, P.; Hartmann, L.; Grubitzsch, H.; Giannini, C.; Laule, M.; Stangl, V.; et al. Severe Aortic Valve Stenosis in Adults is Associated with Increased Levels of Circulating Intermediate Monocytes. J. Cardiovasc. Transl. Res. 2017, 10, 27–34. [Google Scholar] [CrossRef]
- Hewing, B.; Ellerbroek, R.; Au, S.C.; Stangl, V.; Dreger, H.; Laule, M.; Grubitzsch, H.; Knebel, F.; Baumann, G.; Ludwig, A.; et al. Levels of Circulating Intermediate Monocytes Decrease after Aortic Valve Replacement in Patients with Severe Aortic Stenosis. Thromb. Haemost. 2017, 117, 2346–2355. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.; Cho, E.; Kim, M.G.; Jo, S.K.; Cho, W.Y.; Kim, H.K. Proinflammatory CD14(+)CD16(+) monocytes are associated with vascular stiffness in predialysis patients with chronic kidney disease. Kidney Res. Clin. Pract. 2013, 32, 147–152. [Google Scholar] [CrossRef]
- Rogacev, K.S.; Seiler, S.; Zawada, A.M.; Reichart, B.; Herath, E.; Roth, D.; Ulrich, C.; Fliser, D.; Heine, G.H. CD14++CD16+ monocytes and cardiovascular outcome in patients with chronic kidney disease. Eur. Heart J. 2011, 32, 84–92. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1–m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef]
- Jean, G.; Bresson, E.; Terrat, J.C.; Vanel, T.; Hurot, J.M.; Lorriaux, C.; Mayor, B.; Chazot, C. Peripheral vascular calcification in long-haemodialysis patients: Associated factors and survival consequences. Nephrol. Dial. Transplant. 2009, 24, 948–955. [Google Scholar] [CrossRef]
- Zimmermann, J.; Herrlinger, S.; Pruy, A.; Metzger, T.; Wanner, C. Inflammation enhances cardiovascular risk and mortality in hemodialysis patients. Kidney Int. 1999, 55, 648–658. [Google Scholar] [CrossRef] [Green Version]
- Rysz, J.; Banach, M.; Cialkowska-Rysz, A.; Stolarek, R.; Barylski, M.; Drozdz, J.; Okonski, P. Blood serum levels of IL-2, IL-6, IL-8, TNF-alpha and IL-1beta in patients on maintenance hemodialysis. Cell. Mol. Immunol. 2006, 3, 151–154. [Google Scholar]
- Lee, C.T.; Chua, S.; Hsu, C.Y.; Tsai, Y.C.; Ng, H.Y.; Kuo, C.C.; Wu, C.H.; Chen, T.C.; Chiu, T.T.; Lee, Y.T. Biomarkers associated with vascular and valvular calcification in chronic hemodialysis patients. Dis. Markers 2013, 34, 229–235. [Google Scholar] [CrossRef]
- Pecoits-Filho, R.; Bárány, P.; Lindholm, B.; Heimbürger, O.; Stenvinkel, P. Interleukin-6 is an independent predictor of mortality in patients starting dialysis treatment. Nephrol. Dial. Transplant. 2002, 17, 1684–1688. [Google Scholar] [CrossRef]
- Li, G.; Qiao, W.; Zhang, W.; Li, F.; Shi, J.; Dong, N. The shift of macrophages toward M1 phenotype promotes aortic valvular calcification. J. Thorac. Cardiovasc. Surg. 2017, 153, 1318–1327. [Google Scholar] [CrossRef]
- Hénaut, L.; Sanchez-Nino, M.D.; Castillo, G.A.-E.; Sanz, A.B.; Ortiz, A. Targeting local vascular and systemic consequences of inflammation on vascular and cardiac valve calcification. Expert Opin. Ther. Targets 2016, 20, 89–105. [Google Scholar] [CrossRef]
- Shobeiri, N.; Bendeck, M.P. Interleukin-1β Is a Key Biomarker and Mediator of Inflammatory Vascular Calcification. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 179–180. [Google Scholar] [CrossRef]
- Ikeda, K.; Souma, Y.; Akakabe, Y.; Kitamura, Y.; Matsuo, K.; Shimoda, Y.; Ueyama, T.; Matoba, S.; Yamada, H.; Okigaki, M.; et al. Macrophages play a unique role in the plaque calcification by enhancing the osteogenic signals exerted by vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2012, 425, 39–44. [Google Scholar] [CrossRef]
- Wen, C.; Yang, X.; Yan, Z.; Zhao, M.; Yue, X.; Cheng, X.; Zheng, Z.; Guan, K.; Dou, J.; Xu, T.; et al. Nalp3 inflammasome is activated and required for vascular smooth muscle cell calcification. Int. J. Cardiol. 2013, 168, 2242–2247. [Google Scholar] [CrossRef]
- Awan, Z.; Denis, M.; Roubtsova, A.; Essalmani, R.; Marcinkiewicz, J.; Awan, A.; Gram, H.; Seidah, N.G.; Genest, J. Reducing Vascular Calcification by Anti-IL-1β Monoclonal Antibody in a Mouse Model of Familial Hypercholesterolemia. Angiology 2016, 67, 157–167. [Google Scholar] [CrossRef]
- Nadra, I.; Mason, J.C.; Philippidis, P.; Florey, O.; Smythe, C.D.; McCarthy, G.M.; Landis, R.C.; Haskard, D.O. Proinflammatory activation of macrophages by basic calcium phosphate crystals via protein kinase C and MAP kinase pathways: A vicious cycle of inflammation and arterial calcification? Circ. Res. 2005, 96, 1248–1256. [Google Scholar] [CrossRef]
- Nadra, I.; Boccaccini, A.R.; Philippidis, P.; Whelan, L.C.; McCarthy, G.M.; Haskard, D.O.; Landis, R.C. Effect of particle size on hydroxyapatite crystal-induced tumor necrosis factor alpha secretion by macrophages. Atherosclerosis 2008, 196, 98–105. [Google Scholar] [CrossRef]
- Chuang, S.C.; Boeing, H.; Vollset, S.E.; Midttun, Ø.; Ueland, P.M.; Bueno-de-Mesquita, B.; Lajous, M.; Fagherazzi, G.; Boutron-Ruault, M.C.; Kaaks, R.; et al. Cellular immune activity biomarker neopterin is associated hyperlipidemia: Results from a large population-based study. Immun. Ageing 2016, 13, 5. [Google Scholar] [CrossRef]
- Prebble, H.; Cross, S.; Marks, E.; Healy, J.; Searle, E.; Aamir, R.; Butler, A.; Roake, J.; Hock, B.; Anderson, N.; et al. Induced macrophage activation in live excised atherosclerotic plaque. Immunobiology 2018, 223, 526–535. [Google Scholar] [CrossRef]
- Chatrou, M.L.; Cleutjens, J.P.; van der Vusse, G.J.; Roijers, R.B.; Mutsaers, P.H.; Schurgers, L.J. Intra-Section Analysis of Human Coronary Arteries Reveals a Potential Role for Micro-Calcifications in Macrophage Recruitment in the Early Stage of Atherosclerosis. PLoS ONE 2015, 10, e0142335. [Google Scholar] [CrossRef]
- Heiss, A.; DuChesne, A.; Denecke, B.; Grötzinger, J.; Yamamoto, K.; Renné, T.; Jahnen-Dechent, W. Structural basis of calcification inhibition by alpha 2-HS glycoprotein/fetuin-A. Formation of colloidal calciprotein particles. J. Biol. Chem. 2003, 278, 13333–13341. [Google Scholar] [CrossRef]
- Smith, E.R.; Ford, M.L.; Tomlinson, L.A.; Rajkumar, C.; McMahon, L.P.; Holt, S.G. Phosphorylated fetuin-A-containing calciprotein particles are associated with aortic stiffness and a procalcific milieu in patients with pre-dialysis CKD. Nephrol. Dial. Transplant. 2012, 27, 1957–1966. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, M.; Schäfer, C.; Heiss, A.; Gräber, S.; Kinkeldey, A.; Büscher, A.; Schmitt, M.M.; Bornemann, J.; Nimmerjahn, F.; Helming, L.; et al. Clearance of fetuin-A—Containing calciprotein particles is mediated by scavenger receptor-A. Circ. Res. 2012, 111, 575–584. [Google Scholar] [CrossRef]
- Heiss, A.; Pipich, V.; Jahnen-Dechent, W.; Schwahn, D. Fetuin-A is a mineral carrier protein: Small angle neutron scattering provides new insight on Fetuin-A controlled calcification inhibition. Biophys. J. 2010, 99, 3986–3995. [Google Scholar] [CrossRef]
- Kirsch, T.; Wang, W.; Pfander, D. Functional differences between growth plate apoptotic bodies and matrix vesicles. J. Bone Miner. Res. 2003, 18, 1872–1881. [Google Scholar] [CrossRef]
- Grskovic, I.; Kutsch, A.; Frie, C.; Groma, G.; Stermann, J.; Schlötzer-Schrehardt, U.; Niehoff, A.; Moss, S.E.; Rosenbaum, S.; Pöschl, E.; et al. Depletion of annexin A5, annexin A6, and collagen X causes no gross changes in matrix vesicle-mediated mineralization, but lack of collagen X affects hematopoiesis and the Th1/Th2 response. J. Bone Miner. Res. 2012, 27, 2399–2412. [Google Scholar] [CrossRef]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef]
- Harris, H.E.; Andersson, U.; Pisetsky, D.S. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012, 8, 195–202. [Google Scholar] [CrossRef]
- Passmore, M.; Nataatmadja, M.; Fung, Y.L.; Pearse, B.; Gabriel, S.; Tesar, P.; Fraser, J.F. Osteopontin alters endothelial and valvular interstitial cell behaviour in calcific aortic valve stenosis through HMGB1 regulation. Eur. J. Cardiothorac. Surg. 2015, 48, e20–e29. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Bei, J.J.; Liu, C.; Feng, S.B.; Zhao, W.B.; Zhou, Z.; Yu, Z.P.; Du, X.J.; Hu, H.Y. HMGB1 Induces Secretion of Matrix Vesicles by Macrophages to Enhance Ectopic Mineralization. PLoS ONE 2016, 11, e0156686. [Google Scholar] [CrossRef]
- Eghbali-Fatourechi, G.Z.; Lamsam, J.; Fraser, D.; Nagel, D.; Riggs, B.L.; Khosla, S. Circulating osteoblast-lineage cells in humans. N. Engl. J. Med. 2005, 352, 1959–1966. [Google Scholar] [CrossRef]
- Eghbali-Fatourechi, G.Z.; Mödder, U.I.; Charatcharoenwitthaya, N.; Sanyal, A.; Undale, A.H.; Clowes, J.A.; Tarara, J.E.; Khosla, S. Characterization of circulating osteoblast lineage cells in humans. Bone 2007, 40, 1370–1377. [Google Scholar] [CrossRef] [Green Version]
- Fadini, G.P.; Albiero, M.; Menegazzo, L.; Boscaro, E.; de Kreutzenberg, S.V.; Agostini, C.; Cabrelle, A.; Binotto, G.; Rattazzi, M.; Bertacco, E.; et al. Widespread increase in myeloid calcifying cells contributes to ectopic vascular calcification in type 2 diabetes. Circ. Res. 2011, 108, 1112–1121. [Google Scholar] [CrossRef]
- Menegazzo, L.; Albiero, M.; Millioni, R.; Tolin, S.; Arrigoni, G.; Poncina, N.; Tessari, P.; Avogaro, A.; Fadini, G.P. Circulating myeloid calcifying cells have antiangiogenic activity via thrombospondin-1 overexpression. FASEB J. 2013, 27, 4355–4365. [Google Scholar] [CrossRef]
- Albiero, M.; Rattazzi, M.; Menegazzo, L.; Boscaro, E.; Cappellari, R.; Pagnin, E.; Bertacco, E.; Poncina, N.; Dyar, K.; Ciciliot, S.; et al. Myeloid calcifying cells promote atherosclerotic calcification via paracrine activity and allograft inflammatory factor-1 overexpression. Basic Res. Cardiol. 2013, 108, 368. [Google Scholar] [CrossRef]
- Dube, P.R.; Birnbaumer, L.; Vazquez, G. Evidence for constitutive bone morphogenetic protein-2 secretion by M1 macrophages: Constitutive auto/paracrine osteogenic signaling by BMP-2 in M1 macrophages. Biochem. Biophys. Res. Commun. 2017, 491, 154–158. [Google Scholar] [CrossRef]
- Liberman, M.; Johnson, R.C.; Handy, D.E.; Loscalzo, J.; Leopold, J.A. Bone morphogenetic protein-2 activates NADPH oxidase to increase endoplasmic reticulum stress and human coronary artery smooth muscle cell calcification. Biochem. Biophys. Res. Commun. 2011, 413, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Champagne, C.M.; Takebe, J.; Offenbacher, S.; Cooper, L.F. Macrophage cell lines produce osteoinductive signals that include bone morphogenetic protein-2. Bone 2002, 30, 26–31. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Hamczyk, M.R.; Andrés, V. Alternatively activated macrophages exhibit an anticalcifying activity dependent on extracellular ATP/pyrophosphate metabolism. Am. J. Physiol. Cell Physiol. 2016, 310, C788–C799. [Google Scholar] [CrossRef] [Green Version]
- Rattazzi, M.; Rosenfeld, M.E. The multifaceted role of macrophages in cardiovascular calcification. Atherosclerosis 2018, 270, 193–195. [Google Scholar] [CrossRef]
- Tabas, I.; Bornfeldt, K.E. Macrophage Phenotype and Function in Different Stages of Atherosclerosis. Circ. Res. 2016, 118, 653–667. [Google Scholar] [CrossRef] [Green Version]
- Kayashima, Y.; Makhanova, N.; Maeda, N. DBA/2J Haplotype on Distal Chromosome 2 Reduces Mertk Expression, Restricts Efferocytosis, and Increases Susceptibility to Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e82–e91. [Google Scholar] [CrossRef] [Green Version]
- Clinton, S.K.; Underwood, R.; Hayes, L.; Sherman, M.L.; Kufe, D.W.; Libby, P. Macrophage colony-stimulating factor gene expression in vascular cells and in experimental and human atherosclerosis. Am. J. Pathol. 1992, 140, 301–316. [Google Scholar]
- Sekulic, M.; Truskinovsky, A.M. Metaplastic ossification of the temporal artery with osteoclast-like giant cells: A mimicker of giant cell (temporal) arteritis. Eur. J. Ophthalmol. 2017, 27, e99–e103. [Google Scholar] [CrossRef]
- Tseng, W.; Graham, L.S.; Geng, Y.; Reddy, A.; Lu, J.; Effros, R.B.; Demer, L.; Tintut, Y. PKA-induced receptor activator of NF-kappaB ligand (RANKL) expression in vascular cells mediates osteoclastogenesis but not matrix calcification. J. Biol. Chem. 2010, 285, 29925–29931. [Google Scholar] [CrossRef]
- Jeziorska, M.; McCollum, C.; Wooley, D.E. Observations on bone formation and remodelling in advanced atherosclerotic lesions of human carotid arteries. Virchows Arch. 1998, 433, 559–565. [Google Scholar] [CrossRef]
- Han, K.H.; Hennigar, R.A.; O’Neill, W.C. The association of bone and osteoclasts with vascular calcification. Vasc. Med. 2015, 20, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.H.; Mishra, V.; Fishbein, M.C.; Sinha, S.K.; Rajavashisth, T.B. Multinucleated giant cells in atherosclerotic plaques of human carotid arteries: Identification of osteoclast-like cells and their specific proteins in artery wall. Exp. Mol. Pathol. 2015, 99, 654–662. [Google Scholar] [CrossRef]
- Simpson, C.L.; Lindley, S.; Eisenberg, C.; Basalyga, D.M.; Starcher, B.C.; Simionescu, D.T.; Vyavahare, N.R. Toward cell therapy for vascular calcification: Osteoclast-mediated demineralization of calcified elastin. Cardiovasc. Pathol. 2007, 16, 29–37. [Google Scholar] [CrossRef]
- Barinda, A.J.; Ikeda, K.; Hirata, K.I.; Emoto, N. Macrophages Highly Express Carbonic Anhydrase 2 and Play a Significant Role in Demineralization of the Ectopic Calcification. Kobe J. Med. Sci. 2017, 63, E45–E50. [Google Scholar]
- Väänänen, H.K.; Zhao, H.; Mulari, M.; Halleen, J.M. The cell biology of osteoclast function. J. Cell Sci. 2000, 113, 377–381. [Google Scholar]
- Lehenkari, P.; Hentunen, T.A.; Laitala-Leinonen, T.; Tuukkanen, J.; Väänänen, H.K. Carbonic anhydrase II plays a major role in osteoclast differentiation and bone resorption by effecting the steady state intracellular pH and Ca2+. Exp. Cell Res. 1998, 242, 128–137. [Google Scholar] [CrossRef]
- Maxson, M.E.; Grinstein, S. The vacuolar-type H⁺-ATPase at a glance—more than a proton pump. J. Cell Sci. 2014, 127, 4987–4993. [Google Scholar] [CrossRef]
- Barascuk, N.; Skjøt-Arkil, H.; Register, T.C.; Larsen, L.; Byrjalsen, I.; Christiansen, C.; Karsdal, M.A. Human macrophage foam cells degrade atherosclerotic plaques through cathepsin K mediated processes. BMC Cardiovasc. Disord. 2010, 10, 19. [Google Scholar] [CrossRef]
- Tintut, Y.; Abedin, M.; Cho, J.; Choe, A.; Lim, J.; Demer, L.L. Regulation of RANKL-induced osteoclastic differentiation by vascular cells. J. Mol. Cell. Cardiol. 2005, 39, 389–393. [Google Scholar] [CrossRef]
- Mazière, C.; Louvet, L.; Gomila, C.; Kamel, S.; Massy, Z.; Mazière, J.C. Oxidized low density lipoprotein decreases Rankl-induced differentiation of osteoclasts by inhibition of Rankl signaling. J. Cell. Physiol. 2009, 221, 572–578. [Google Scholar] [CrossRef]
- van Meel, E.; Boonen, M.; Zhao, H.; Oorschot, V.; Ross, F.P.; Kornfeld, S.; Klumperman, J. Disruption of the Man-6-P targeting pathway in mice impairs osteoclast secretory lysosome biogenesis. Traffic 2011, 12, 912–924. [Google Scholar] [CrossRef]
- Lei, Y.; Iwashita, M.; Choi, J.; Aikawa, M.; Aikawa, E. N-acetylglucosamine-1-Phosphate Transferase Suppresses Lysosomal Hydrolases in Dysfunctional Osteoclasts: A Potential Mechanism for Vascular Calcification. J. Cardiovasc. Dev. Dis. 2015, 2, 31–47. [Google Scholar] [CrossRef] [Green Version]
- Rogers, M.A.; Aikawa, M.; Aikawa, E. Macrophage Heterogeneity Complicates Reversal of Calcification in Cardiovascular Tissues. Circ. Res. 2017, 121, 5–7. [Google Scholar] [CrossRef]
- Chinetti-Gbaguidi, G.; Daoudi, M.; Rosa, M.; Vinod, M.; Louvet, L.; Copin, C.; Fanchon, M.; Vanhoutte, J.; Derudas, B.; Belloy, L.; et al. Human Alternative Macrophages Populate Calcified Areas of Atherosclerotic Lesions and Display Impaired RANKL-Induced Osteoclastic Bone Resorption Activity. Circ. Res. 2017, 121, 19–30. [Google Scholar] [CrossRef]
- Nagy, E.; Lei, Y.; Martínez-Martínez, E.; Body, S.C.; Schlotter, F.; Creager, M.; Assmann, A.; Khabbaz, K.; Libby, P.; Hansson, G.K.; et al. Interferon-γ Released by Activated CD8(+) T Lymphocytes Impairs the Calcium Resorption Potential of Osteoclasts in Calcified Human Aortic Valves. Am. J. Pathol. 2017, 187, 1413–1425. [Google Scholar] [CrossRef]
- Merino, A.; Portolés, J.; Selgas, R.; Ojeda, R.; Buendia, P.; Ocaña, J.; Bajo, M.A.; del Peso, G.; Carracedo, J.; Ramírez, R.; et al. Effect of different dialysis modalities on microinflammatory status and endothelial damage. Clin. J. Am. Soc. Nephrol. 2010, 5, 227–234. [Google Scholar] [CrossRef]
- Ramírez, R.; Carracedo, J.; Merino, A.; Soriano, S.; Ojeda, R.; Alvarez-Lara, M.A.; Martín-Malo, A.; Aljama, P. CD14+CD16+ monocytes from chronic kidney disease patients exhibit increased adhesion ability to endothelial cells. Contrib. Nephrol. 2011, 171, 57–61. [Google Scholar]
- Rogacev, K.S.; Zawada, A.M.; Emrich, I.; Seiler, S.; Böhm, M.; Fliser, D.; Woollard, K.J.; Heine, G.H. Lower Apo A-I and lower HDL-C levels are associated with higher intermediate CD14++CD16+ monocyte counts that predict cardiovascular events in chronic kidney disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2120–2127. [Google Scholar] [CrossRef]
- Yang, J.; Fang, P.; Yu, D.; Zhang, L.; Zhang, D.; Jiang, X.; Yang, W.Y.; Bottiglieri, T.; Kunapuli, S.P.; Yu, J.; et al. Chronic Kidney Disease Induces Inflammatory CD40+ Monocyte Differentiation via Homocysteine Elevation and DNA Hypomethylation. Circ. Res. 2016, 119, 1226–1241. [Google Scholar] [CrossRef]
- Gouroju, S.; Rao, P.V.L.N.; Bitla, A.R.; Vinapamula, K.S.; Manohar, S.M.; Vishnubhotla, S. Role of Gut-derived Uremic Toxins on Oxidative Stress and Inflammation in Patients with Chronic Kidney Disease. Indian J. Nephrol. 2017, 27, 359–364. [Google Scholar]
- Castillo-Rodríguez, E.; Pizarro-Sánchez, S.; Sanz, A.B.; Ramos, A.M.; Sanchez-Niño, M.D.; Martin-Cleary, C.; Fernandez-Fernandez, B.; Ortiz, A. Inflammatory Cytokines as Uremic Toxins: “Ni Son Todos Los Que Estan, Ni Estan Todos Los Que Son”. Toxins 2017, 9, 114. [Google Scholar] [CrossRef]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef]
- Adeney, K.L.; Siscovick, D.S.; Ix, J.H.; Seliger, S.L.; Shlipak, M.G.; Jenny, N.S.; Kestenbaum, B.R. Association of serum phosphate with vascular and valvular calcification in moderate CKD. J. Am. Soc. Nephrol. 2009, 20, 381–387. [Google Scholar] [CrossRef]
- Kestenbaum, B.; Sampson, J.N.; Rudser, K.D.; Patterson, D.J.; Seliger, S.L.; Young, B.; Sherrard, D.J.; Andress, D.L. Serum phosphate levels and mortality risk among people with chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 520–528. [Google Scholar] [CrossRef]
- Block, G.A.; Klassen, P.S.; Lazarus, J.M.; Ofsthun, N.; Lowrie, E.G.; Chertow, G.M. Mineral metabolism, mortality, and morbidity in maintenance hemodialysis. J. Am. Soc. Nephrol. 2004, 15, 2208–2218. [Google Scholar] [CrossRef]
- Block, G.A.; Hulbert-Shearon, T.E.; Levin, N.W.; Port, F.K. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: A national study. Am. J. Kidney Dis. 1998, 31, 607–617. [Google Scholar] [CrossRef]
- Tentori, F.; Blayney, M.; Albert, J.M.; Gillespie, B.W.; Kerr, P.G.; Bommer, J.; Young, E.W.; Akizawa, T.; Akiba, T.; Pisoni, R.L.; et al. Mortality risk for dialysis patients with different levels of serum calcium, phosphorus, and PTH: The Dialysis Outcomes and Practice Patterns Study (DOPPS). Am. J. Kidney Dis. 2008, 52, 519–530. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work Group. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef]
- Holle, J.; Querfeld, U.; Kirchner, M.; Anninos, A.; Okun, J.; Thurn-Valsassina, D.; Bayazit, A.; Niemirska, A.; Canpolat, N.; Bulut, I.K.; et al. Indoxyl sulfate associates with cardiovascular phenotype in children with chronic kidney disease. Pediatr. Nephrol. 2019. [Google Scholar] [CrossRef]
- Chiu, C.A.; Lu, L.F.; Yu, T.H.; Hung, W.C.; Chung, F.M.; Tsai, I.T.; Yang, C.Y.; Hsu, C.C.; Lu, Y.C.; Wang, C.P.; et al. Increased levels of total P-Cresylsulphate and indoxyl sulphate are associated with coronary artery disease in patients with diabetic nephropathy. Rev. Diabet. Stud. 2010, 7, 275–284. [Google Scholar] [CrossRef]
- Goto, S.; Kitamura, K.; Kono, K.; Nakai, K.; Fujii, H.; Nishi, S. Association between AST-120 and abdominal aortic calcification in predialysis patients with chronic kidney disease. Clin. Exp. Nephrol. 2013, 17, 365–371. [Google Scholar] [CrossRef]
- Nakamura, T.; Kawagoe, Y.; Matsuda, T.; Ueda, Y.; Shimada, N.; Ebihara, I.; Koide, H. Oral ADSORBENT AST-120 decreases carotid intima-media thickness and arterial stiffness in patients with chronic renal failure. Kidney Blood Press. Res. 2004, 27, 121–126. [Google Scholar] [CrossRef]
- Six, I.; Maizel, J.; Barreto, F.C.; Rangrez, A.Y.; Dupont, S.; Slama, M.; Tribouilloy, C.; Choukroun, G.; Mazière, J.C.; Bode-Boeger, S.; et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc. Res. 2012, 96, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Higuchi, Y.; Yagi, Y.; Nishijima, F.; Yamato, H.; Ishii, H.; Osaka, M.; Yoshida, M. Reduction of indoxyl sulfate by AST-120 attenuates monocyte inflammation related to chronic kidney disease. J. Leukoc. Biol. 2013, 93, 837–845. [Google Scholar] [CrossRef]
- Pletinck, A.; Glorieux, G.; Schepers, E.; Cohen, G.; Gondouin, B.; Van Landschoot, M.; Eloot, S.; Rops, A.; Van de Voorde, J.; De Vriese, A.; et al. Protein-bound uremic toxins stimulate crosstalk between leukocytes and vessel wall. J. Am. Soc. Nephrol. 2013, 24, 1981–1994. [Google Scholar] [CrossRef]
- Ito, S.; Osaka, M.; Edamatsu, T.; Itoh, Y.; Yoshida, M. Crucial Role of the Aryl Hydrocarbon Receptor (AhR) in Indoxyl Sulfate-Induced Vascular Inflammation. J. Atheroscler. Thromb. 2016, 23, 960–975. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J. Biol. Chem. 2010, 285, 38869–38875. [Google Scholar] [CrossRef]
- Sriramarao, P.; Steffner, P.; Gehlsen, K.R. Biochemical evidence for a homophilic interaction of the alpha 3 beta 1 integrin. J. Biol. Chem. 1993, 268, 22036–22041. [Google Scholar]
- Sheppard, D. In vivo functions of integrins: Lessons from null mutations in mice. Matrix Biol. 2000, 19, 203–209. [Google Scholar] [CrossRef]
- Inami, Y.; Hamada, C.; Seto, T.; Hotta, Y.; Aruga, S.; Inuma, J.; Azuma, K.; Io, H.; Kaneko, K.; Watada, H.; et al. Effect of AST-120 on Endothelial Dysfunction in Adenine-Induced Uremic Rats. Int. J. Nephrol. 2014, 2014, 164125. [Google Scholar] [CrossRef]
- Adelibieke, Y.; Shimizu, H.; Muteliefu, G.; Bolati, D.; Niwa, T. Indoxyl sulfate induces endothelial cell senescence by increasing reactive oxygen species production and p53 activity. J. Ren. Nutr. 2012, 22, 86–89. [Google Scholar] [CrossRef]
- Peng, Y.S.; Lin, Y.T.; Chen, Y.; Hung, K.Y.; Wang, S.M. Effects of indoxyl sulfate on adherens junctions of endothelial cells and the underlying signaling mechanism. J. Cell. Biochem. 2012, 113, 1034–1043. [Google Scholar] [CrossRef]
- Li, C.; Ding, X.Y.; Xiang, D.M.; Xu, J.; Huang, X.L.; Hou, F.F.; Zhou, Q.G. Enhanced M1 and Impaired M2 Macrophage Polarization and Reduced Mitochondrial Biogenesis via Inhibition of AMP Kinase in Chronic Kidney Disease. Cell. Physiol. Biochem. 2015, 36, 358–372. [Google Scholar] [CrossRef]
- Nakano, T.; Katsuki, S.; Chen, M.; Decano, J.L.; Halu, A.; Lee, L.H.; Pestana, D.V.S.; Kum, A.S.T.; Kuromoto, R.K.; Golden, W.S.; et al. Uremic Toxin Indoxyl Sulfate Promotes Proinflammatory Macrophage Activation Via the Interplay of OATP2B1 and Dll4-Notch Signaling. Circulation 2019, 139, 78–96. [Google Scholar] [CrossRef]
- Wakamatsu, T.; Yamamoto, S.; Ito, T.; Sato, Y.; Matsuo, K.; Takahashi, Y.; Kaneko, Y.; Goto, S.; Kazama, J.J.; Gejyo, F.; et al. Indoxyl Sulfate Promotes Macrophage IL-1β Production by Activating Aryl Hydrocarbon Receptor/NF-κ/MAPK Cascades, but the NLRP3 inflammasome Was Not Activated. Toxins 2018, 10, 124. [Google Scholar] [CrossRef]
- Matsuo, K.; Yamamoto, S.; Wakamatsu, T.; Takahashi, Y.; Kawamura, K.; Kaneko, Y.; Goto, S.; Kazama, J.J.; Narita, I. Increased Proinflammatory Cytokine Production and Decreased Cholesterol Efflux Due to Downregulation of ABCG1 in Macrophages Exposed to Indoxyl Sulfate. Toxins 2015, 7, 3155–3166. [Google Scholar] [CrossRef]
- Barisione, C.; Garibaldi, S.; Furfaro, A.L.; Nitti, M.; Palmieri, D.; Passalacqua, M.; Garuti, A.; Verzola, D.; Parodi, A.; Ameri, P.; et al. Moderate Increase of Indoxyl Sulfate Promotes Monocyte Transition into Profibrotic Macrophages. PLoS ONE 2016, 11, e0149276. [Google Scholar] [CrossRef]
- Mozar, A.; Haren, N.; Chasseraud, M.; Louvet, L.; Mazière, C.; Wattel, A.; Mentaverri, R.; Morlière, P.; Kamel, S.; Brazier, M.; et al. High extracellular inorganic phosphate concentration inhibits RANK-RANKL signaling in osteoclast-like cells. J. Cell. Physiol. 2008, 215, 47–54. [Google Scholar] [CrossRef]
- Mozar, A.; Louvet, L.; Godin, C.; Mentaverri, R.; Brazier, M.; Kamel, S.; Massy, Z.A. Indoxyl sulphate inhibits osteoclast differentiation and function. Nephrol. Dial. Transplant. 2012, 27, 2176–2181. [Google Scholar] [CrossRef]
- Villa-Bellosta, R.; Hamczyk, M.R.; Andrés, V. Novel phosphate-activated macrophages prevent ectopic calcification by increasing extracellular ATP and pyrophosphate. PLoS ONE 2017, 12, e0174998. [Google Scholar] [CrossRef]
- Jing, Y.J.; Ni, J.W.; Ding, F.H.; Fang, Y.H.; Wang, X.Q.; Wang, H.B.; Chen, X.N.; Chen, N.; Zhan, W.W.; Lu, L.; et al. p-Cresyl sulfate is associated with carotid arteriosclerosis in hemodialysis patients and promotes atherogenesis in apoE-/- mice. Kidney Int. 2016, 89, 439–449. [Google Scholar] [CrossRef]
- Wang, C.P.; Lu, L.F.; Yu, T.H.; Hung, W.C.; Chiu, C.A.; Chung, F.M.; Yeh, L.R.; Chen, H.J.; Lee, Y.J.; Houng, J.Y. Serum levels of total p-cresylsulphate are associated with angiographic coronary atherosclerosis severity in stable angina patients with early stage of renal failure. Atherosclerosis 2010, 211, 579–583. [Google Scholar] [CrossRef]
- Wu, I.W.; Hsu, K.H.; Hsu, H.J.; Lee, C.C.; Sun, C.Y.; Tsai, C.J.; Wu, M.S. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients—A prospective cohort study. Nephrol. Dial. Transplant. 2012, 27, 1169–1175. [Google Scholar] [CrossRef]
- Shiba, T.; Kawakami, K.; Sasaki, T.; Makino, I.; Kato, I.; Kobayashi, T.; Uchida, K.; Kaneko, K. Effects of intestinal bacteria-derived p-cresyl sulfate on Th1-type immune response in vivo and in vitro. Toxicol. Appl. Pharmacol. 2014, 274, 191–199. [Google Scholar] [CrossRef]
- Shiba, T.; Makino, I.; Kawakami, K.; Kato, I.; Kobayashi, T.; Kaneko, K. p-Cresyl sulfate suppresses lipopolysaccharide-induced anti-bacterial immune responses in murine macrophages in vitro. Toxicol. Lett. 2016, 245, 24–30. [Google Scholar] [CrossRef]
- De Deyn, P.P.; Vanholder, R.; Eloot, S.; Glorieux, G. Guanidino compounds as uremic (neuro) toxins. Semin. Dial. 2009, 22, 340–345. [Google Scholar] [CrossRef]
- Schepers, E.; Glorieux, G.; Dou, L.; Cerini, C.; Gayrard, N.; Louvet, L.; Maugard, C.; Preus, P.; Rodriguez-Ortiz, M.; Argiles, A.; et al. Guanidino compounds as cause of cardiovascular damage in chronic kidney disease: An in vitro evaluation. Blood Purif. 2010, 30, 277–287. [Google Scholar] [CrossRef]
- Schepers, E.; Glorieux, G.; Dhondt, A.; Leybaert, L.; Vanholder, R. Role of symmetric dimethylarginine in vascular damage by increasing ROS via store-operated calcium influx in monocytes. Nephrol. Dial. Transplant. 2009, 24, 1429–1435. [Google Scholar] [CrossRef]
- Tan, H.Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 2795090. [Google Scholar] [CrossRef]
- Glorieux, G.L.; Dhondt, A.W.; Jacobs, P.; Van Langeraert, J.; Lameire, N.H.; De Deyn, P.P.; Vanholder, R.C. In vitro study of the potential role of guanidines in leukocyte functions related to atherogenesis and infection. Kidney Int. 2004, 65, 2184–2192. [Google Scholar] [CrossRef] [Green Version]
- Schepers, E.; Barreto, D.V.; Liabeuf, S.; Glorieux, G.; Eloot, S.; Barreto, F.C.; Massy, Z.; Vanholder, R.; European Uremic Toxin Work Group. Symmetric dimethylarginine as a proinflammatory agent in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 2374–2383. [Google Scholar] [CrossRef]
- Krzanowski, M.; Krzanowska, K.; Gajda, M.; Dumnicka, P.; Kopeć, G.; Guzik, B.; Woziwodzka, K.; Dziewierz, A.; Litwin, J.A.; Sułowicz, W. Asymmetric dimethylarginine as a useful risk marker of radial artery calcification in patients with advanced kidney disease. Pol. Arch. Intern. Med. 2018, 128, 157–165. [Google Scholar]
- Cagirci, G.; Cay, S.; Canga, A.; Karakurt, O.; Yazihan, N.; Kilic, H.; Topaloğlu, S.; Aras, D.; Demir, A.D.; Akdemir, R. Association between plasma asymmetrical dimethylarginine activity and severity of aortic valve stenosis. J. Cardiovasc. Med. 2011, 12, 96–101. [Google Scholar] [CrossRef]
- Kobayashi, S.; Oka, M.; Maesato, K.; Ikee, R.; Mano, T.; Hidekazu, M.; Ohtake, T. Coronary artery calcification, ADMA, and insulin resistance in CKD patients. Clin. J. Am. Soc. Nephrol. 2008, 3, 1289–1295. [Google Scholar] [CrossRef]
- Bode-Böger, S.M.; Scalera, F.; Kielstein, J.T.; Martens-Lobenhoffer, J.; Breithardt, G.; Fobker, M.; Reinecke, H. Symmetrical dimethylarginine: A new combined parameter for renal function and extent of coronary artery disease. J. Am. Soc. Nephrol. 2006, 17, 1128–1134. [Google Scholar] [CrossRef]
- van Guldener, C. Why is homocysteine elevated in renal failure and what can be expected from homocysteine-lowering? Nephrol. Dial. Transplant. 2006, 21, 1161–1166. [Google Scholar] [CrossRef] [Green Version]
- Heinz, J.; Kropf, S.; Luley, C.; Dierkes, J. Homocysteine as a risk factor for cardiovascular disease in patients treated by dialysis: A meta-analysis. Am. J. Kidney Dis. 2009, 54, 478–489. [Google Scholar] [CrossRef]
- Mallamaci, F.; Zoccali, C.; Tripepi, G.; Fermo, I.; Benedetto, F.A.; Cataliotti, A.; Bellanuova, I.; Malatino, L.S.; Soldarini, A.; Investigators, C. Hyperhomocysteinemia predicts cardiovascular outcomes in hemodialysis patients. Kidney Int. 2002, 61, 609–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, P.; Zhang, D.; Cheng, Z.; Yan, C.; Jiang, X.; Kruger, W.D.; Meng, S.; Arning, E.; Bottiglieri, T.; Choi, E.T.; et al. Hyperhomocysteinemia potentiates hyperglycemia-induced inflammatory monocyte differentiation and atherosclerosis. Diabetes 2014, 63, 4275–4290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Jiang, X.; Fang, P.; Yan, Y.; Song, J.; Gupta, S.; Schafer, A.I.; Durante, W.; Kruger, W.D.; Yang, X.; et al. Hyperhomocysteinemia promotes inflammatory monocyte generation and accelerates atherosclerosis in transgenic cystathionine beta-synthase-deficient mice. Circulation 2009, 120, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Dai, J.; Remick, D.G.; Wang, X. Homocysteine mediated expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human monocytes. Circ. Res. 2003, 93, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Kloor, D.; Osswald, H. S-Adenosylhomocysteine hydrolase as a target for intracellular adenosine action. Trends Pharmacol. Sci. 2004, 25, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Ovechkin, A.V.; Lominadze, D.; Moshal, K.S.; Tyagi, S.C. Mitochondrial mechanism of microvascular endothelial cells apoptosis in hyperhomocysteinemia. J. Cell. Biochem. 2006, 98, 1150–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, K.; Kanbay, M.; Lanaspa, M.A.; Johnson, R.J.; Ejaz, A.A. Serum uric acid and acute kidney injury: A mini review. J. Adv. Res. 2017, 8, 529–536. [Google Scholar] [CrossRef]
- Geraci, G.; Mulè, G.; Morreale, M.; Cusumano, C.; Castiglia, A.; Gervasi, F.; D’Ignoto, F.; Mogavero, M.; Geraci, C.; Cottone, S. Association between uric acid and renal function in hypertensive patients: Which role for systemic vascular involvement? J. Am. Soc. Hypertens. 2016, 10, 559–569. [Google Scholar] [CrossRef]
- Bassols, J.; Martínez-Calcerrada, J.M.; Prats-Puig, A.; Carreras-Badosa, G.; Díaz-Roldán, F.; Osiniri, I.; Riera-Pérez, E.; de Zegher, F.; Ibáñez, L.; López-Bermejo, A. Uric acid, carotid intima-media thickness and body composition in prepubertal children. Pediatr. Obes. 2016, 11, 375–382. [Google Scholar] [CrossRef]
- Andrés, M.; Quintanilla, M.A.; Sivera, F.; Sánchez-Payá, J.; Pascual, E.; Vela, P.; Ruiz-Nodar, J.M. Silent Monosodium Urate Crystal Deposits Are Associated With Severe Coronary Calcification in Asymptomatic Hyperuricemia: An Exploratory Study. Arthritis Rheumatol. 2016, 68, 1531–1539. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, S.H.; Choi, A.R.; Kim, S.; Choi, H.Y.; Kim, H.J.; Park, H.C. Asymptomatic hyperuricemia is independently associated with coronary artery calcification in the absence of overt coronary artery disease: A single-center cross-sectional study. Medicine 2017, 96, e6565. [Google Scholar] [CrossRef] [PubMed]
- Kiss, L.Z.; Bagyura, Z.; Csobay-Novák, C.; Lux, Á.; Polgár, L.; Jermendy, Á.; Soós, P.; Szelid, Z.; Maurovich-Horvat, P.; Becker, D.; et al. Serum Uric Acid Is Independently Associated with Coronary Calcification in an Asymptomatic Population. J. Cardiovasc. Transl. Res. 2019, 12, 204–210. [Google Scholar] [CrossRef]
- Grossman, C.; Shemesh, J.; Koren-Morag, N.; Bornstein, G.; Ben-Zvi, I.; Grossman, E. Serum uric acid is associated with coronary artery calcification. J. Clin. Hypertens. 2014, 16, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Crişan, T.O.; Cleophas, M.C.P.; Novakovic, B.; Erler, K.; van de Veerdonk, F.L.; Stunnenberg, H.G.; Netea, M.G.; Dinarello, C.A.; Joosten, L.A.B. Uric acid priming in human monocytes is driven by the AKT-PRAS40 autophagy pathway. Proc. Natl. Acad. Sci. USA 2017, 114, 5485–5490. [Google Scholar] [CrossRef]
- Liang, W.Y.; Zhu, X.Y.; Zhang, J.W.; Feng, X.R.; Wang, Y.C.; Liu, M.L. Uric acid promotes chemokine and adhesion molecule production in vascular endothelium via nuclear factor-kappa B signaling. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.M.; Barber, J.S.; Sacre, S.M.; Davies, K.A.; Ghezzi, P.; Mullen, L.M. Precipitation of Soluble Uric Acid Is Necessary for in vitro activation of the NLRP3 inflammasome in primary human monocytes. J. Rheumatol. 2019, 46, 1141–1150. [Google Scholar] [CrossRef]
- Liu, Y.; Duan, C.; Chen, H.; Wang, C.; Liu, X.; Qiu, M.; Tang, H.; Zhang, F.; Zhou, X.; Yang, J. Inhibition of COX-2/mPGES-1 and 5-LOX in macrophages by leonurine ameliorates monosodium urate crystal-induced inflammation. Toxicol. Appl. Pharmacol. 2018, 351, 1–11. [Google Scholar] [CrossRef]
- Aroor, A.R.; Jia, G.; Habibi, J.; Sun, Z.; Ramirez-Perez, F.I.; Brady, B.; Chen, D.; Martinez-Lemus, L.A.; Manrique, C.; Nistala, R.; et al. Uric acid promotes vascular stiffness, maladaptive inflammatory responses and proteinuria in western diet fed mice. Metabolism 2017, 74, 32–40. [Google Scholar] [CrossRef]
- Andrews, E.S.; Perrenoud, L.; Nowak, K.L.; You, Z.; Pasch, A.; Chonchol, M.; Kendrick, J.; Jalal, D. Examining the effects of uric acid-lowering on markers vascular of calcification and CKD-MBD.; A post-hoc analysis of a randomized clinical trial. PLoS ONE 2018, 13, e0205831. [Google Scholar] [CrossRef]
- Massy, Z.A.; Pietrement, C.; Touré, F. Reconsidering the Lack of Urea Toxicity in Dialysis Patients. Semin. Dial. 2016, 29, 333–337. [Google Scholar] [CrossRef]
- Velasquez, M.T.; Ramezani, A.; Raj, D.S. Urea and protein carbamylation in ESRD: Surrogate markers or partners in crime? Kidney Int. 2015, 87, 1092–1094. [Google Scholar] [CrossRef]
- Ok, E.; Basnakian, A.G.; Apostolov, E.O.; Barri, Y.M.; Shah, S.V. Carbamylated low-density lipoprotein induces death of endothelial cells: A link to atherosclerosis in patients with kidney disease. Kidney Int. 2005, 68, 173–178. [Google Scholar] [CrossRef]
- Carracedo, J.; Merino, A.; Briceño, C.; Soriano, S.; Buendía, P.; Calleros, L.; Rodriguez, M.; Martín-Malo, A.; Aljama, P.; Ramírez, R. Carbamylated low-density lipoprotein induces oxidative stress and accelerated senescence in human endothelial progenitor cells. FASEB J. 2011, 25, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Apostolov, E.O.; Shah, S.V.; Ok, E.; Basnakian, A.G. Carbamylated low-density lipoprotein induces monocyte adhesion to endothelial cells through intercellular adhesion molecule-1 and vascular cell adhesion molecule-1. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 826–832. [Google Scholar] [CrossRef]
- Asci, G.; Basci, A.; Shah, S.V.; Basnakian, A.; Toz, H.; Ozkahya, M.; Duman, S.; Ok, E. Carbamylated low-density lipoprotein induces proliferation and increases adhesion molecule expression of human coronary artery smooth muscle cells. Nephrology 2008, 13, 480–486. [Google Scholar] [CrossRef]
- Wang, Z.; Nicholls, S.J.; Rodriguez, E.R.; Kummu, O.; Hörkkö, S.; Barnard, J.; Reynolds, W.F.; Topol, E.J.; DiDonato, J.A.; Hazen, S.L. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 2007, 13, 1176–1184. [Google Scholar] [CrossRef]
- Holzer, M.; Gauster, M.; Pfeifer, T.; Wadsack, C.; Fauler, G.; Stiegler, P.; Koefeler, H.; Beubler, E.; Schuligoi, R.; Heinemann, A.; et al. Protein carbamylation renders high-density lipoprotein dysfunctional. Antioxid. Redox Signal. 2011, 14, 2337–2346. [Google Scholar] [CrossRef]
- Sun, J.T.; Yang, K.; Lu, L.; Zhu, Z.B.; Zhu, J.Z.; Ni, J.W.; Han, H.; Chen, N.; Zhang, R.Y. Increased carbamylation level of HDL in end-stage renal disease: Carbamylated-HDL attenuated endothelial cell function. Am. J. Physiol. Ren. Physiol. 2016, 310, F511–F517. [Google Scholar] [CrossRef]
- Kerr, D.N. Hypercalcemia and metastatic calcification. Cardiovasc. Res. 1997, 36, 293–297. [Google Scholar] [CrossRef]
- Bas, A.; Lopez, I.; Perez, J.; Rodriguez, M.; Aguilera-Tejero, E. Reversibility of calcitriol-induced medial artery calcification in rats with intact renal function. J. Bone Miner. Res. 2006, 21, 484–490. [Google Scholar] [CrossRef]
- Mizobuchi, M.; Finch, J.L.; Martin, D.R.; Slatopolsky, E. Differential effects of vitamin D receptor activators on vascular calcification in uremic rats. Kidney Int. 2007, 72, 709–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, T.M.; Tang, W.; Dascalos, S.; Watson, K.E.; Demer, L.L.; Shavelle, R.M.; Detrano, R.C. Ethnic origin and serum levels of 1alpha,25-dihydroxyvitamin D3 are independent predictors of coronary calcium mass measured by electron-beam computed tomography. Circulation 1997, 96, 1477–1481. [Google Scholar] [CrossRef] [PubMed]
- Watson, K.E.; Abrolat, M.L.; Malone, L.L.; Hoeg, J.M.; Doherty, T.; Detrano, R.; Demer, L.L. Active serum vitamin D levels are inversely correlated with coronary calcification. Circulation 1997, 96, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Jeziorska, M.; McCollum, C.; Woolley, D.E. Calcification in atherosclerotic plaque of human carotid arteries: Associations with mast cells and macrophages. J. Pathol. 1998, 185, 10–17. [Google Scholar] [CrossRef]
- Franczyk, A.; Stolarz-Skrzypek, K.; Wesołowska, A.; Czarnecka, D. Vitamin D and vitamin D receptor activators in treatment of hypertension and cardiovascular disease. Cardiovasc. Hematol. Disord. Drug Targets 2014, 14, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Parra, E.; Rojas-Rivera, J.; Tuñón, J.; Praga, M.; Ortiz, A.; Egido, J. Vitamin D receptor activation and cardiovascular disease. Nephrol. Dial. Transplant. 2012, 27, 17–21. [Google Scholar] [CrossRef]
- Li, X.; Speer, M.Y.; Yang, H.; Bergen, J.; Giachelli, C.M. Vitamin D receptor activators induce an anticalcific paracrine program in macrophages: Requirement of osteopontin. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 321–326. [Google Scholar] [CrossRef]
- Almeida, A.C.S.F.; Siqueira, M.C.; Bonan, N.B.; Dambiski, A.; Bertuzzo, G.; Moreno-Amaral, A.N.; Barreto, F.C. Vitamin D levels reverberate in monocytes modulation in hemodialysis patients. J. Cell. Physiol. 2019, 234, 16275–16280. [Google Scholar] [CrossRef]
- Honkanen, E.; Grönhagen-Riska, C.; Teppo, A.M.; Maury, C.P.; Meri, S. Acute-phase proteins during hemodialysis: Correlations with serum interleukin-1 beta levels and different dialysis membranes. Nephron 1991, 57, 283–287. [Google Scholar] [CrossRef]
- Guo, L.L.; Pan, Y.; Zhu, X.J.; Tan, L.Y.; Xu, Q.J.; Jin, H.M. Conventional, but not high-purity, dialysate-induced monocyte apoptosis is mediated by activation of PKC-delta and inflammatory factors release. Nephrol. Dial. Transplant. 2011, 26, 1516–1522. [Google Scholar] [CrossRef]
- Liakopoulos, V.; Jeron, A.; Shah, A.; Bruder, D.; Mertens, P.R.; Gorny, X. Hemodialysis-related changes in phenotypical features of monocytes. Sci. Rep. 2018, 8, 13964. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Yang, H.N.; Kim, M.G.; Choi, H.M.; Jo, S.K.; Cho, W.Y.; Kim, H.K. Microinflammation in hemodialysis patients is associated with increased CD14CD16(+) pro-inflammatory monocytes: Possible modification by on-line hemodiafiltration. Blood Purif. 2011, 31, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Zickler, D.; Luecht, C.; Willy, K.; Chen, L.; Witowski, J.; Girndt, M.; Fiedler, R.; Storr, M.; Kamhieh-Milz, J.; Schoon, J.; et al. Tumour necrosis factor-alpha in uraemic serum promotes osteoblastic transition and calcification of vascular smooth muscle cells via extracellular signal-regulated kinases and activator protein 1/c-FOS-mediated induction of interleukin 6 expression. Nephrol. Dial. Transplant. 2017, 33, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Hénaut, L.; Massy, Z.A. New insights into the key role of interleukin 6 in vascular calcification of chronic kidney disease. Nephrol Dial. Transplant. 2018, 33, 543–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elewa, U.; Sanchez-Niño, M.D.; Martin-Cleary, C.; Fernandez-Fernandez, B.; Egido, J.; Ortiz, A. Cardiovascular risk biomarkers in CKD: The inflammation link and the road less traveled. Int. Urol. Nephrol. 2012, 44, 1731–1744. [Google Scholar] [CrossRef]
- Heine, G.H.; Ortiz, A.; Massy, Z.A.; Lindholm, B.; Wiecek, A.; Martínez-Castelao, A.; Covic, A.; Goldsmith, D.; Süleymanlar, G.; London, G.M.; et al. Monocyte subpopulations and cardiovascular risk in chronic kidney disease. Nat. Rev. Nephrol. 2012, 8, 362–369. [Google Scholar] [CrossRef]
- Hutchison, C.A.; Heyne, N.; Airia, P.; Schindler, R.; Zickler, D.; Cook, M.; Cockwell, P.; Grima, D. Immunoglobulin free light chain levels and recovery from myeloma kidney on treatment with chemotherapy and high cut-off haemodialysis. Nephrol. Dial. Transplant. 2012, 27, 3823–3828. [Google Scholar] [CrossRef]
- Girndt, M.; Fiedler, R.; Martus, P.; Pawlak, M.; Storr, M.; Bohler, T.; Glomb, M.A.; Liehr, K.; Henning, C.; Templin, M.; et al. High cut-off dialysis in chronic haemodialysis patients. Eur. J. Clin. Investig. 2015, 45, 1333–1340. [Google Scholar] [CrossRef]
- Fiedler, R.; Neugebauer, F.; Ulrich, C.; Wienke, A.; Gromann, C.; Storr, M.; Böhler, T.; Seibert, E.; Girndt, M. Randomized controlled pilot study of 2 weeks’ treatment with high cutoff membrane for hemodialysis patients with elevated C-reactive protein. Artif. Organs 2012, 36, 886–893. [Google Scholar] [CrossRef]
- Kneis, C.; Beck, W.; Boenisch, O.; Klefisch, F.; Deppisch, R.; Zickler, D.; Schindler, R. Elimination of middle-sized uremic solutes with high-flux and high-cut-off membranes: A randomized in vivo study. Blood Purif. 2013, 36, 287–294. [Google Scholar] [CrossRef]
- Trojanowicz, B.; Ulrich, C.; Fiedler, R.; Storr, M.; Boehler, T.; Martus, P.; Pawlak, M.; Glomb, M.A.; Henning, C.; Templin, M.; et al. Impact of serum and dialysates obtained from chronic hemodialysis patients maintained on high cut-off membranes on inflammation profile in human THP-1 monocytes. Hemodial. Int. 2017, 21, 348–358. [Google Scholar] [CrossRef]
- Zickler, D.; Willy, K.; Girndt, M.; Fiedler, R.; Martus, P.; Storr, M.; Schindler, R. High cut-off dialysis in chronic haemodialysis patients reduces serum procalcific activity. Nephrol. Dial. Transplant. 2016, 31, 1706–1712. [Google Scholar] [CrossRef] [Green Version]
- Boschetti-de-Fierro, A.; Voigt, M.; Storr, M.; Krause, B. MCO Membranes: Enhanced Selectivity in High-Flux Class. Sci. Rep. 2015, 5, 18448. [Google Scholar] [CrossRef]
- Zickler, D.; Schindler, R.; Willy, K.; Martus, P.; Pawlak, M.; Storr, M.; Hulko, M.; Boehler, T.; Glomb, M.A.; Liehr, K.; et al. Medium Cut-Off (MCO) Membranes Reduce Inflammation in Chronic Dialysis Patients-A Randomized Controlled Clinical Trial. PLoS ONE 2017, 12, e0169024. [Google Scholar] [CrossRef]
- Willy, K.; Hulko, M.; Storr, M.; Speidel, R.; Gauss, J.; Schindler, R.; Zickler, D. In Vitro Dialysis of Cytokine-Rich Plasma with High and Medium Cut-Off Membranes Reduces Its Procalcific Activity. Artif. Organs 2017, 41, 803–809. [Google Scholar] [CrossRef]
- Trojanowicz, B.; Ulrich, C.; Fiedler, R.; Martus, P.; Storr, M.; Boehler, T.; Werner, K.; Hulko, M.; Zickler, D.; Willy, K.; et al. Modulation of leucocytic angiotensin-converting enzymes expression in patients maintained on high-permeable haemodialysis. Nephrol. Dial. Transplant. 2018, 33, 34–43. [Google Scholar] [CrossRef]
- Molostvov, G.; James, S.; Fletcher, S.; Bennett, J.; Lehnert, H.; Bland, R.; Zehnder, D. Extracellular calcium-sensing receptor is functionally expressed in human artery. Am. J. Physiol. Ren. Physiol. 2007, 293, F946–F955. [Google Scholar] [CrossRef]
- Hénaut, L.; Boudot, C.; Massy, Z.A.; Lopez-Fernandez, I.; Dupont, S.; Mary, A.; Drüeke, T.B.; Kamel, S.; Brazier, M.; Mentaverri, R. Calcimimetics increase CaSR expression and reduce mineralization in vascular smooth muscle cells: Mechanisms of action. Cardiovasc. Res. 2014, 101, 256–265. [Google Scholar] [CrossRef]
- Mendoza, F.J.; Martinez-Moreno, J.; Almaden, Y.; Rodriguez-Ortiz, M.E.; Lopez, I.; Estepa, J.C.; Henley, C.; Rodriguez, M.; Aguilera-Tejero, E. Effect of calcium and the calcimimetic AMG 641 on matrix-Gla protein in vascular smooth muscle cells. Calcif. Tissue Int. 2011, 88, 169–178. [Google Scholar] [CrossRef]
- Ivanovski, O.; Nikolov, I.G.; Joki, N.; Caudrillier, A.; Phan, O.; Mentaverri, R.; Maizel, J.; Hamada, Y.; Nguyen-Khoa, T.; Fukagawa, M.; et al. The calcimimetic R-568 retards uremia-enhanced vascular calcification and atherosclerosis in apolipoprotein E deficient (apoE-/-) mice. Atherosclerosis 2009, 205, 55–62. [Google Scholar] [CrossRef]
- Raggi, P.; Chertow, G.M.; Torres, P.U.; Csiky, B.; Naso, A.; Nossuli, K.; Moustafa, M.; Goodman, W.G.; Lopez, N.; Downey, G.; et al. The ADVANCE study: A randomized study to evaluate the effects of cinacalcet plus low-dose vitamin D on vascular calcification in patients on hemodialysis. Nephrol. Dial. Transplant. 2011, 26, 1327–1339. [Google Scholar] [CrossRef]
- Chertow, G.M.; Block, G.A.; Correa-Rotter, R.; Drüeke, T.B.; Floege, J.; Goodman, W.G.; Herzog, C.A.; Kubo, Y.; London, G.M.; Mahaffey, K.W.; et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N. Engl. J. Med. 2012, 367, 2482–2494. [Google Scholar]
- House, M.G.; Kohlmeier, L.; Chattopadhyay, N.; Kifor, O.; Yamaguchi, T.; Leboff, M.S.; Glowacki, J.; Brown, E.M. Expression of an extracellular calcium-sensing receptor in human and mouse bone marrow cells. J. Bone Miner. Res. 1997, 12, 1959–1970. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Olozak, I.; Chattopadhyay, N.; Butters, R.R.; Kifor, O.; Scadden, D.T.; Brown, E.M. Expression of extracellular calcium (Ca2+o)-sensing receptor in human peripheral blood monocytes. Biochem. Biophys. Res. Commun. 1998, 246, 501–506. [Google Scholar] [CrossRef]
- Olszak, I.T.; Poznansky, M.C.; Evans, R.H.; Olson, D.; Kos, C.; Pollak, M.R.; Brown, E.M.; Scadden, D.T. Extracellular calcium elicits a chemokinetic response from monocytes in vitro and in vivo. J. Clin. Investig. 2000, 105, 1299–1305. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Rossol, M.; Pierer, M.; Raulien, N.; Quandt, D.; Meusch, U.; Rothe, K.; Schubert, K.; Schöneberg, T.; Schaefer, M.; Krügel, U.; et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 2012, 3, 1329. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Zhang, X.; Zhao, M.; Chi, J.; Liu, Y.; Lin, F.; Fu, Y.; Ma, D.; Yin, X. Activation in M1 but not M2 Macrophages Contributes to Cardiac Remodeling after Myocardial Infarction in Rats: A Critical Role of the Calcium Sensing Receptor/NRLP3 Inflammasome. Cell. Physiol. Biochem. 2015, 35, 2483–2500. [Google Scholar] [CrossRef]
- Heine, G.H.; Ulrich, C.; Seibert, E.; Seiler, S.; Marell, J.; Reichart, B.; Krause, M.; Schlitt, A.; Köhler, H.; Girndt, M. CD14(++)CD16+ monocytes but not total monocyte numbers predict cardiovascular events in dialysis patients. Kidney Int. 2008, 73, 622–629. [Google Scholar] [CrossRef]
- Yamamoto, D.; Suzuki, S.; Ishii, H.; Hirayama, K.; Harada, K.; Aoki, T.; Shibata, Y.; Negishi, Y.; Tatami, Y.; Sumi, T.; et al. Predictors of abdominal aortic calcification progression in patients with chronic kidney disease without hemodialysis. Atherosclerosis 2016, 253, 15–21. [Google Scholar] [CrossRef]

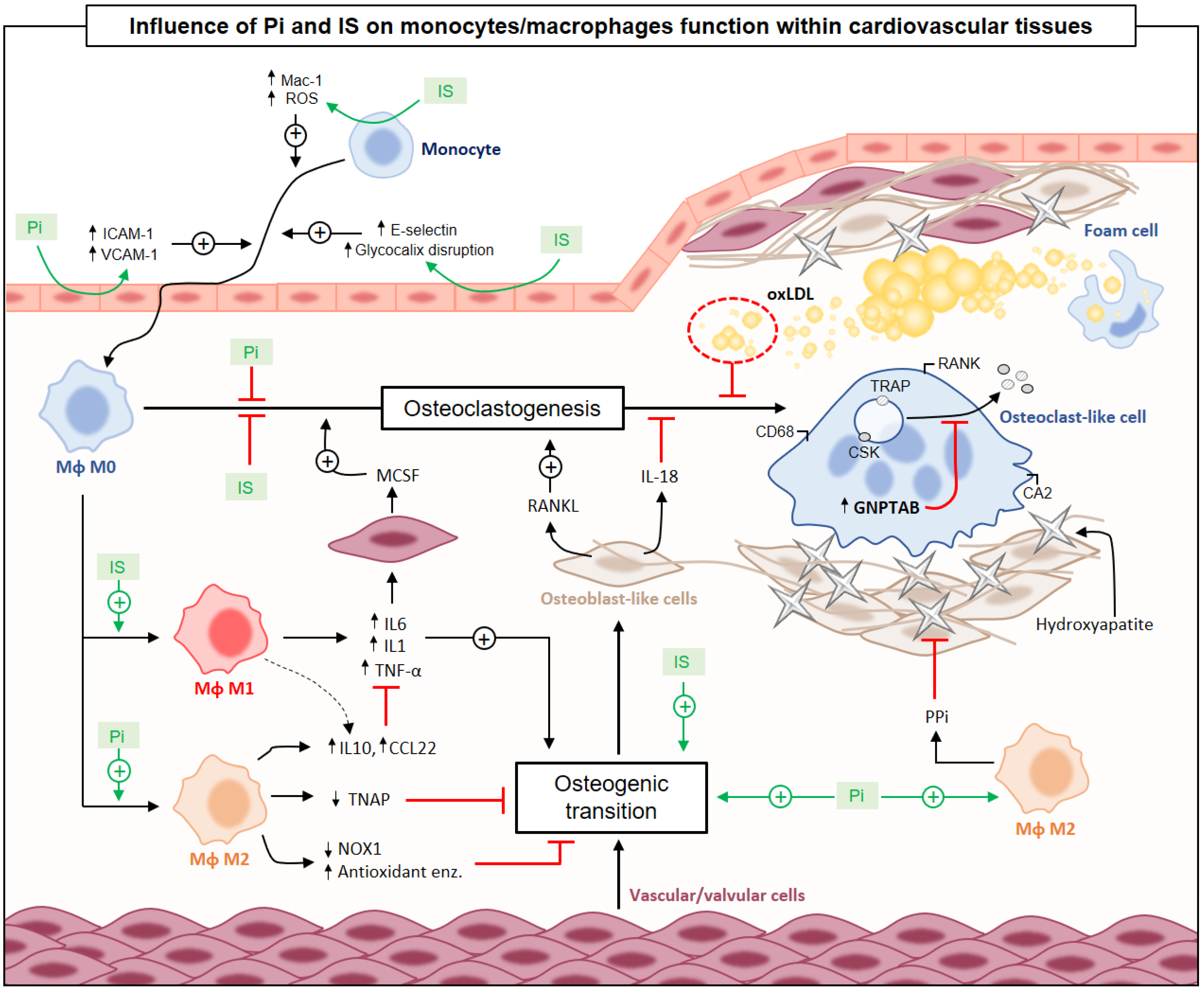

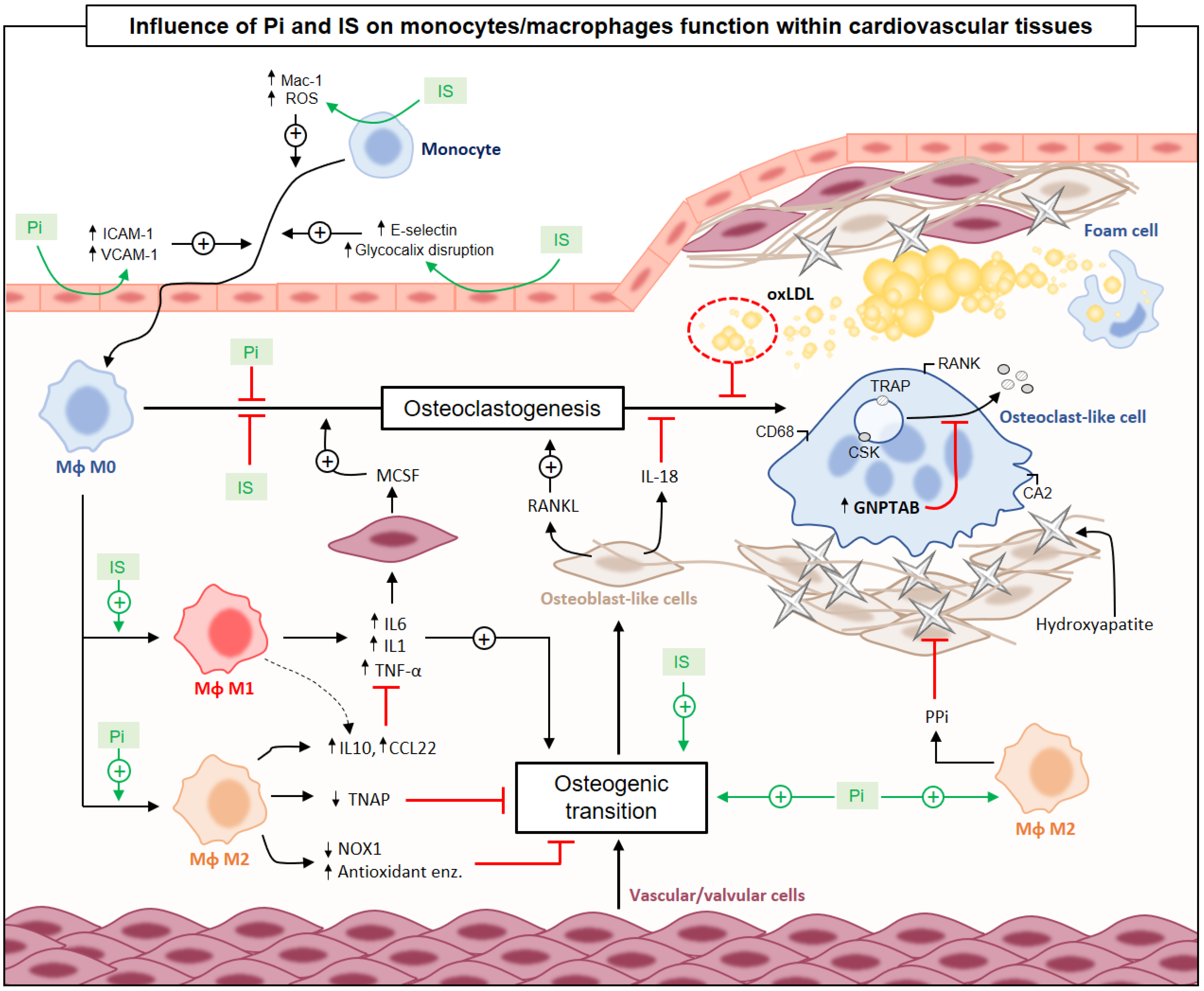
{kind=link}
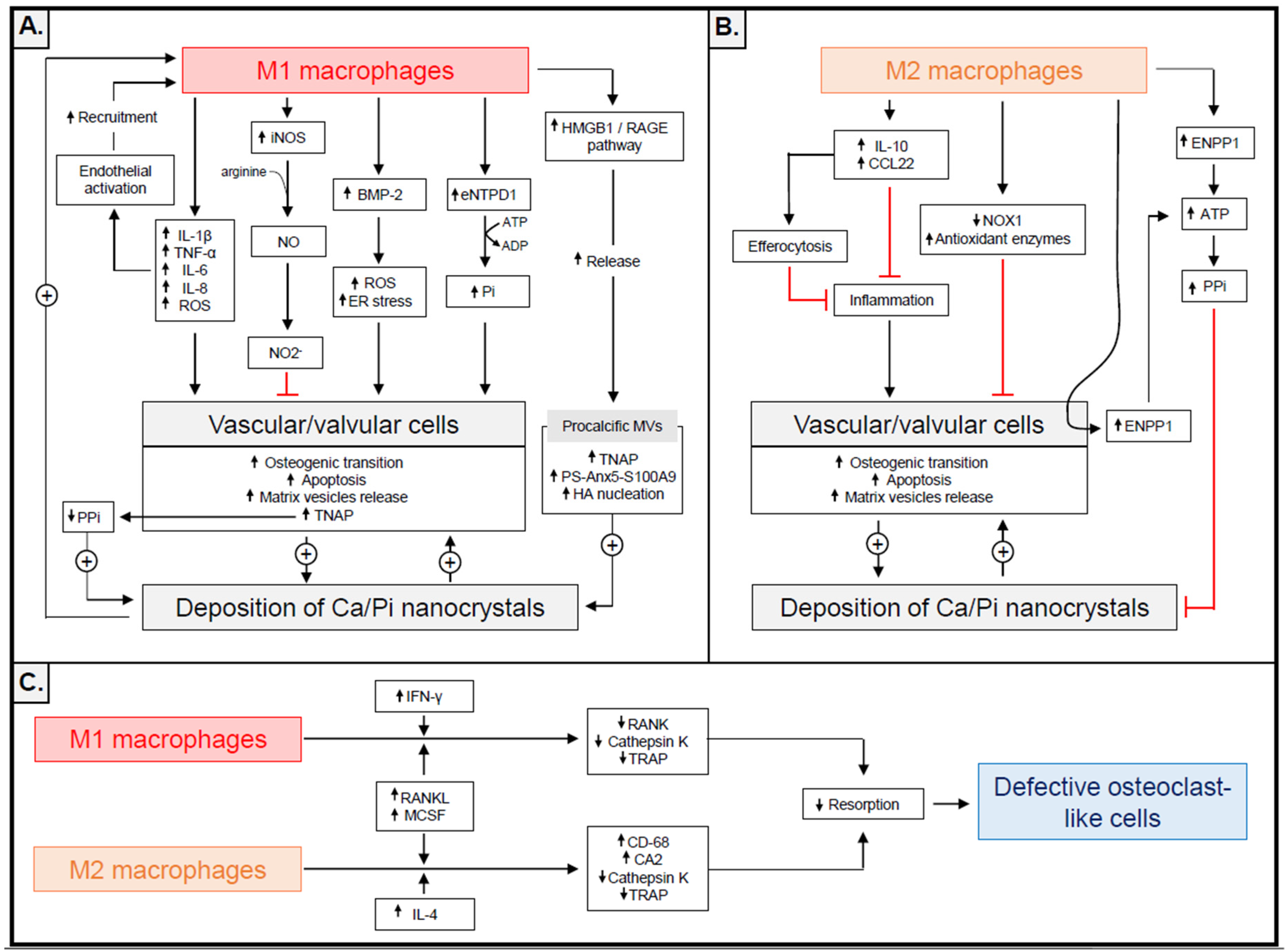
{kind=link}
| Uraemic Toxins | Action | Signalling | Experimental Model | Potential Effect on CVC | Ref |
|---|---|---|---|---|---|
| Phosphate | VCAM ICAM | ND | CKD mice | Procalcific | [116] |
| Osteoclast differentiation TRAP resorption | RANKL-induced NFΚB, AP1 and Sp1/Sp3 via Na/Pi co-transporters | PBMC, RAW 264.7 | Procalcific | [131] | |
| ARG1 and arginine degradation PGC1β, HIF-1 NOX1 Antioxidant enzymes, antioxidant metabolites ATP, PPi eNTPD1, TNAP | ND | Mice BMDMs | Anticalcific | [133] | |
| Indoxyl sulphate | MAC1 ROS | P38 phosphorylation Translocation of NADPH oxidase subunit p47 phox | PBMCs from CKD mice | Procalcific | [117] |
| THP1 adhesion to IL1-β-activated HUVECs | THP1 and HUVECs | ||||
| Adhesion, extravasation, glycocalix disruption | ND | Rat circulating leukocytes | Procalcific | [118] | |
| TNF-α-induced leukocyte adhesion though E-selectin | Intake via AhR AP1 activity | Non-CKD mice | Procalcific | [119] | |
| JNK, P38 and NFΚB NADPH oxidase | THP1 and HUVECs CKD mice | Procalcific | [120] | ||
| Endothelial cell senescence | ROS and P53 | HUVECs | Procalcific | [124] | |
| Adherens junction between endothelial cells | ROS which activates ERK1/2 pathway and subsequent MLCK and MLC phosphorylation | Bovine pulmonary artery endothelial cells | Procalcific | [125] | |
| IL-1β, TNF-α and MCP1 | Ubiquitin proteasome pathway Notch signalling | PBMCs Ldlr-/- mice with CKD | Procalcific | [127] | |
| Pro-IL1β | Intake via AhR NFΚB and MAPK activation | THP1-derived macrophages | Procalcific | [128] | |
| Polarization toward low inflammatory pro-fibrotic macrophages: IL-6, CCL2, Cox2 CD163, IL-10, PPARγ, TIMP1 | Intake via Ahr Nrf2 activation | THP1 | Procalcific | [130] | |
| Viability, cholesterol efflux IL-1β, TNF-α and ROS | ND | THP1-derived macrophages | Procalcific | [129] | |
| Osteoclast differentiation, resorption | JNK, P38, AKT, ERK1/2 DNA binding activity of AP1 and NFΚB | RAW 264.7 and PBMCs | Procalcific | [132] | |
| Paracresyl sulphate | TNF-α, MCP1 and ROS Monocytes/endothelial cells interactions Atherogenesis and infiltration E-selectin, ICAM-1 and VCAM-1 | Nox1, Nox4 and P22 | HUVECs, HAEC, THP1 and peritoneal macrophages ApoE-/- mice with CKD | Procalcific | [134] |
| IFN-γ, IL-4 Th1 cells, Th2 cells | ND | Mouse splenocytes | Anticalcific | [137] | |
| IL12 p70, IL-10, CD40 | ND | RAW 264.7 and peritoneal macrophages | Anticalcific | [138] |
| Uraemic Toxins | Action | Signalling | Experimental Model | Potential Effect on CVC | Refs | |
|---|---|---|---|---|---|---|
| Guanidino compounds | SDMA | ROS | Ca2+-entry via SOCS | Human PBMCs | Procalcific | [140,141] |
| IL-6 and TNF-α | NFΚB pathway | THP1 | Procalcific | [144] | ||
| GPA | ROS | ND | Human monocytes | Procalcific | [140] | |
| MG | TNF-α | ND | Human monocytes | Procalcific | [143] | |
| ROS | ND | Human monocytes | Procalcific | [140] | ||
| GAA | TNF-α | ND | Human monocytes | Procalcific | [143] | |
| RANK-L induced osteoclastogenesis | ND | RAW 264.7 | Anticalcific | [140] | ||
| ADMA | Endothelial cell senescence | ROS and P53 | HUVECs | Procalcific | [124] | |
| No impact on inflammation | ND | THP1 | None | [144] | ||
| RANK-L induced osteoclastogenesis | ND | RAW 264.7 | Anticalcific | [140] | ||
| ROS | ND | Human monocytes | Procalcific | [140] | ||
| GSA | TNF-α | ND | Human monocytes | Anticalcific | [143] | |
| GBA | RANK-L induced osteoclastogenesis | ND | RAW 264.7 | Anticalcific | [140] | |
| ROS | ND | Human monocytes | Procalcific | [140] | ||
| G | RANK-L induced osteoclastogenesis | ND | RAW 264.7 | Anticalcific | [140] | |
| ROS | ND | Human monocytes | Procalcific | [140] | ||
| Homocysteine | IL-8 and MCP1 | PKC/calmodulin NADPH oxidase, ROS p38, ERK1/2 and NFΚB activation | PBMCs | Procalcific | [154] | |
| Ly-6C subset accumulation within atherosclerotic lesions MCP1, TNF-α | NADPH oxidase-mediated oxidative stress | Tg-hCBS apoE-/- Cbs-/- mice | Procalcific | [153] | ||
| Atherosclerotic lesions Ly-6C subset TNF-α M1 and M2 polarization | DNA methyltransferase activity | Tg-hCBS apoE-/- Cbs-/- mice Primary mice splenocytes | Procalcific | [152] | ||
| CD40 / CD40 intermediate monocytes | DNA methyltransferase activity DNA hypomethylation of the NFΚB consensus element in the CD40 promoter | PBMCs | Procalcific | [102] | ||
| Endothelial cell apoptosis | ROS Transmembrane mitochondrial potential Cytochrome C release Caspase 3 and 9 activation, PARP cleavage | Microvascular endothelial cells (MVECs) | Procalcific | [156] | ||
| Uric acid | IL1RA IL1-β | AKT/PRAS40 pathway mTOR signalling Autophagy through LC3I / LC3II | PBMCs | Procalcific | [164] | |
| Viability MCP1, Il-8, VCAM-1, ICAM-1 THP1 migration and adhesion to HUVECs | NFΚB activation | THP1, HUVECs Rats with hyperuricemia | Procalcific | [165] | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hénaut, L.; Candellier, A.; Boudot, C.; Grissi, M.; Mentaverri, R.; Choukroun, G.; Brazier, M.; Kamel, S.; Massy, Z.A. New Insights into the Roles of Monocytes/Macrophages in Cardiovascular Calcification Associated with Chronic Kidney Disease. Toxins 2019, 11, 529. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11090529
Hénaut L, Candellier A, Boudot C, Grissi M, Mentaverri R, Choukroun G, Brazier M, Kamel S, Massy ZA. New Insights into the Roles of Monocytes/Macrophages in Cardiovascular Calcification Associated with Chronic Kidney Disease. Toxins. 2019; 11(9):529. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11090529
Chicago/Turabian StyleHénaut, Lucie, Alexandre Candellier, Cédric Boudot, Maria Grissi, Romuald Mentaverri, Gabriel Choukroun, Michel Brazier, Saïd Kamel, and Ziad A. Massy. 2019. "New Insights into the Roles of Monocytes/Macrophages in Cardiovascular Calcification Associated with Chronic Kidney Disease" Toxins 11, no. 9: 529. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins11090529