
Perfringolysin O: The Underrated Clostridium perfringens Toxin?

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Genetics
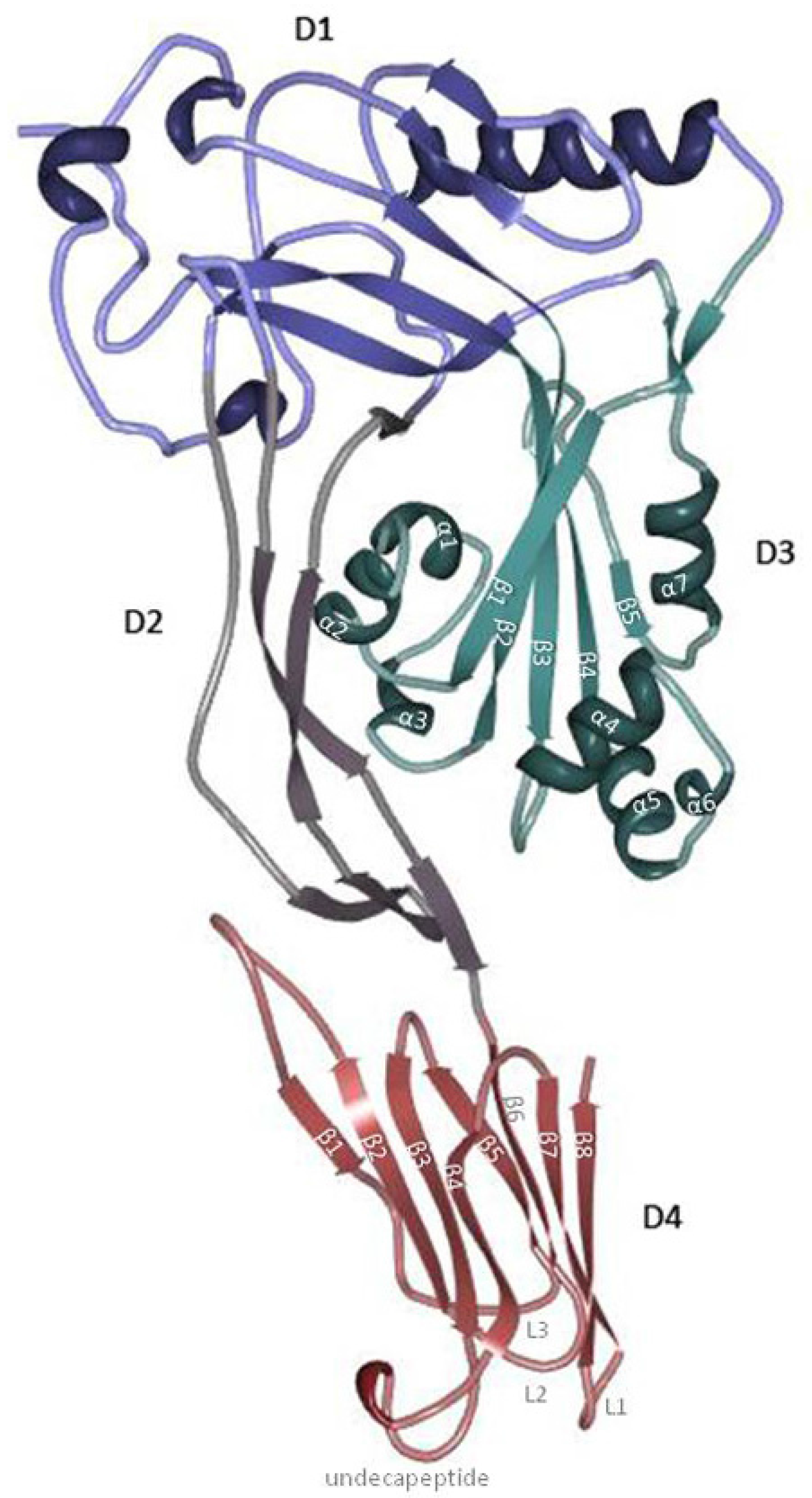
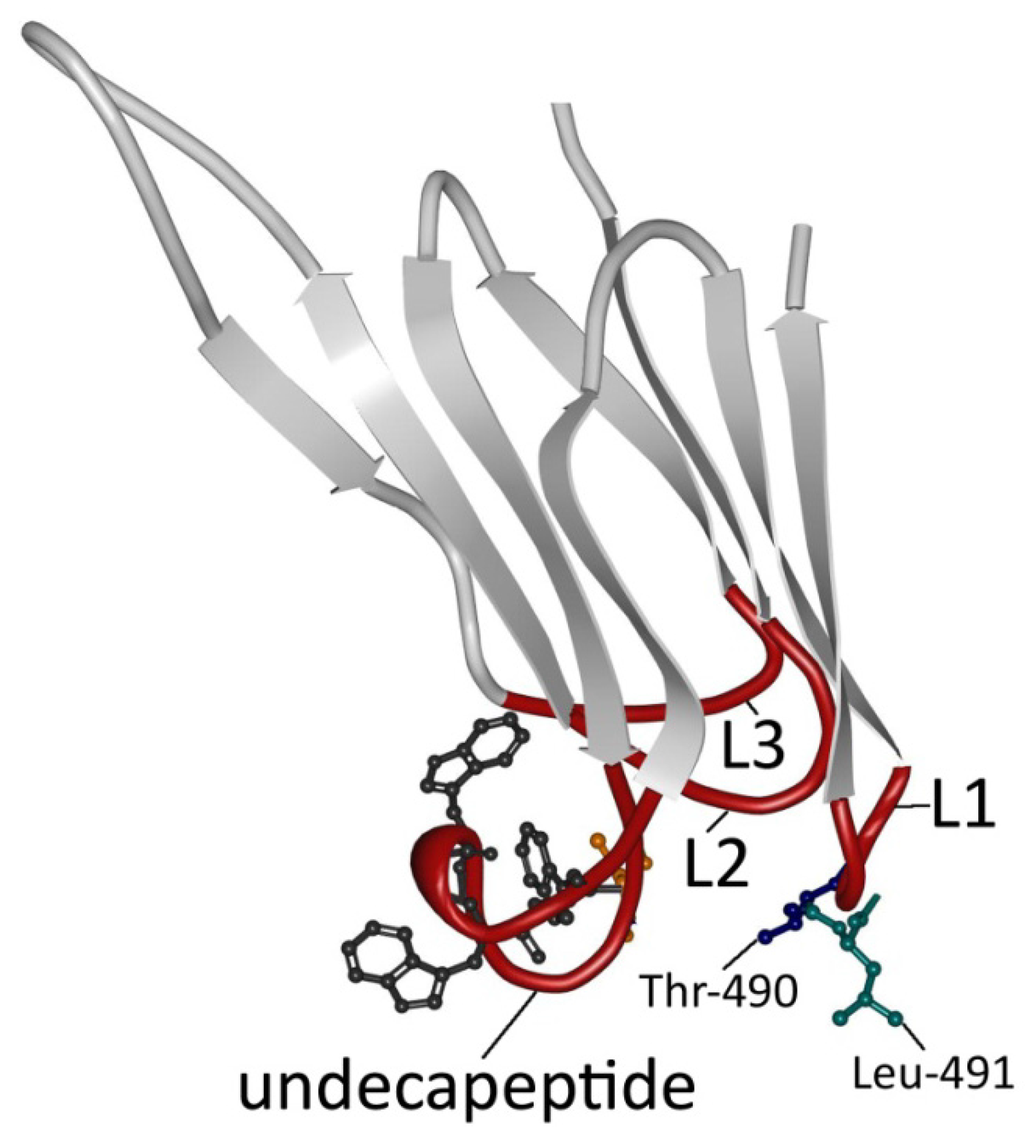
3. PFO Structure
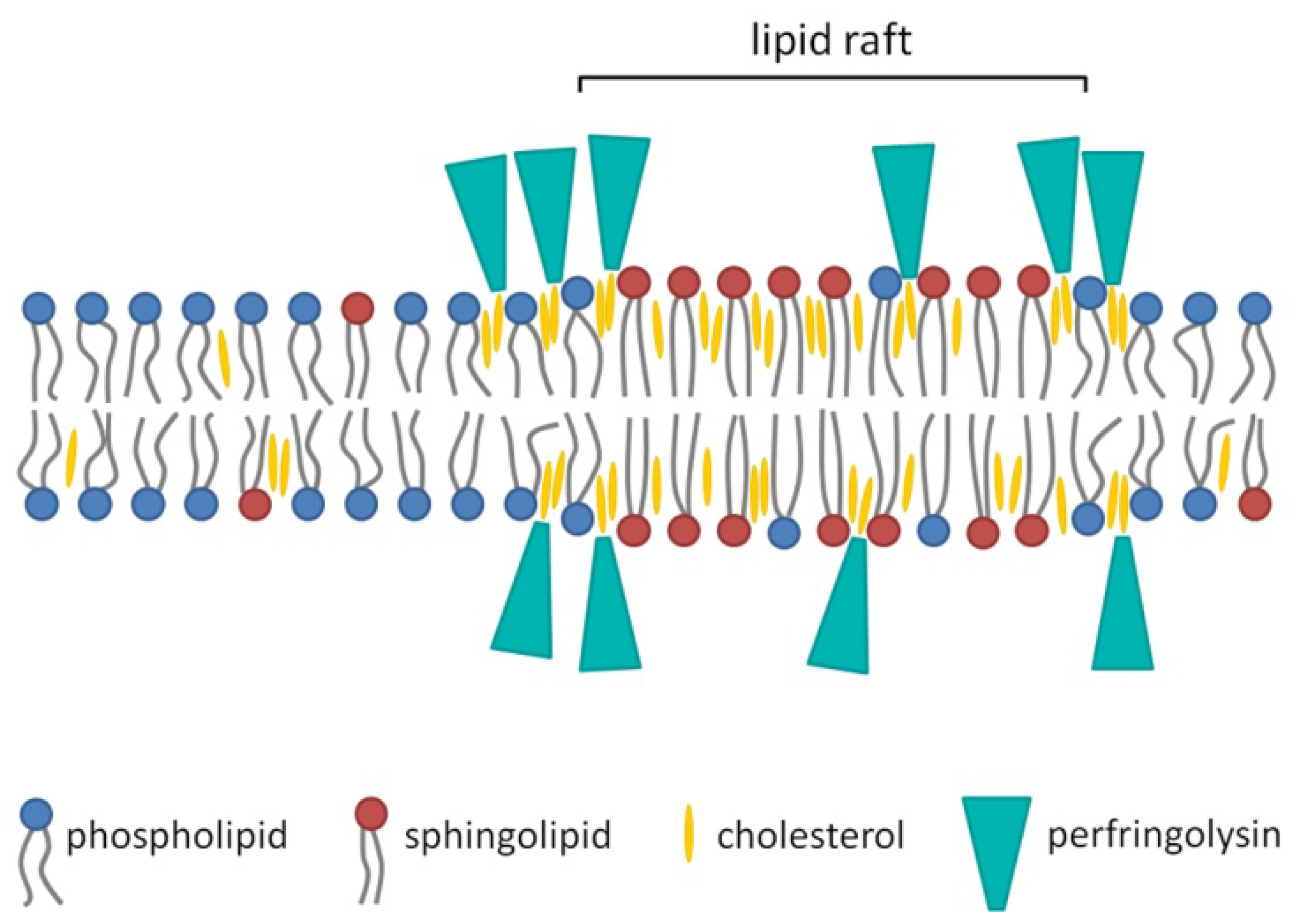
4. Membrane Binding



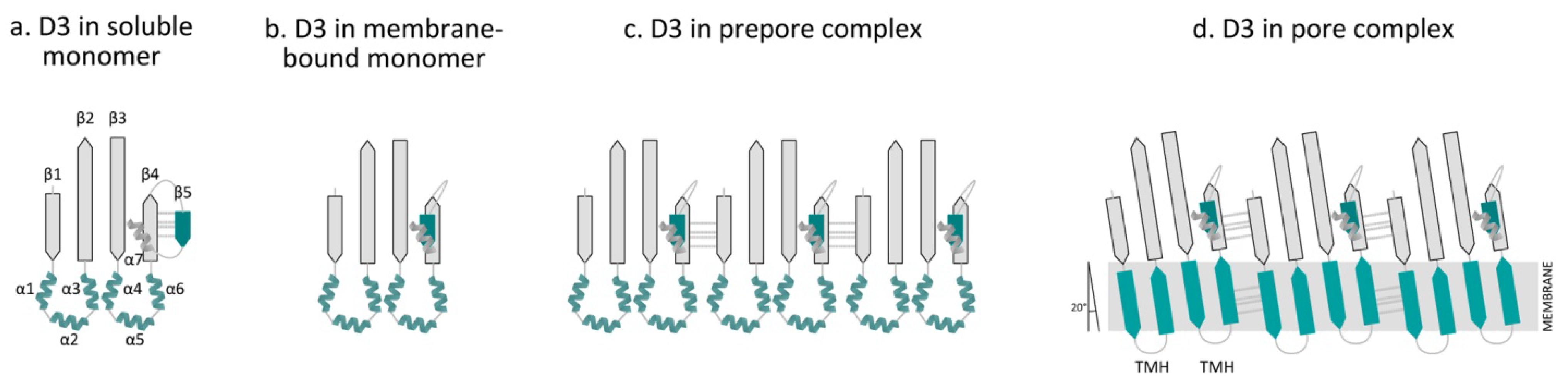
5. Molecular Mechanism of Pore Formation


6. Genetic Regulation
7. The Role of PFO in Disease
8. Remaining Questions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rood, J.I. Virulence genes of Clostridium perfringens. Annu. Rev. Microbiol. 1998, 52, 333–360. [Google Scholar]
- Lebrun, M.; Filee, P.; Mousset, B.; Desmecht, D.; Galleni, M.; Mainil, J.G.; Linden, A. The expression of Clostridium perfringens consensus β2 toxin is associated with bovine enterotoxaemia syndrome. Vet. Microbiol. 2007, 120, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Morris, W.E.; Dunleavy, M.V.; Diodati, J.; Berra, G.; Fernandez-Miyakawa, M.E. Effects of Clostridium perfringens α and ε toxins in the bovine gut. Anaerobe 2012, 18, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Songer, J.G. Clostridial enteric diseases of domestic animals. Clin. Microbiol. Rev. 1996, 9, 216–234. [Google Scholar] [PubMed]
- Songer, J.G.; Miskimmins, D.W. Clostridium perfringens type E enteritis in calves: Two cases and a brief review of the literature. Anaerobe 2004, 10, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Ellemor, D.M.; Boyd, R.L.; Emmins, J.J.; Rood, J.I. Synergistic effects of α-toxin and perfringolysin O in Clostridium perfringens-mediated gas gangrene. Infect. Immun. 2001, 69, 7904–7910. [Google Scholar] [CrossRef] [PubMed]
- Verherstraeten, S.; Goossens, E.; Valgaeren, B.; Pardon, B.; Timbermont, L.; Vermeulen, K.; Schauvliege, S.; Haesebrouck, F.; Ducatelle, R.; Deprez, P.; et al. The synergistic necrohemorrhagic action of Clostridium perfringens perfringolysin and α toxin in the bovine intestine and against bovine endothelial cells. Vet. Res. 2013, 44, 45. [Google Scholar]
- Fernandez-Miyakawa, M.E.; Jost, B.H.; Billington, S.J.; Uzal, F.A. Lethal effects of Clostridium perfringens ε toxin are potentiated by α and perfringolysin-O toxins in a mouse model. Vet. Microbiol. 2008, 127, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Heuck, A.P.; Savva, C.G.; Holzenburg, A.; Johnson, A.E. Conformational changes that effect oligomerization and initiate pore formation are triggered throughout perfringolysin O upon binding to cholesterol. J. Biol. Chem. 2007, 282, 22629–22637. [Google Scholar] [CrossRef] [PubMed]
- Hotze, E.M.; Tweten, R.K. Membrane assembly of the cholesterol-dependent cytolysin pore complex. Biochim. Biophys. Acta 2012, 1818, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R.; Bouvet, P. Genetic characteristics of toxigenic Clostridia and toxin gene evolution. Toxicon 2013, 75, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.J.; Fernandez-Miyakawa, M.E.; Sayeed, S.; Poon, R.; Adams, V.; Rood, J.I.; Uzal, F.A.; McClane, B.A. Dissecting the contributions of Clostridium perfringens type C toxins to lethality in the mouse intravenous injection model. Infect. Immun. 2006, 74, 5200–5210. [Google Scholar] [CrossRef] [PubMed]
- Marvaud, J.C.; Stiles, B.G.; Chenal, A.; Gillet, D.; Gibert, M.; Smith, L.A.; Popoff, M.R. Clostridium perfringens ι toxin. Mapping of the Ia domain involved in docking with Ib and cellular internalization. J. Biol. Chem. 2002, 277, 43659–43666. [Google Scholar]
- Nagahama, M.; Hayashi, S.; Morimitsu, S.; Sakurai, J. Biological activities and pore formation of Clostridium perfringens β toxin in HL 60 cells. J. Biol. Chem. 2003, 278, 36934–36941. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R. ε toxin: A fascinating pore-forming toxin. FEBS J. 2011, 278, 4602–4615. [Google Scholar] [CrossRef] [PubMed]
- Myers, G.S.; Rasko, D.A.; Cheung, J.K.; Ravel, J.; Seshadri, R.; DeBoy, R.T.; Ren, Q.; Varga, J.; Awad, M.M.; Brinkac, L.M.; et al. Skewed genomic variability in strains of the toxigenic bacterial pathogen. Clostridium perfringens. Genome Res. 2006, 16, 1031–1040. [Google Scholar]
- Rood, J.I.; Cole, S.T. Molecular genetics and pathogenesis of Clostridium perfringens. Microbiol. Rev. 1991, 55, 621–648. [Google Scholar] [PubMed]
- Uzal, F.A.; Freedman, J.C.; Shrestha, A.; Theoret, J.R.; Garcia, J.; Awad, M.M.; Adams, V.; Moore, R.J.; Rood, J.I.; McClane, B.A. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 2014, 9, 361–377. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, A.; Miyamoto, K.; Kuwahara, T.; Miki, Y.; Kaneko, I.; Li, J.; McClane, B.A.; Akimoto, S. Genetic characterization of type A enterotoxigenic Clostridium perfringens strains. PLoS ONE 2009, 4, e5598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, M.; Li, J.; McClane, B.A. Genotypic and phenotypic characterization of Clostridium perfringens isolates from Darmbrand cases in post-World War II Germany. Infect. Immun. 2012, 80, 4354–4363. [Google Scholar] [CrossRef]
- Tweten, R.K. Cloning and expression in Escherichia coli of the perfringolysin O (θ-toxin) gene from Clostridium perfringens and characterization of the gene product. Infect. Immun. 1988, 56, 3228–3234. [Google Scholar] [PubMed]
- Shimizu, T.; Okabe, A.; Minami, J.; Hayashi, H. An upstream regulatory sequence stimulates expression of the perfringolysin O gene of Clostridium perfringens. Infect. Immun. 1991, 59, 137–142. [Google Scholar]
- Canard, B.; Cole, S.T. Genome organization of the anaerobic pathogen Clostridium perfringens. Proc. Natl. Acad. Sci. USA 1989, 86, 6676–6680. [Google Scholar] [CrossRef] [PubMed]
- Tweten, R.K. Nucleotide sequence of the gene for perfringolysin O (θ-toxin) from Clostridium perfringens: Significant homology with the genes for streptolysin O and pneumolysin. Infect. Immun. 1988, 56, 3235–3240. [Google Scholar] [PubMed]
- Sawires, Y.S.; Songer, J.G. Clostridium perfringens: Insight into virulence evolution and population structure. Anaerobe 2006, 12, 23–43. [Google Scholar] [CrossRef] [PubMed]
- Rooney, A.P.; Swezey, J.L.; Friedman, R.; Hecht, D.W.; Maddox, C.W. Analysis of core housekeeping and virulence genes reveals cryptic lineages of Clostridium perfringens that are associated with distinct disease presentations. Genetics 2006, 172, 2081–2092. [Google Scholar] [CrossRef] [PubMed]
- Sandkvist, M. Type II secretion and pathogenesis. Infect. Immun. 2001, 69, 3523–3535. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.T.; Schneewind, O. Protein secretion and the pathogenesis of bacterial infections. Genes Dev. 2001, 15, 1725–1752. [Google Scholar] [CrossRef]
- Solovyova, A.S.; Nollmann, M.; Mitchell, T.J.; Byron, O. The solution structure and oligomerization behavior of two bacterial toxins: Pneumolysin and perfringolysin O. Biophys. J. 2004, 87, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Rossjohn, J.; Feil, S.C.; McKinstry, W.J.; Tweten, R.K.; Parker, M.W. Structure of a cholesterol-binding, thiol-activated cytolysin and a model of its membrane form. Cell 1997, 89, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Heuck, A.P.; Tweten, R.K.; Johnson, A.E. Structural insights into the membrane-anchoring mechanism of a cholesterol-dependent cytolysin. Nat. Struct. Biol. 2002, 9, 823–827. [Google Scholar] [PubMed]
- Dang, T.X.; Milligan, R.A.; Tweten, R.K.; Wilson-Kubalek, E.M. Helical crystallization on nickel-lipid nanotubes: Perfringolysin O as a model protein. J. Struct. Biol. 2005, 152, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Heuck, A.P.; Tweten, R.K.; Johnson, A.E. Assembly and topography of the prepore complex in cholesterol-dependent cytolysins. J. Biol. Chem. 2003, 278, 31218–31225. [Google Scholar] [CrossRef] [PubMed]
- Czajkowsky, D.M.; Hotze, E.M.; Shao, Z.; Tweten, R.K. Vertical collapse of a cytolysin prepore moves its transmembrane β-hairpins to the membrane. EMBO J. 2004, 23, 3206–3215. [Google Scholar] [CrossRef] [PubMed]
- Alouf, J.E.; Geoffroy, C.; Pattus, F.; Verger, R. Surface properties of bacterial sulfhydryl-activated cytolytic toxins. Interaction with monomolecular films of phosphatidylcholine and various sterols. Eur. J. Biochem. 1984, 141, 205–210. [Google Scholar]
- Flanagan, J.J.; Tweten, R.K.; Johnson, A.E.; Heuck, A.P. Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry 2009, 48, 3977–3987. [Google Scholar] [CrossRef] [PubMed]
- Nollmann, M.; Gilbert, R.; Mitchell, T.; Sferrazza, M.; Byron, O. The role of cholesterol in the activity of pneumolysin, a bacterial protein toxin. Biophys. J. 2004, 86, 3141–3151. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.G.; Wallace, K.R.; Wright, G.P. The inhibitory effects of cholesterol and related sterols on haemolysis by streptolysin O. Br. J. Exp. Pathol. 1953, 34, 174–180. [Google Scholar] [PubMed]
- Mitsui, K.; Saeki, Y.; Hase, J. Effects of cholesterol evulsion on susceptibility to perfringolysin O of human erythrocytes. Biochim. Biophys. Acta 1982, 686, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Shany, S.; Bernheimer, A.W.; Grushoff, P.S.; Kim, K.S. Evidence for membrane cholesterol as the common binding site for cereolysin, streptolysin O and saponin. Mol. Cell Biochem. 1974, 3, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Giddings, K.S.; Johnson, A.E.; Tweten, R.K. Redefining cholesterol’s role in the mechanism of the cholesterol-dependent cytolysins. Proc. Natl. Acad. Sci. USA 2003, 100, 11315–11320. [Google Scholar] [CrossRef] [PubMed]
- Giddings, K.S.; Zhao, J.; Sims, P.J.; Tweten, R.K. Human CD59 is a receptor for the cholesterol-dependent cytolysin intermedilysin. Nat. Struct. Mol. Biol. 2004, 11, 1173–1178. [Google Scholar] [CrossRef] [PubMed]
- Heuck, A.P.; Hotze, E.M.; Tweten, R.K.; Johnson, A.E. Mechanism of membrane insertion of a multimeric β-barrel protein: Perfringolysin O creates a pore using ordered and coupled conformational changes. Mol. Cell 2000, 6, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Sekino-Suzuki, N.; Nakamura, M.; Mitsui, K.I.; Ohno-Iwashita, Y. Contribution of individual tryptophan residues to the structure and activity of θ-toxin (perfringolysin O), a cholesterol-binding cytolysin. Eur. J. Biochem. 1996, 241, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sekino, N.; Iwamoto, M.; Ohno-Iwashita, Y. Interaction of θ-toxin (perfringolysin O), a cholesterol-binding cytolysin, with liposomal membranes: Change in the aromatic side chains upon binding and insertion. Biochemistry 1995, 34, 6513–6520. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Ohno-Iwashita, Y.; Ando, S. Role of the essential thiol group in the thiol-activated cytolysin from Clostridium perfringens. Eur. J. Biochem. 1987, 167, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Soltani, C.E.; Hotze, E.M.; Johnson, A.E.; Tweten, R.K. Structural elements of the cholesterol-dependent cytolysins that are responsible for their cholesterol-sensitive membrane interactions. Proc. Natl. Acad. Sci. USA 2007, 104, 20226–20231. [Google Scholar] [CrossRef]
- Iwamoto, M.; Ohno-Iwashita, Y.; Ando, S. Effect of isolated C-terminal fragment of θ-toxin (perfringolysin O) on toxin assembly and membrane lysis. Eur. J. Biochem. 1990, 194, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Dowd, K.J.; Tweten, R.K. The cholesterol-dependent cytolysin signature motif: A critical element in the allosteric pathway that couples membrane binding to pore assembly. PLoS Pathog. 2012, 8, e1002787. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Sekino-Suzuki, N.; Mitsui, K.; Ohno-Iwashita, Y. Contribution of tryptophan residues to the structural changes in perfringolysin O during interaction with liposomal membranes. J. Biochem. 1998, 123, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Farrand, A.J.; LaChapelle, S.; Hotze, E.M.; Johnson, A.E.; Tweten, R.K. Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc. Natl. Acad. Sci. USA 2010, 107, 4341–4346. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, R.; Tweten, R.K.; Johnson, A.E. The domains of a cholesterol-dependent cytolysin undergo a major FRET-detected rearrangement during pore formation. Proc. Natl. Acad. Sci. USA 2005, 102, 7139–7144. [Google Scholar] [CrossRef]
- LaChapelle, S.; Tweten, R.K.; Hotze, E.M. Intermedilysin-receptor interactions during assembly of the pore complex: Assembly intermediates increase host cell susceptibility to complement-mediated lysis. J. Biol. Chem. 2009, 284, 12719–12726. [Google Scholar] [CrossRef] [PubMed]
- Ohno-Iwashita, Y.; Shimada, Y.; Waheed, A.A.; Hayashi, M.; Inomata, M.; Nakamura, M.; Maruya, M.; Iwashita, S. Perfringolysin O, a cholesterol-binding cytolysin, as a probe for lipid rafts. Anaerobe 2004, 10, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Ohno-Iwashita, Y.; Iwamoto, M.; Ando, S.; Iwashita, S. Effect of lipidic factors on membrane cholesterol topology—Mode of binding of θ-toxin to cholesterol in liposomes. Biochim. Biophys. Acta 1992, 1109, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Maruya, M.; Iwashita, S.; Ohno-Iwashita, Y. The C-terminal domain of perfringolysin O is an essential cholesterol-binding unit targeting to cholesterol-rich microdomains. Eur. J. Biochem. 2002, 269, 6195–6203. [Google Scholar] [CrossRef] [PubMed]
- Waheed, A.A.; Shimada, Y.; Heijnen, H.F.; Nakamura, M.; Inomata, M.; Hayashi, M.; Iwashita, S.; Slot, J.W.; Ohno-Iwashita, Y. Selective binding of perfringolysin O derivative to cholesterol-rich membrane microdomains (rafts). Proc. Natl. Acad. Sci. USA 2001, 98, 4926–4931. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.D.; Johnson, A.E.; London, E. How interaction of perfringolysin O with membranes is controlled by sterol structure, lipid structure, and physiological low pH: Insights into the origin of perfringolysin O-lipid raft interaction. J. Biol. Chem. 2008, 283, 4632–4642. [Google Scholar] [CrossRef]
- Nelson, L.D.; Chiantia, S.; London, E. Perfringolysin O association with ordered lipid domains: Implications for transmembrane protein raft affinity. Biophys. J. 2010, 99, 3255–3263. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Wang, T.; Li, H.; London, E. Decreasing Transmembrane Segment Length Greatly Decreases Perfringolysin O Pore Size. J. Membr. Biol. 2015. [Google Scholar]
- Ramachandran, R.; Tweten, R.K.; Johnson, A.E. Membrane-dependent conformational changes initiate cholesterol-dependent cytolysin oligomerization and intersubunit β-strand alignment. Nat. Struct. Mol. Biol. 2004, 11, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.K.; Tweten, R.K.; Johnson, A.E. Disulfide-bond scanning reveals assembly state and β-strand tilt angle of the PFO β-barrel. Nat. Chem. Biol. 2013, 9, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Hotze, E.M.; Wilson-Kubalek, E.; Farrand, A.J.; Bentsen, L.; Parker, M.W.; Johnson, A.E.; Tweten, R.K. Monomer-monomer interactions propagate structural transitions necessary for pore formation by the cholesterol-dependent cytolysins. J. Biol. Chem. 2012, 287, 24534–24543. [Google Scholar] [CrossRef] [PubMed]
- Rossjohn, J.; Polekhina, G.; Feil, S.C.; Morton, C.J.; Tweten, R.K.; Parker, M.W. Structures of perfringolysin O suggest a pathway for activation of cholesterol-dependent cytolysins. J. Mol. Biol. 2007, 367, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Shatursky, O.; Heuck, A.P.; Shepard, L.A.; Rossjohn, J.; Parker, M.W.; Johnson, A.E.; Tweten, R.K. The mechanism of membrane insertion for a cholesterol-dependent cytolysin: A novel paradigm for pore-forming toxins. Cell 1999, 99, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Shepard, L.A.; Shatursky, O.; Johnson, A.E.; Tweten, R.K. The mechanism of pore assembly for a cholesterol-dependent cytolysin: Formation of a large prepore complex precedes the insertion of the transmembrane β-hairpins. Biochemistry 2000, 39, 10284–10293. [Google Scholar] [CrossRef] [PubMed]
- Shepard, L.A.; Heuck, A.P.; Hamman, B.D.; Rossjohn, J.; Parker, M.W.; Ryan, K.R.; Johnson, A.E.; Tweten, R.K. Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O: An α-helical to β-sheet transition identified by fluorescence spectroscopy. Biochemistry 1998, 37, 14563–14574. [Google Scholar] [CrossRef] [PubMed]
- Hotze, E.M.; Heuck, A.P.; Czajkowsky, D.M.; Shao, Z.; Johnson, A.E.; Tweten, R.K. Monomer-monomer interactions drive the prepore to pore conversion of a β-barrel-forming cholesterol-dependent cytolysin. J. Biol. Chem. 2002, 277, 11597–11605. [Google Scholar] [CrossRef] [PubMed]
- Tweten, R.K. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect. Immun. 2005, 73, 6199–6209. [Google Scholar] [CrossRef] [PubMed]
- White, S.H.; Wimley, W.C. Membrane protein folding and stability: Physical principles. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 319–365. [Google Scholar] [CrossRef] [PubMed]
- Kacprzyk-Stokowiec, A.; Kulma, M.; Traczyk, G.; Kwiatkowska, K.; Sobota, A.; Dadlez, M. Crucial role of perfringolysin O D1 domain in orchestrating structural transitions leading to membrane-perforating pores: A hydrogen-deuterium exchange study. J. Biol. Chem. 2014, 289, 28738–28752. [Google Scholar] [CrossRef] [PubMed]
- Wade, K.R.; Hotze, E.M.; Kuiper, M.J.; Morton, C.J.; Parker, M.W.; Tweten, R.K. An intermolecular electrostatic interaction controls the prepore-to-pore transition in a cholesterol-dependent cytolysin. Proc. Natl. Acad. Sci. USA 2015, 112, 2204–2209. [Google Scholar] [CrossRef] [PubMed]
- Tilley, S.J.; Orlova, E.V.; Gilbert, R.J.; Andrew, P.W.; Saibil, H.R. Structural basis of pore formation by the bacterial toxin pneumolysin. Cell 2005, 121, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Wuster, A.; Babu, M.M. Conservation and evolutionary dynamics of the agr cell-to-cell communication system across firmicutes. J. Bacteriol. 2008, 190, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, K.; Yuan, Y.; Hassan, S.; Wang, R.; Wang, Y.; Shimizu, T. Virulence gene regulation by the agr system in Clostridium perfringens. J. Bacteriol. 2009, 191, 3919–3927. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.E.; Chen, J.; Li, J.; McClane, B.A. Use of an EZ-Tn5-based random mutagenesis system to identify a novel toxin regulatory locus in Clostridium perfringens strain 13. PLoS ONE 2009, 4, e6232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schauder, S.; Shokat, K.; Surette, M.G.; Bassler, B.L. The LuxS family of bacterial autoinducers: Biosynthesis of a novel quorum-sensing signal molecule. Mol. Microbiol. 2001, 41, 463–476. [Google Scholar] [CrossRef]
- Ba-Thein, W.; Lyristis, M.; Ohtani, K.; Nisbet, I.T.; Hayashi, H.; Rood, J.I.; Shimizu, T. The virR/virS locus regulates the transcription of genes encoding extracellular toxin production in Clostridium perfringens. J. Bacteriol. 1996, 178, 2514–2520. [Google Scholar] [PubMed]
- Lyristis, M.; Bryant, A.E.; Sloan, J.; Awad, M.M.; Nisbet, I.T.; Stevens, D.L.; Rood, J.I. Identification and molecular analysis of a locus that regulates extracellular toxin production in Clostridium perfringens. Mol. Microbiol. 1994, 12, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Okumura, K.; Ohtani, K.; Hayashi, H.; Shimizu, T. Characterization of genes regulated directly by the VirR/VirS system in Clostridium perfringens. J. Bacteriol. 2008, 190, 7719–7727. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Ba-Thein, W.; Tamaki, M.; Hayashi, H. The virR gene, a member of a class of two-component response regulators, regulates the production of perfringolysin O, collagenase, and hemagglutinin in Clostridium perfringens. J. Bacteriol. 1994, 176, 1616–1623. [Google Scholar] [PubMed]
- Ohtani, K.; Hayashi, H.; Shimizu, T. The luxS gene is involved in cell-cell signalling for toxin production in Clostridium perfringens. Mol. Microbiol. 2002, 44, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.K.; Awad, M.M.; McGowan, S.; Rood, J.I. Functional analysis of the VirSR phosphorelay from Clostridium perfringens. PLoS ONE 2009, 4, e5849. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.K.; Rood, J.I. The VirR response regulator from Clostridium perfringens binds independently to two imperfect direct repeats located upstream of the pfoA promoter. J. Bacteriol. 2000, 182, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.K.; Dupuy, B.; Deveson, D.S.; Rood, J.I. The spatial organization of the VirR boxes is critical for VirR-mediated expression of the perfringolysin O gene, pfoA, from Clostridium perfringens. J. Bacteriol. 2004, 186, 3321–3330. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.M.; Rood, J.I. Perfringolysin O expression in Clostridium perfringens is independent of the upstream pfoR gene. J. Bacteriol. 2002, 184, 2034–2038. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, M.; Falkow, S. The Prokaryotes: A Handbook on the Biology of Bacteria, 3rd ed.; Springer: New York, NY, USA, 2006; pp. 698–770. [Google Scholar]
- Titball, R.W. Gas gangrene: An open and closed case. Microbiology 2005, 151, 2821–2828. [Google Scholar] [CrossRef] [PubMed]
- Bryant, A.E.; Stevens, D.L. Clostridial myonecrosis: New insights in pathogenesis and management. Curr. Infect. Dis. Rep. 2010, 12, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Beran, G.W. Handbook of Zoonoses, 2nd ed.; CRC Press: Boca Raton, FL, USA, 1994; pp. 127–138. [Google Scholar]
- Ninomiya, M.; Matsushita, O.; Minami, J.; Sakamoto, H.; Nakano, M.; Okabe, A. Role of α-toxin in Clostridium perfringens infection determined by using recombinants of C. perfringens and Bacillus subtilis. Infect. Immun. 1994, 62, 5032–5039. [Google Scholar]
- Awad, M.M.; Bryant, A.E.; Stevens, D.L.; Rood, J.I. Virulence studies on chromosomal α-toxin and θ-toxin mutants constructed by allelic exchange provide genetic evidence for the essential role of α-toxin in Clostridium perfringens-mediated gas gangrene. Mol. Microbiol. 1995, 15, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Ellemor, D.M.; Baird, R.N.; Awad, M.M.; Boyd, R.L.; Rood, J.I.; Emmins, J.J. Use of genetically manipulated strains of Clostridium perfringens reveals that both α-toxin and θ-toxin are required for vascular leukostasis to occur in experimental gas gangrene. Infect. Immun. 1999, 67, 4902–4907. [Google Scholar] [PubMed]
- Stevens, D.L.; Tweten, R.K.; Awad, M.M.; Rood, J.I.; Bryant, A.E. Clostridial gas gangrene: Evidence that α and θ toxins differentially modulate the immune response and induce acute tissue necrosis. J. Infect. Dis. 1997, 176, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.L.; Bryant, A.E. Pathogenesis of Clostridium perfringens infection: Mechanisms and mediators of shock. Clin. Infect. Dis. 1997, 25 (Suppl. 2), S160–S164. [Google Scholar] [CrossRef]
- Asmuth, D.M.; Olson, R.D.; Hackett, S.P.; Bryant, A.E.; Tweten, R.K.; Tso, J.Y.; Zollman, T.; Stevens, D.L. Effects of Clostridium perfringens recombinant and crude phospholipase C and θ-toxin on rabbit hemodynamic parameters. J. Infect. Dis. 1995, 172, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Bryant, A.E.; Bergstrom, R.; Zimmerman, G.A.; Salyer, J.L.; Hill, H.R.; Tweten, R.K.; Sato, H.; Stevens, D.L. Clostridium perfringens invasiveness is enhanced by effects of θ toxin upon PMNL structure and function: The roles of leukocytotoxicity and expression of CD11/CD18 adherence glycoprotein. FEMS Immunol. Med. Microbiol. 1993, 7, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Bryant, A.E.; Chen, R.Y.; Nagata, Y.; Wang, Y.; Lee, C.H.; Finegold, S.; Guth, P.H.; Stevens, D.L. Clostridial gas gangrene. I. Cellular and molecular mechanisms of microvascular dysfunction induced by exotoxins of Clostridium perfringens. J. Infect. Dis. 2000, 182, 799–807. [Google Scholar]
- Hickey, M.J.; Kwan, R.Y.; Awad, M.M.; Kennedy, C.L.; Young, L.F.; Hall, P.; Cordner, L.M.; Lyras, D.; Emmins, J.J.; Rood, J.I. Molecular and cellular basis of microvascular perfusion deficits induced by Clostridium perfringens and Clostridium septicum. PLoS Pathog. 2008, 4, e1000045. [Google Scholar] [CrossRef] [PubMed]
- Bryant, A.E.; Chen, R.Y.; Nagata, Y.; Wang, Y.; Lee, C.H.; Finegold, S.; Guth, P.H.; Stevens, D.L. Clostridial gas gangrene. II. Phospholipase C-induced activation of platelet gpIIbIIIa mediates vascular occlusion and myonecrosis in Clostridium perfringens gas gangrene. J. Infect. Dis. 2000, 182, 808–815. [Google Scholar]
- Bryant, A.E.; Bayer, C.R.; Hayes-Schroer, S.M.; Stevens, D.L. Activation of platelet gpIIbIIIa by phospholipase C from Clostridium perfringens involves store-operated calcium entry. J. Infect. Dis. 2003, 187, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Bryant, A.E.; Bayer, C.R.; Aldape, M.J.; Wallace, R.J.; Titball, R.W.; Stevens, D.L. Clostridium perfringens phospholipase C-induced platelet/leukocyte interactions impede neutrophil diapedesis. J. Med. Microbiol. 2006, 55, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.L.; Troyer, B.E.; Merrick, D.T.; Mitten, J.E.; Olson, R.D. Lethal effects and cardiovascular effects of purified α- and θ-toxins from Clostridium perfringens. J. Infect. Dis. 1988, 157, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Bryant, A.E.; Stevens, D.L. Phospholipase C and perfringolysin O from Clostridium perfringens upregulate endothelial cell-leukocyte adherence molecule 1 and intercellular leukocyte adherence molecule 1 expression and induce interleukin-8 synthesis in cultured human umbilical vein endothelial cells. Infect. Immun. 1996, 64, 358–362. [Google Scholar] [PubMed]
- Whatley, R.E.; Nelson, P.; Zimmerman, G.A.; Stevens, D.L.; Parker, C.J.; McIntyre, T.M.; Prescott, S.M. The regulation of platelet-activating factor production in endothelial cells. The role of calcium and protein kinase C. J. Biol. Chem. 1989, 264, 6325–6333. [Google Scholar]
- Bunting, M.; Lorant, D.E.; Bryant, A.E.; Zimmerman, G.A.; McIntyre, T.M.; Stevens, D.L.; Prescott, S.M. α toxin from Clostridium perfringens induces proinflammatory changes in endothelial cells. J. Clin. Invest. 1997, 100, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.L.; Bryant, A.E. The role of clostridial toxins in the pathogenesis of gas gangrene. Clin. Infect. Dis. 2002, 35, S93–S100. [Google Scholar] [CrossRef] [PubMed]
- Stevens, D.L.; Mitten, J.; Henry, C. Effects of α and θ toxins from Clostridium perfringens on human polymorphonuclear leukocytes. J. Infect. Dis. 1987, 156, 324–333. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, D.K.; Melville, S.B. The anaerobic pathogen Clostridium perfringens can escape the phagosome of macrophages under aerobic conditions. Cell. Microbiol. 2000, 2, 505–519. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, D.K.; Melville, S.B. Effects of Clostridium perfringens α-toxin (PLC) and perfringolysin O (PFO) on cytotoxicity to macrophages, on escape from the phagosomes of macrophages, and on persistence of C. perfringens in host tissues. Infect. Immun. 2004, 72, 5204–5215. [Google Scholar] [CrossRef]
- Sayeed, S.; Uzal, F.A.; Fisher, D.J.; Saputo, J.; Vidal, J.E.; Chen, Y.; Gupta, P.; Rood, J.I.; McClane, B.A. β toxin is essential for the intestinal virulence of Clostridium perfringens type C disease isolate CN3685 in a rabbit ileal loop model. Mol. Microbiol. 2008, 67, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Valgaeren, B.R.; Pardon, B.; Verherstraeten, S.; Goossens, E.; Timbermont, L.; Haesebrouck, F.; Ducatelle, R.; Deprez, P.R.; van Immerseel, F. Intestinal clostridial counts have no diagnostic value in the diagnosis of enterotoxaemia in veal calves. Vet. Rec. 2013, 172, 237. [Google Scholar] [CrossRef] [PubMed]
- Manteca, C.; Daube, G.; Pirson, V.; Limbourg, B.; Kaeckenbeeck, A.; Mainil, J.G. Bacterial intestinal flora associated with enterotoxaemia in Belgian Blue calves. Vet. Microbiol. 2001, 81, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Muylaert, A.; Lebrun, M.; Duprez, J.N.; Labrozzo, S.; Theys, H.; Taminiau, B.; Mainil, J. Enterotoxaemia-like syndrome and Clostridium perfringens in veal calves. Vet. Rec. 2010, 167, 64–65. [Google Scholar] [CrossRef] [PubMed]
- Pardon, B.; De Bleecker, K.; Hostens, M.; Callens, J.; Dewulf, J.; Deprez, P. Longitudinal study on morbidity and mortality in white veal calves in Belgium. BMC Vet. Res. 2012, 8, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebrun, M.; Mainil, J.G.; Linden, A. Cattle enterotoxaemia and Clostridium perfringens: Description, diagnosis and prophylaxis. Vet. Rec. 2010, 167, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Morris, W.E.; Venzano, A.J.; Elizondo, A.; Vilte, D.A.; Mercado, E.C.; Fernandez-Miyakawa, M.E. Necrotic enteritis in young calves. J. Vet. Diagn. Invest. 2011, 23, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Valgaeren, B.; Pardon, B.; Goossens, E.; Verherstraeten, S.; Schauvliege, S.; Timbermont, L.; Ducatelle, R.; Deprez, P.; van Immerseel, F. Lesion development in a new intestinal loop model indicates the involvement of a shared Clostridium perfringens virulence factor in haemorrhagic enteritis in calves. J. Comp. Pathol. 2013, 149, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, J.; Nomura, S.; Fujii, Y.; Oshita, Y. Effect of Clostridium perfringens α toxin on the isolated rat vas deferens. Toxicon 1985, 23, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A.; Songer, J.G. Diagnosis of Clostridium perfringens intestinal infections in sheep and goats. J. Vet. Diagn. Invest. 2008, 20, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, S.; Fernandez-Miyakawa, M.E.; Fisher, D.J.; Adams, V.; Poon, R.; Rood, J.I.; Uzal, F.A.; McClane, B.A. ε-toxin is required for most Clostridium perfringens type D vegetative culture supernatants to cause lethality in the mouse intravenous injection model. Infect. Immun. 2005, 73, 7413–7421. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.P.; Adams, V.; Beingesser, J.; Hughes, M.L.; Poon, R.; Lyras, D.; Hill, A.; McClane, B.A.; Rood, J.I.; Uzal, F.A. ε toxin is essential for the virulence of Clostridium perfringens type D infection in sheep, goats, and mice. Infect. Immun. 2013, 81, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Shewell, L.K.; Harvey, R.M.; Higgins, M.A.; Day, C.J.; Hartley-Tassell, L.E.; Chen, A.Y.; Gillen, C.M.; James, D.B.; Alonzo, F., 3rd; Torres, V.J.; et al. The cholesterol-dependent cytolysins pneumolysin and streptolysin O require binding to red blood cell glycans for hemolytic activity. Proc. Natl. Acad. Sci. USA 2014, 111, E5312–E5320. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verherstraeten, S.; Goossens, E.; Valgaeren, B.; Pardon, B.; Timbermont, L.; Haesebrouck, F.; Ducatelle, R.; Deprez, P.; Wade, K.R.; Tweten, R.; et al. Perfringolysin O: The Underrated Clostridium perfringens Toxin? Toxins 2015, 7, 1702-1721. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7051702
Verherstraeten S, Goossens E, Valgaeren B, Pardon B, Timbermont L, Haesebrouck F, Ducatelle R, Deprez P, Wade KR, Tweten R, et al. Perfringolysin O: The Underrated Clostridium perfringens Toxin? Toxins. 2015; 7(5):1702-1721. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7051702
Chicago/Turabian StyleVerherstraeten, Stefanie, Evy Goossens, Bonnie Valgaeren, Bart Pardon, Leen Timbermont, Freddy Haesebrouck, Richard Ducatelle, Piet Deprez, Kristin R. Wade, Rodney Tweten, and et al. 2015. "Perfringolysin O: The Underrated Clostridium perfringens Toxin?" Toxins 7, no. 5: 1702-1721. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins7051702