
Toxin-Antitoxin Modules Are Pliable Switches Activated by Multiple Protease Pathways


Abstract
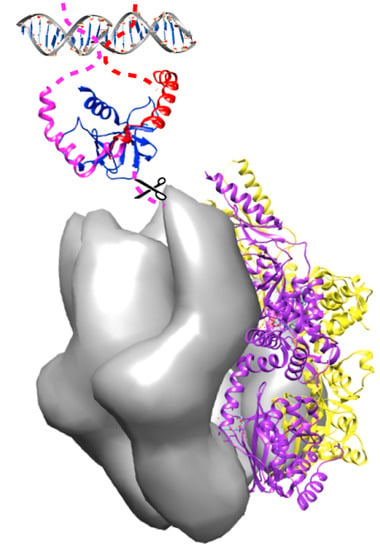
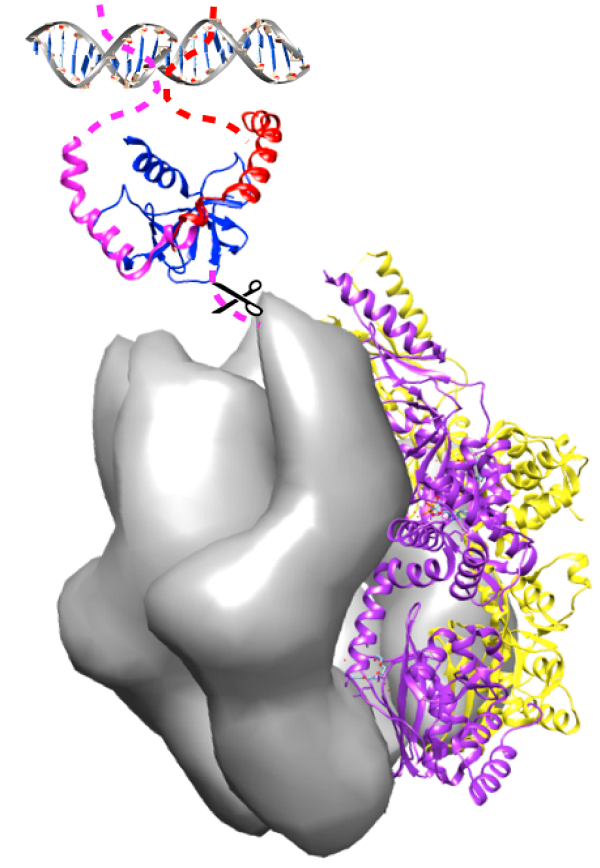
:
1. Introduction
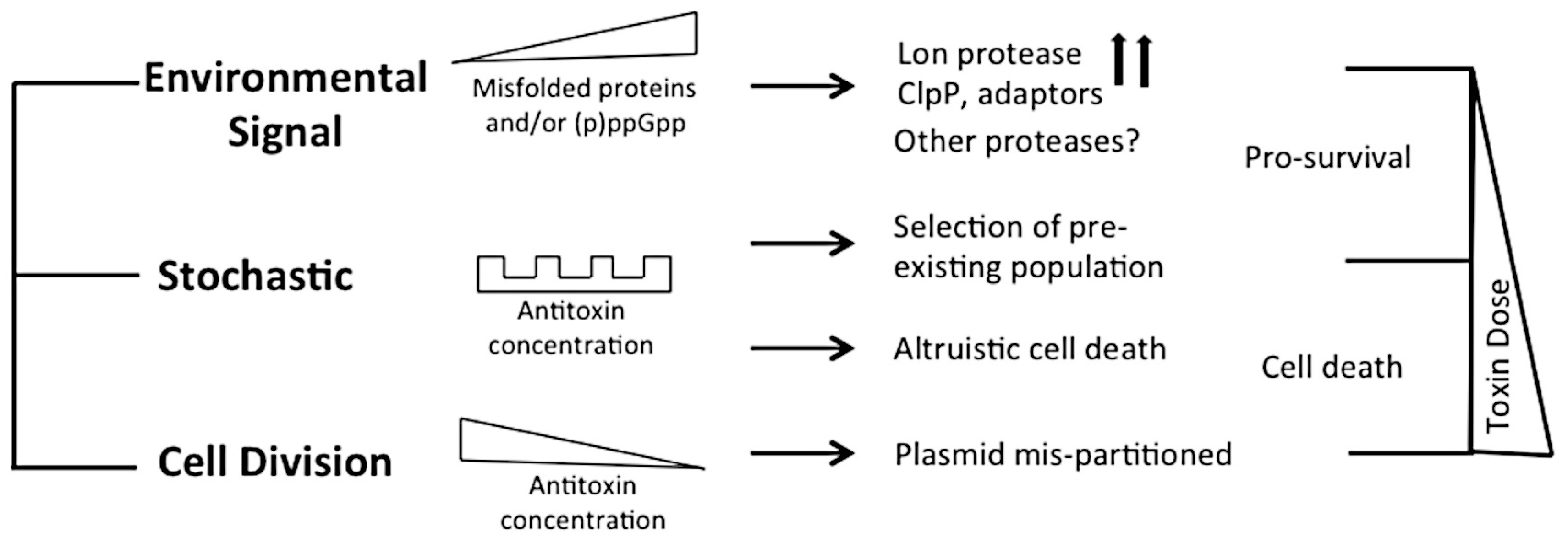
2. Activation and Phenotypic Outcomes of TA Modules
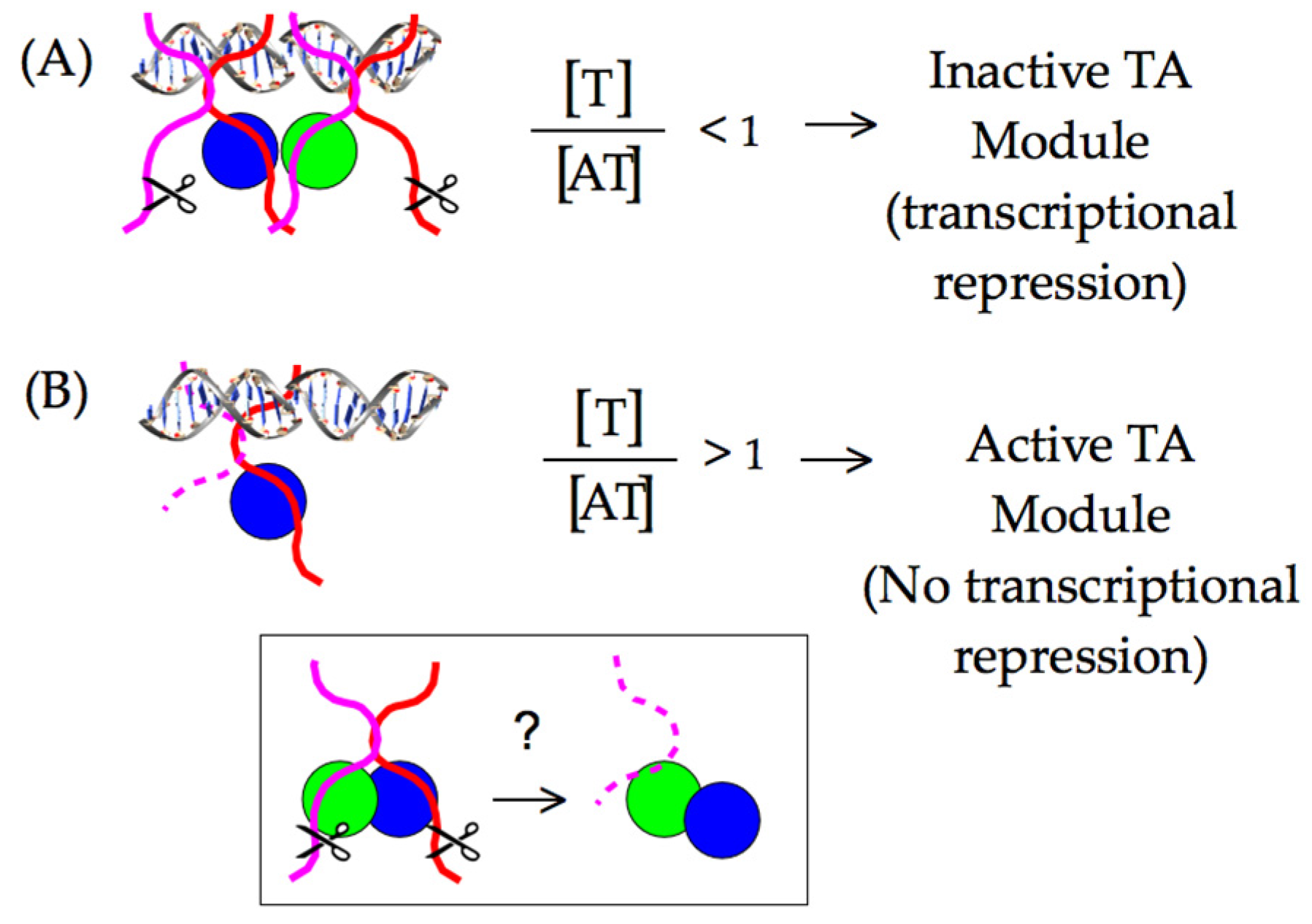
3. Proteases Shape the TA Module Proteome
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res. 2013, 41, 4360–4377. [Google Scholar] [CrossRef] [PubMed]
- Van Melderen, L.; Saavedra de Bast, M. Bacterial toxin-antitoxin systems: More than selfish entities? PLoS Genet. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Park, J.H.; Inouye, M. Toxin-antitoxin systems in bacteria and archaea. Annu. Rev. Genet. 2011, 45, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.T.; Balsa, D.; Espinosa, M. One cannot rule them all: Are bacterial toxins-antitoxins druggable? FEMS Microbiol. Rev. 2015, 39, 522–540. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.K. Combatting bacterial persister cells. Biotechnol. Bioeng. 2016, 113, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Hayes, F.; Kedzierska, B. Regulating toxin-antitoxin expression: Controlled detonation of intracellular molecular timebombs. Toxins 2014, 6, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, S.J.; Poppenberger, B.; Rozhon, W. Toxin-antitoxin systems: Biology, identification, and application. Mob. Genet. Elem. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Brzozowska, I.; Zielenkiewicz, U. Regulation of toxin-antitoxin systems by proteolysis. Plasmid 2013, 70, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.P.; Gerdes, K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005, 33, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Buts, L.; Lah, J.; Dao-Thi, M.H.; Wyns, L.; Loris, R. Toxin-antitoxin modules as bacterial metabolic stress managers. Trends Biochem. Sci. 2005, 30, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Christensen, S.K.; Lobner-Olesen, A. Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 2005, 3, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wood, T.K. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 2011, 77, 5577–5583. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Maisonneuve, E. Bacterial persistence and toxin-antitoxin loci. Annu. Rev. Microbiol. 2012, 66, 103–123. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.T.; Espinosa, M.; Yeo, C.C. Keeping the wolves at bay: Antitoxins of prokaryotic type II toxin-antitoxin systems. Front. Mol. Biosci. 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Page, R.; Peti, W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat. Chem. Biol. 2016, 12, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Rocker, A.; Meinhart, A. Type II toxin: Antitoxin systems. More than small selfish entities? Curr. Genet. 2016, 62, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Schuster, C.F.; Bertram, R. Toxin-antitoxin systems of Staphylococcus aureus. Toxins 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Loris, R.; Garcia-Pino, A. Disorder- and dynamics-based regulatory mechanisms in toxin-antitoxin modules. Chem. Rev. 2014, 114, 6933–6947. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K. Toxin-antitoxin modules may regulate synthesis of macromolecules during nutritional stress. J. Bacteriol. 2000, 182, 561–572. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Kasari, V.; Mets, T.; Tenson, T.; Kaldalu, N. Transcriptional cross-activation between toxin-antitoxin systems of Escherichia coli. BMC Microbiol. 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Boggild, A.; Sofos, N.; Andersen, K.R.; Feddersen, A.; Easter, A.D.; Passmore, L.A.; Brodersen, D.E. The crystal structure of the intact E. coli RelBE toxin-antitoxin complex provides the structural basis for conditional cooperativity. Structure 2012, 20, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Dalton, K.M.; Crosson, S. A conserved mode of protein recognition and binding in a ParD-ParE toxin-antitoxin complex. Biochemistry 2010, 49, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Son, W.S.; Lee, B.J. Structural overview of toxin-antitoxin systems in infectious bacteria: A target for developing antimicrobial agents. Biochim. Biophys. Acta 2013, 1834, 1155–1167. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.M.; Cruz, J.W.; Ouyang, M.; Woychik, N.A. Bacterial toxin RelE mediates frequent codon-independent mrna cleavage from the 5′ end of coding regions in vivo. J. Biol. Chem. 2011, 286, 14770–14778. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, M.; Austin, S. Prevalence and significance of plasmid maintenance functions in the virulence plasmids of pathogenic bacteria. Infect. Immun. 2011, 79, 2502–2509. [Google Scholar] [CrossRef] [PubMed]
- Grady, R.; Hayes, F. Axe-Txe, a broad-spectrum proteic toxin-antitoxin system specified by a multidrug-resistant, clinical isolate of Enterococcus faecium. Mol. Microbiol. 2003, 47, 1419–1432. [Google Scholar] [CrossRef] [PubMed]
- Kamada, K.; Hanaoka, F. Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin. Mol. Cell 2005, 19, 497–509. [Google Scholar] [CrossRef] [PubMed]
- Bertram, R.; Schuster, C.F. Post-transcriptional regulation of gene expression in bacterial pathogens by toxin-antitoxin systems. Front. Cell. Infect. Microbiol. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, E.; Shakespeare, L.J.; Jorgensen, M.G.; Gerdes, K. Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. USA 2011, 108, 13206–13211. [Google Scholar] [CrossRef] [PubMed]
- Engelberg-Kulka, H.; Amitai, S.; Kolodkin-Gal, I.; Hazan, R. Bacterial programmed cell death and multicellular behavior in bacteria. PLoS Genet. 2006, 2, 1518–1526. [Google Scholar] [CrossRef] [PubMed]
- Erental, A.; Kalderon, Z.; Saada, A.; Smith, Y.; Engelberg-Kulka, H. Apoptosis-like death, an extreme SOS response in Escherichia coli. mBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Erental, A.; Sharon, I.; Engelberg-Kulka, H. Two programmed cell death systems in Escherichia coli: An apoptotic-like death is inhibited by the mazEF-mediated death pathway. PLoS Biol. 2012, 10. [Google Scholar] [CrossRef] [PubMed]
- Bernard, P.; Couturier, M. Cell killing by the F plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. J. Mol. Biol. 1992, 226, 735–745. [Google Scholar] [CrossRef]
- Jensen, R.B.; Grohmann, E.; Schwab, H.; Diaz-Orejas, R.; Gerdes, K. Comparison of ccd of F, parDE of RP4, and parD of R1 using a novel conditional replication control system of plasmid R1. Mol. Microbiol. 1995, 17, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Kolodkin-Gal, I.; Verdiger, R.; Shlosberg-Fedida, A.; Engelberg-Kulka, H. A differential effect of E. coli toxin-antitoxin systems on cell death in liquid media and biofilm formation. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Lehnherr, H.; Yarmolinsky, M.B. Addiction protein Phd of plasmid prophage P1 is a substrate of the ClpXP serine protease of Escherichia coli. Proc. Natl. Acad. Sci. USA 1995, 92, 3274–3277. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.C.; Strom, A.R.; Helinski, D.R. The parDE operon of the broad-host-range plasmid RK2 specifies growth inhibition associated with plasmid loss. J. Mol. Biol. 1994, 237, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.S.; Rawlings, D.E. The poison-antidote stability system of the broad-host-range Thiobacillus ferrooxidans plasmid Ptf-Fc2. Mol. Microbiol. 1997, 26, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Yamaichi, Y.; Waldor, M.K. The three Vibrio cholerae chromosome II-encoded ParE toxins degrade chromosome I following loss of chromosome II. J. Bacteriol. 2011, 193, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Holden, D.W. Microbiology. Persisters unmasked. Science 2015, 347, 30–32. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Benedik, M.J.; Wood, T.K. Orphan toxin OrtT (YdcX) of Escherichia coli reduces growth during the stringent response. Toxins 2015, 7, 299–321. [Google Scholar] [CrossRef] [PubMed]
- Keren, I.; Mulcahy, L.R.; Lewis, K. Persister eradication: Lessons from the world of natural products. Methods Enzymol. 2012, 517, 387–406. [Google Scholar] [PubMed]
- Kim, Y.; Wang, X.; Zhang, X.S.; Grigoriu, S.; Page, R.; Peti, W.; Wood, T.K. Escherichia coli toxin/antitoxin pair MqsR/MqsA regulate toxin CspD. Environ. Microbiol. 2010, 12, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, E.; Castro-Camargo, M.; Gerdes, K. (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 2013, 154, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.Q. Persistence: Mechanisms for triggering and enhancing phenotypic variability. Curr. Opin. Genet. Dev. 2011, 21, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial persistence as a phenotypic switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef] [PubMed]
- Fasani, R.A.; Savageau, M.A. Molecular mechanisms of multiple toxin-antitoxin systems are coordinated to govern the persister phenotype. Proc. Natl. Acad. Sci. USA 2013, 110, E2528–E2537. [Google Scholar] [CrossRef] [PubMed]
- Cheverton, A.M.; Gollan, B.; Przydacz, M.; Wong, C.T.; Mylona, A.; Hare, S.A.; Helaine, S. A salmonella toxin promotes persister formation through acetylation of tRNA. Mol. Cell 2016, in press. [Google Scholar]
- Allison, K.R.; Brynildsen, M.P.; Collins, J.J. Heterogeneous bacterial persisters and engineering approaches to eliminate them. Curr. Opin. Microbiol. 2011, 14, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, B.; Nair, R.; Bermejo-Rodriguez, C.; Preston, M.A.; Agu, C.A.; Wang, X.; Bernal, J.A.; Sherratt, D.J.; de la Cueva-Mendez, G. Toxin Kid uncouples DNA replication and cell division to enforce retention of plasmid R1 in Escherichia coli cells. Proc. Natl. Acad. Sci. USA 2014, 111, 2734–2739. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.J.; Jakob, U. Oxidative stress protection by polyphosphate-new roles for an old player. Curr. Opin. Microbiol. 2015, 24C, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kint, C.I.; Verstraeten, N.; Fauvart, M.; Michiels, J. New-found fundamentals of bacterial persistence. Trends Microbiol. 2012, 20, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Persister cells. Annu. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.K.; Knabel, S.J.; Kwan, B.W. Bacterial persister cell formation and dormancy. Appl. Environ. Microbiol. 2013, 79, 7116–7121. [Google Scholar] [CrossRef] [PubMed]
- Gefen, O.; Gabay, C.; Mumcuoglu, M.; Engel, G.; Balaban, N.Q. Single-cell protein induction dynamics reveals a period of vulnerability to antibiotics in persister bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 6145–6149. [Google Scholar] [CrossRef] [PubMed]
- Vogwill, T.; Comfort, A.C.; Furio, V.; MaClean, R.C. Persistence and resistance as complementary bacterial adaptations to antibiotics. J. Evol. Biol. 2016, 29, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Hofsteenge, N.; van Nimwegen, E.; Silander, O.K. Quantitative analysis of persister fractions suggests different mechanisms of formation among environmental isolates of E. coli. BMC Microbiol. 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Keren, I.; Kaldalu, N.; Spoering, A.; Wang, Y.; Lewis, K. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 2004, 230, 13–18. [Google Scholar] [CrossRef]
- Vazquez-Laslop, N.; Lee, H.; Neyfakh, A.A. Increased persistence in Escherichia coli caused by controlled expression of toxins or other unrelated proteins. J. Bacteriol. 2006, 188, 3494–3497. [Google Scholar] [CrossRef] [PubMed]
- Kwan, B.W.; Valenta, J.A.; Benedik, M.J.; Wood, T.K. Arrested protein synthesis increases persister-like cell formation. Antimicrob. Agents Chemother. 2013, 57, 1468–1473. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Maisonneuve, E. Remarkable functional convergence: Alarmone ppGpp mediates persistence by activating type I and II toxin-antitoxins. Mol. Cell 2015, 59, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Meredith, H.R.; Srimani, J.K.; Lee, A.J.; Lopatkin, A.J.; You, L. Collective antibiotic tolerance: Mechanisms, dynamics and intervention. Nat. Chem. Biol. 2015, 11, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Fasani, R.A.; Savageau, M.A. Unrelated toxin-antitoxin systems cooperate to induce persistence. J. R. Soc. Interface 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Rotem, E.; Loinger, A.; Ronin, I.; Levin-Reisman, I.; Gabay, C.; Shoresh, N.; Biham, O.; Balaban, N.Q. Regulation of phenotypic variability by a threshold-based mechanism underlies bacterial persistence. Proc. Natl. Acad. Sci. USA 2010, 107, 12541–12546. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, N.; Knapen, W.J.; Kint, C.I.; Liebens, V.; van den Bergh, B.; Dewachter, L.; Michiels, J.E.; Fu, Q.; David, C.C.; Fierro, A.C.; et al. Obg and membrane depolarization are part of a microbial bet-hedging strategy that leads to antibiotic tolerance. Mol. Cell 2015, 59, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Corrigan, R.M.; Bellows, L.E.; Wood, A.; Grundling, A. ppGpp negatively impacts ribosome assembly affecting growth and antimicrobial tolerance in gram-positive bacteria. Proc. Natl. Acad. Sci. USA 2016, 113, E1710–E1719. [Google Scholar] [CrossRef] [PubMed]
- Armalyte, J.; Jurenaite, M.; Beinoraviciute, G.; Teiserskas, J.; Suziedeliene, E. Characterization of Escherichia coli DinJ-YafQ toxin-antitoxin system using insights from mutagenesis data. J. Bacteriol. 2012, 194, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.J.; Wade, W.D.; Akierman, S.; Vacchi-Suzzi, C.; Stremick, C.A.; Turner, R.J.; Ceri, H. The chromosomal toxin gene YafQ is a determinant of multidrug tolerance for Escherichia coli growing in a biofilm. Antimicrob. Agents Chemother. 2009, 53, 2253–2258. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.N.; Morozov, A.; Planzos, P.; Zelaya, H.M. The fatty acid signaling molecule cis-2-decenoic acid increases metabolic activity and reverts persister cells to an antimicrobial-susceptible state. Appl. Environ. Microbiol. 2014, 80, 6976–6991. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Bahar, A.A.; Syed, H.; Ren, D. Reverting antibiotic tolerance of Pseudomonas aeruginosa PAO1 persister cells by (z)-4-bromo-5-(bromomethylene)-3-methylfuran-2(5h)-one. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.A.; VanBogelen, R.A.; Neidhardt, F.C. Lon gene product of Escherichia coli is a heat-shock protein. J. Bacteriol. 1984, 159, 283–287. [Google Scholar] [PubMed]
- Breidenstein, E.B.; Hancock, R.E. Armand-frappier outstanding student award—Role of ATP-dependent proteases in antibiotic resistance and virulence. Can. J. Microbiol. 2013, 59, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jonas, K.; Liu, J.; Chien, P.; Laub, M.T. Proteotoxic stress induces a cell-cycle arrest by stimulating Lon to degrade the replication initiator dnaA. Cell 2013, 154, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Valdez-Cruz, N.A.; Ramirez, O.T.; Trujillo-Roldan, M.A. Molecular responses of Escherichia coli caused by heat stress and recombinant protein production during temperature induction. Bioeng. Bugs 2011, 2, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Cortes, T.; Schubert, O.T.; Rose, G.; Arnvig, K.B.; Comas, I.; Aebersold, R.; Young, D.B. Genome-wide mapping of transcriptional start sites defines an extensive leaderless transcriptome in mycobacterium tuberculosis. Cell Rep. 2013, 5, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Engelberg-Kulka, H.; Sat, B.; Reches, M.; Amitai, S.; Hazan, R. Bacterial programmed cell death systems as targets for antibiotics. Trends microbiol. 2004, 12, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Korch, S.B.; Hill, T.M. Ectopic overexpression of wild-type and mutant Hipa genes in Escherichia coli: Effects on macromolecular synthesis and persister formation. J. Bacteriol. 2006, 188, 3826–3836. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.; Zhang, Z.; Khodursky, A.; Kaldalu, N.; Kurg, K.; Lewis, K. Persisters: A distinct physiological state of E. coli. BMC Microbiol. 2006, 6. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; McCarthy, D.J.; Chen, Y.; Okoniewski, M.; Smyth, G.K.; Huber, W.; Robinson, M.D. Count-based differential expression analysis of RNA sequencing data using R and bioconductor. Nat. Protoc. 2013, 8, 1765–1786. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Soheili, S.; Ghafourian, S.; Sekawi, Z.; Neela, V.K.; Sadeghifard, N.; Taherikalani, M.; Khosravi, A.; Ramli, R.; Hamat, R.A. The MazEF toxin-antitoxin system as an attractive target in clinical isolates of Enterococcus faecium and Enterococcus faecalis. Drug Des. Dev. Ther. 2015, 9, 2553–2561. [Google Scholar]
- Singh, R.; Jiang, X. Expression of stress and virulence genes in Escherichia coli O157:H7 heat shocked in fresh dairy compost. J. Food Prot. 2015, 78, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Van Melderen, L.; Aertsen, A. Regulation and quality control by Lon-dependent proteolysis. Res. Microbiol. 2009, 160, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Goeders, N.; Van Melderen, L. Toxin-antitoxin systems as multilevel interaction systems. Toxins 2014, 6, 304–324. [Google Scholar] [CrossRef] [PubMed]
- Drobnak, I.; De Jonge, N.; Haesaerts, S.; Vesnaver, G.; Loris, R.; Lah, J. Energetic basis of uncoupling folding from binding for an intrinsically disordered protein. J. Am. Chem. Soc. 2013, 135, 1288–1294. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, S. Proteases and their tagets in Escherichia coli. Annu. Rev. Genet. 1996, 30, 465–506. [Google Scholar] [CrossRef] [PubMed]
- Narberhaus, F.; Obrist, M.; Fuhrer, F.; Langklotz, S. Degradation of cytoplasmic substrates by FtsH, a membrane-anchored protease with many talents. Res. Microbiol. 2009, 160, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Olivares, A.O.; Baker, T.A.; Sauer, R.T. Mechanistic insights into bacterial AAA+ proteases and protein-remodelling machines. Nat. Rev. Microbiol. 2016, 14, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Sauer, R.T.; Baker, T.A. AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 2011, 80, 587–612. [Google Scholar] [CrossRef] [PubMed]
- Schilling, O.; Overall, C.M. Proteomic discovery of protease substrates. Curr. Opin. Chem. Biol. 2007, 11, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Pruteanu, M.; Baker, T.A. Proteolysis in the SOS response and metal homeostasis in Escherichia coli. Res. Microbiol. 2009, 160, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Katz, C.; Rasouly, A.; Gur, E.; Shenhar, Y.; Biran, D.; Ron, E.Z. Temperature-dependent proteolysis as a control element in Escherichia coli metabolism. Res. Microbiol. 2009, 160, 684–686. [Google Scholar] [CrossRef] [PubMed]
- Neher, S.B.; Villen, J.; Oakes, E.C.; Bakalarski, C.E.; Sauer, R.T.; Gygi, S.P.; Baker, T.A. Proteomic profiling of ClpXP substrates after DNA damage reveals extensive instability within SOS regulon. Mol. Cell 2006, 22, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Donegan, N.P.; Thompson, E.T.; Fu, Z.; Cheung, A.L. Proteolytic regulation of toxin-antitoxin systems by ClpCP in staphylococcus aureus. J. Bacteriol. 2010, 192, 1416–1422. [Google Scholar] [CrossRef] [PubMed]
- Donegan, N.P.; Marvin, J.S.; Cheung, A.L. Role of adaptor Trfa and ClpPC in controlling levels of SsrA-tagged proteins and antitoxins in Staphylococcus aureus. J. Bacteriol. 2014, 196, 4140–4151. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.; Frank, E.G.; Levine, A.S.; Woodgate, R. Lon-mediated proteolysis of the Escherichia coli UmuD mutagenesis protein: In vitro degradation and identification of residues required for proteolysis. Genes Dev. 1998, 12, 3889–3899. [Google Scholar] [CrossRef] [PubMed]
- Gur, E. The Lon AAA+ protease. Subcell Biochem. 2013, 66, 35–51. [Google Scholar] [PubMed]
- Flynn, J.M.; Neher, S.B.; Kim, Y.I.; Sauer, R.T.; Baker, T.A. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol. Cell 2003, 11, 671–683. [Google Scholar] [CrossRef]
- Shao, Y.; Harrison, E.M.; Bi, D.; Tai, C.; He, X.; Ou, H.Y.; Rajakumar, K.; Deng, Z. Tadb: A web-based resource for Type 2 toxin-antitoxin loci in bacteria and archaea. Nucleic Acids Res. 2011, 39, D606–D611. [Google Scholar] [CrossRef] [PubMed]
- Brzozowska, I.; Zielenkiewicz, U. The ClpXP protease is responsible for the degradation of the epsilon antidote to the zeta toxin of the streptococcal pSM19035 plasmid. J. Biol. Chem. 2014, 289, 7514–7523. [Google Scholar] [CrossRef] [PubMed]
- Van Melderen, L.; Thi, M.H.; Lecchi, P.; Gottesman, S.; Couturier, M.; Maurizi, M.R. ATP-dependent degradation of CcdA by Lon protease. Effects of secondary structure and heterologous subunit interactions. J. Biol. Chem. 1996, 271, 27730–27738. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.K.; Gerdes, K. Delayed-relaxed response explained by hyperactivation of rele. Mol. Microbiol. 2004, 53, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Janssen, B.D.; Garza-Sanchez, F.; Hayes, C.S. YoeB toxin is activated during thermal stress. Microbiologyopen 2015, 4, 682–697. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.K.; Maenhaut-Michel, G.; Mine, N.; Gottesman, S.; Gerdes, K.; Van Melderen, L. Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: Involvement of the YefM-YoeB toxin-antitoxin system. Mol. Microbiol. 2004, 51, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, M.; Borch, J.; Gerdes, K. RelB and RelEof Escherichia coli form a tight complex that represses transcription via the ribbon-helix-helix motif in RelB. J. Mol. Biol. 2009, 394, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Prysak, M.H.; Mozdzierz, C.J.; Cook, A.M.; Zhu, L.; Zhang, Y.; Inouye, M.; Woychik, N.A. Bacterial toxin YafQ is an endoribonuclease that associates with the ribosome and blocks translation elongation through sequence-specific and frame-dependent mRNA cleavage. Mol. Microbiol. 2009, 71, 1071–1087. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.S.; Rawlings, D.E. Efficiency of the pTF-Fc2 pas poison-antidote stability system in Escherichia coli is affected by the host strain, and antidote degradation requires the Lon protease. J. Bacteriol. 1998, 180, 5458–5462. [Google Scholar] [PubMed]
- Winther, K.S.; Gerdes, K. Regulation of enteric VapBC transcription: Induction by VapC toxin dimer-breaking. Nucleic Acids Res. 2012, 40, 4347–4357. [Google Scholar] [CrossRef] [PubMed]
- Aizenman, E.; Engelberg-Kulka, H.; Glaser, G. An Escherichia coli chromosomal “addiction module” regulated by guanosine [corrected] 3′,5′-bispyrophosphate: A model for programmed bacterial cell death. Proc. Natl. Acad. Sci. USA 1996, 93, 6059–6063. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.K.; Gerdes, K. RelE toxins from bacteria and archaea cleave mRNAs on translating ribosomes, which are rescued by tmRNA. Mol. Microbiol. 2003, 48, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Diago-Navarro, E.; Hernandez-Arriaga, A.M.; Kubik, S.; Konieczny, I.; Diaz-Orejas, R. Cleavage of the antitoxin of the ParD toxin-antitoxin system is determined by the ClpAP protease and is modulated by the relative ratio of the toxin and the antitoxin. Plasmid 2013, 70, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Christensen-Dalsgaard, M.; Jorgensen, M.G.; Gerdes, K. Three new RelE-homologous mRNA interferases of Escherichia coli differentially induced by environmental stresses. Mol. Microbiol. 2010, 75, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.; Vulic, M.; Min, J.; Yen, T.J.; Schumacher, M.A.; Brennan, R.G.; Lewis, K. Regulation of the Escherichia coli HipBA toxin-antitoxin system by proteolysis. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Van Melderen, L.; Bernard, P.; Couturier, M. Lon-dependent proteolysis of CcdA is the key control for activation of CcdB in plasmid-free segregant bacteria. Mol. Microbiol. 1994, 11, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lord, D.M.; Hong, S.H.; Peti, W.; Benedik, M.J.; Page, R.; Wood, T.K. Type II toxin/antitoxin MqsR/MqsA controls Type V toxin/antitoxin GhoT/GhoS. Environ. Microbiol. 2013, 15, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, M.G.; Pandey, D.P.; Jaskolska, M.; Gerdes, K. HicA of Escherichia coli defines a novel family of translation-independent mRNA interferases in bacteria and archaea. J. Bacteriol. 2009, 191, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Leplae, R.; Geeraerts, D.; Hallez, R.; Guglielmini, J.; Dreze, P.; Van Melderen, L. Diversity of bacterial Type II toxin-antitoxin systems: A comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011, 39, 5513–5525. [Google Scholar] [CrossRef] [PubMed]
- Arbing, M.A.; Handelman, S.K.; Kuzin, A.P.; Verdon, G.; Wang, C.; Su, M.; Rothenbacher, F.P.; Abashidze, M.; Liu, M.; Hurley, J.M.; et al. Crystal structures of Phd-Doc, HigA, and YeeU establish multiple evolutionary links between microbial growth-regulating toxin-antitoxin systems. Structure 2010, 18, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The pFam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, L.C.; Garrido, S.S.; Marchetto, R. Btoxdb: A comprehensive database of protein structural data on toxin-antitoxin systems. Comput. Biol. Med. 2015, 58, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. Weblogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- QIAGEN. Available online: https://www.qiagenbioinformatics.com/ (accessed on 7 July 2016).
- Smith, C.K.; Baker, T.A.; Sauer, R.T. Lon and Clp family proteases and chaperones share homologous substrate-recognition domains. Proc. Natl. Acad. Sci. USA 1999, 96, 6678–6682. [Google Scholar] [CrossRef] [PubMed]
- Ogle, C.T.; Mather, W.H. Proteolytic crosstalk in multi-protease networks. Phys. Biol. 2016, 13. [Google Scholar] [CrossRef] [PubMed]
- Iosefson, O.; Nager, A.R.; Baker, T.A.; Sauer, R.T. Coordinated gripping of substrate by subunits of a AAA+ proteolytic machine. Nat. Chem. Biol. 2015, 11, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Su, S.C.; Su, M.Y.; Liang, P.H.; Feng, C.C.; Wu, S.H.; Chang, C.I. Structural insights into the allosteric operation of the Lon AAA+ protease. Structure 2016, 24, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Wohlever, M.L.; Baker, T.A.; Sauer, R.T. A mutation in the N domain of Escherichia coli Lon stabilizes dodecamers and selectively alters degradation of model substrates. J. Bacteriol. 2013, 195, 5622–5628. [Google Scholar] [CrossRef] [PubMed]
- Vieux, E.F.; Wohlever, M.L.; Chen, J.Z.; Sauer, R.T.; Baker, T.A. Distinct quaternary structures of the AAA+ lon protease control substrate degradation. Proc. Natl. Acad. Sci. USA 2013, 110, E2002–E2008. [Google Scholar] [CrossRef] [PubMed]
- Su, S.C.; Lin, C.C.; Tai, H.C.; Chang, M.Y.; Ho, M.R.; Babu, C.S.; Liao, J.H.; Wu, S.H.; Chang, Y.C.; Lim, C.; et al. Structural basis for the magnesium-dependent activation and hexamerization of the Lon AAA+ protease. Structure 2016, 24, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Ingmer, H. Antibiotics: Killing the survivors. Nature 2013, 503, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Conlon, B.P.; Nakayasu, E.S.; Fleck, L.E.; LaFleur, M.D.; Isabella, V.M.; Coleman, K.; Leonard, S.N.; Smith, R.D.; Adkins, J.N.; Lewis, K. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 2013, 503, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Li, D.H.; Chung, Y.S.; Gloyd, M.; Joseph, E.; Ghirlando, R.; Wright, G.D.; Cheng, Y.Q.; Maurizi, M.R.; Guarne, A.; Ortega, J. Acyldepsipeptide antibiotics induce the formation of a structured axial channel in ClpP: A model for the ClpX/ClpA-bound state of ClpP. Chem. Biol. 2010, 17, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Moreira, W.; Ngan, G.J.; Low, J.L.; Poulsen, A.; Chia, B.C.; Ang, M.J.; Yap, A.; Fulwood, J.; Lakshmanan, U.; Lim, J.; et al. Target mechanism-based whole-cell screening identifies bortezomib as an inhibitor of caseinolytic protease in mycobacteria. mBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Hazan, R.; Engelberg-Kulka, H. Escherichia coli MazEF-mediated cell death as a defense mechanism that inhibits the spread of phage P1. Mol. Genet. Genom. 2004, 272, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Sat, B.; Hazan, R.; Fisher, T.; Khaner, H.; Glaser, G.; Engelberg-Kulka, H. Programmed cell death in Escherichia coli: Some antibiotics can trigger MazEF lethality. J. Bacteriol. 2001, 183, 2041–2045. [Google Scholar] [CrossRef] [PubMed]
- Christensen, S.K.; Mikkelsen, M.; Pedersen, K.; Gerdes, K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. USA 2001, 98, 14328–14333. [Google Scholar] [CrossRef] [PubMed]
- Micevski, D.; Dougan, D.A. Proteolytic regulation of stress response pathways in Escherichia coli. Subcell Biochem. 2013, 66, 105–128. [Google Scholar] [PubMed]
- Yang, N.; Lan, L. Pseudomonas aeruginosa Lon and ClpXP proteases: Roles in linking carbon catabolite repression system with quorum-sensing system. Curr. Genet. 2016, 62, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Goff, S.A.; Goldberg, A.L. Production of abnormal proteins in E. coli stimulates transcription of Lon and other heat shock genes. Cell 1985, 41, 587–595. [Google Scholar] [CrossRef]
- Tsilibaris, V.; Maenhaut-Michel, G.; van Melderen, L. Biological roles of the Lon ATP-dependent protease. Res. Microbiol. 2006, 157, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Suzuki, C.K. Functional mechanics of the ATP-dependent Lon protease—Lessons from endogenous protein and synthetic peptide substrates. Biochim. Biophys. Acta 2008, 1784, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Nazir, A.; Harinarayanan, R. Inactivation of cell division protein FtsZ by SulA makes Lon indispensable for the viability of a ppGpp0 strain of Escherichia coli. J. Bacteriol. 2015, 198, 688–700. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, A.; Nomura, K.; Ohtomo, R.; Kato, J.; Ikeda, T.; Takiguchi, N.; Ohtake, H.; Kornberg, A. Role of inorganic polyphosphate in promoting ribosomal protein degradation by the Lon protease in E. coli. Science 2001, 293, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Chen, Y.D.; Chang, Y.Y.; Lin, Y.C.; Chang, C.F.; Huang, S.J.; Wu, S.H.; Hsu, C.H. Structural basis for DNA-mediated allosteric regulation facilitated by the AAA+ module of Lon protease. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Osbourne, D.O.; Soo, V.W.; Konieczny, I.; Wood, T.K. Polyphosphate, cyclic AMP, guanosine tetraphosphate, and c-di-GMP reduce in vitro Lon activity. Bioengineered 2014, 5, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Lazinski, D.; Rowe, S.; Camilli, A.; Lewis, K. Genetic basis of persister tolerance to aminoglycosides in Escherichia coli. mBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, O.; Schuenemann, V.J.; Hand, N.J.; Silhavy, T.J.; Martin, J.; Lupas, A.N.; Djuranovic, S. PrlF and YhaV encode a new toxin-antitoxin system in Escherichia coli. J. Mol. Biol. 2007, 372, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Snyder, W.B.; Silhavy, T.J. Enhanced export of beta-galactosidase fusion proteins in PrlF mutants is Lon dependent. J. Bacteriol. 1992, 174, 5661–5668. [Google Scholar] [PubMed]
- Aakre, C.D.; Phung, T.N.; Huang, D.; Laub, M.T. A bacterial toxin inhibits DNA replication elongation through a direct interaction with the beta sliding clamp. Mol. Cell 2013, 52, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Bordes, P.; Cirinesi, A.M.; Ummels, R.; Sala, A.; Sakr, S.; Bitter, W.; Genevaux, P. SecB-like chaperone controls a toxin-antitoxin stress-responsive system in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2011, 108, 8438–8443. [Google Scholar] [CrossRef] [PubMed]
- Sala, A.; Calderon, V.; Bordes, P.; Genevaux, P. Tac from Mycobacterium tuberculosis: A paradigm for stress-responsive toxin-antitoxin systems controlled by SecB-like chaperones. Cell Stress Chaperones 2013, 18, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Ainelo, A.; Tamman, H.; Leppik, M.; Remme, J.; Horak, R. The toxin GraT inhibits ribosome biogenesis. Mol. Microbiol. 2016, 100, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Tamman, H.; Ainelo, A.; Tagel, M.; Horak, R. Stability of the GraA antitoxin depends on the growth phase, ATP level and global regulator MexT. J. Bacteriol. 2015, 198, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.F.; Zhou, Y.; Gottesman, S. Redundant in vivo proteolytic activities of Escherichia coli Lon and the ClpyQ (HslUV) protease. J. Bacteriol. 1999, 181, 3681–3687. [Google Scholar] [PubMed]
- Christensen, S.K.; Pedersen, K.; Hansen, F.G.; Gerdes, K. Toxin-antitoxin loci as stress-response-elements: ChpAK/MazF and ChpBK cleave translated RNAs and are counteracted by tmRNA. J. Mol. Biol. 2003, 332, 809–819. [Google Scholar] [CrossRef]
- Malik, M.; Capecci, J.; Drlica, K. Lon protease is essential for paradoxical survival of Escherichia coli exposed to high concentrations of quinolone. Antimicrob. Agents Chemother. 2009, 53, 3103–3105. [Google Scholar] [CrossRef] [PubMed]
- Breidenstein, E.B.; Bains, M.; Hancock, R.E. Involvement of the Lon protease in the SOS response triggered by ciprofloxacin in Pseudomonas aeruginosa PAO1. Antimicrob. Agents Chemother. 2012, 56, 2879–2887. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.J.; Hergenrother, P.J. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2012, 20, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Tanouchi, Y.; Lee, A.J.; Meredith, H.; You, L. Programmed cell death in bacteria and implications for antibiotic therapy. Trends Microbiol. 2013, 21, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Schuster, C.F.; Bertram, R. Toxin-antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 2013, 340, 73–85. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}

| TADB ¥ | Pfam ▯ | BToxDB ❖ | TA Module | Protease | Consensus § |
|---|---|---|---|---|---|
| Phd superfamily (PhD/YefM fold) | |||||
| 27 | 2119 | 5 | Antitoxin: PhD (Pfam02604) Toxin: Doc | ClpXP [37] |  |
| 8 | N.D. | 4 | Antitoxin: YefM Toxin: YoeB | Lon [104,105] |  |
| N.D. | N.D. | none | Antitoxin: Axe Toxin: Txe | ClpCP, adaptor TrfA [96,97] | no sequence identified |
| RelB Superfamily (Pfam 04221) | |||||
| 214 | 666 | 16 | Antitoxin: RelB Toxin: RelE | Lon [106,107] | No consensus for logo |
| Antitoxin: DinJ Toxin: YafQ | Lon, ClpXP [108] | ||||
| PasA superfamily | |||||
| 1 | N.D. | none | Antitoxin: PasA Toxin: PasBC | Lon [109] | only one sequence identified |
| 60 | 376 | 1 | Antitoxin: ParD (Pfam 03693) Toxin: ParE | Lon [38] | No consensus for logo |
| VapB/MazE superfamily | |||||
| 385 | 250 | 7 | Antitoxin: VapB (Pfam 09957) Toxin: VapC | Lon [110] |  |
| 92 | 1960 | 9 | Antitoxin: MazE (ChpB) (Pfam 04014) Toxin: MazF (ChpA) | ClpCP with TrfA; Lon, ClpAP [96,97,111,112] |  |
| 1 | N.D. | 2 | Antitoxin: PemI/Kis Toxin: PemK/Kid | ClpAP [113] | only one sequence identified |
| 112 | N.D. | 2 | Antitoxin: HigA Toxin: HigB, ParE/RelE, HipA, GinD, Zeta | Lon [114] | No consensus for logo |
| 1 | N.D. | 11 | Antitoxin: HipB Toxin: HipA | Lon [115] | only one sequence identified |
| Unclassified antitoxins | |||||
| 9 | 139 | 14 | Antitoxin: CcdA (Pfam 07362) Toxin: CcdB | Lon [116] |  |
| 2 | 7 | 2 | Antitoxin: Epsilon * (Pfam 08998) Toxin: Zeta | ClpXP [102] |  |
| 1 | 124 | 1 | Antitoxin: MqsA (Pfam 15731) Toxin: MqsR | Lon, ClpXP [44,117] | Atypical antitoxin No consensus for logo |
| 62 | 951 | none | Antitoxin: HicB (Pfam 15919) Toxin: HicA | Lon [118] |  |
| 3 | N.D. | none | Antitoxin: PrlF(MazE) Toxin: YhaV | unknown | only three sequences identified |
| 1 | N.D. | none | Antitoxin: MosA Toxin: MosT | unknown | only one sequence identified |
| 2 | N.D. | none | Antitoxin: YeeU Toxin: YeeV | unknown | only two sequences identified |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muthuramalingam, M.; White, J.C.; Bourne, C.R. Toxin-Antitoxin Modules Are Pliable Switches Activated by Multiple Protease Pathways. Toxins 2016, 8, 214. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins8070214
Muthuramalingam M, White JC, Bourne CR. Toxin-Antitoxin Modules Are Pliable Switches Activated by Multiple Protease Pathways. Toxins. 2016; 8(7):214. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins8070214
Chicago/Turabian StyleMuthuramalingam, Meenakumari, John C. White, and Christina R. Bourne. 2016. "Toxin-Antitoxin Modules Are Pliable Switches Activated by Multiple Protease Pathways" Toxins 8, no. 7: 214. https://0-doi-org.brum.beds.ac.uk/10.3390/toxins8070214