
Potential of Drug Efficacy Evaluation in Lung and Kidney Cancer Models Using Organ-on-a-Chip Technology

 ,
, 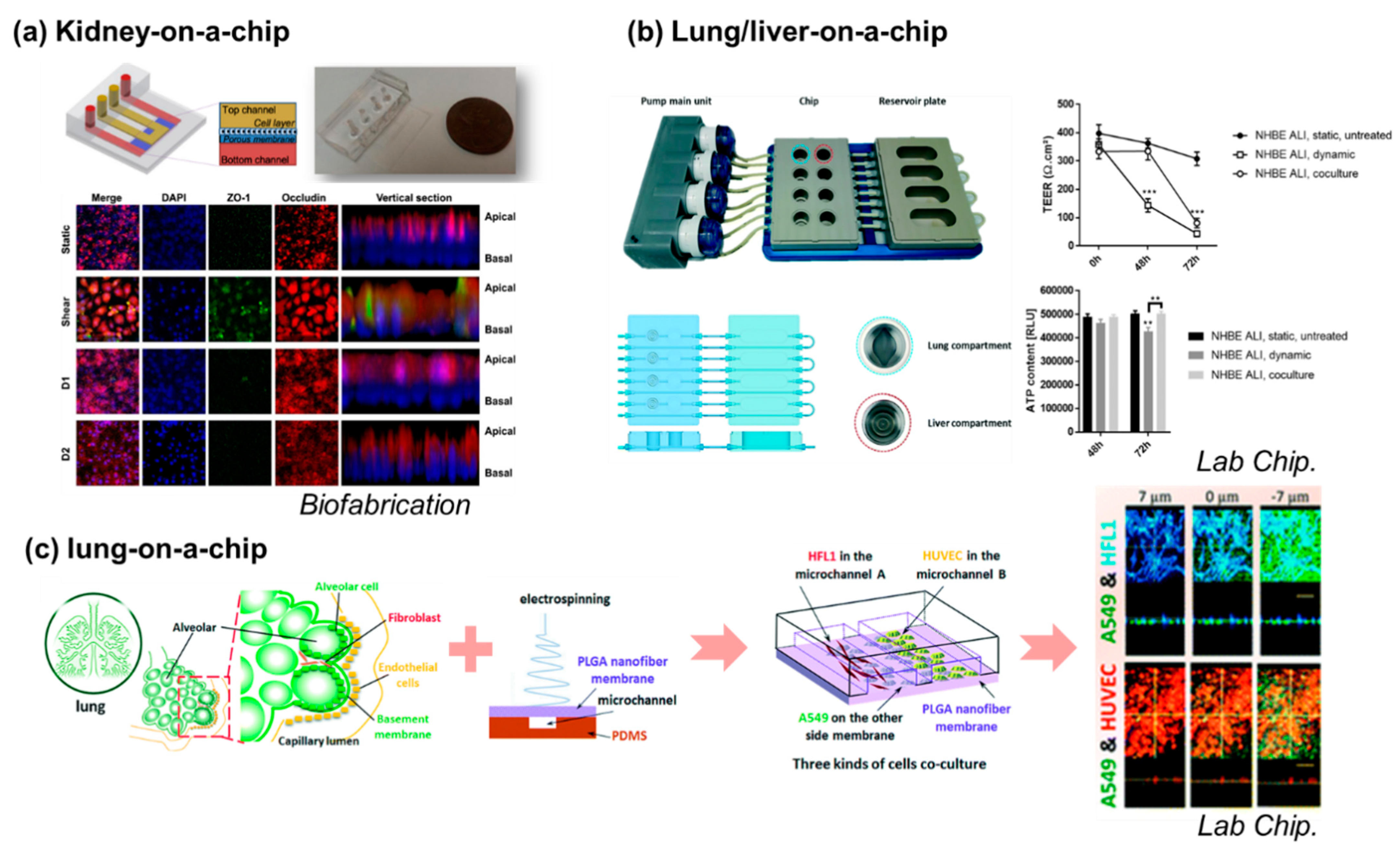{kind=link}
Abstract
:1. Introduction
2. Conventional Preclinical Methods
2.1. Two-Dimensional Cell Cultures
2.2. Animal Studies
3. Organ-on-a-Chip (OoC): Its Basic Elements and Comparison with Other Models
3.1. Basic Elements of OoC
3.1.1. Cell Sourcing
3.1.2. Blood Supply
3.1.3. Elements of the ECM
3.1.4. Scaling of Organs
3.2. Comparison with Other Advanced Cell Culture Techniques
3.2.1. Conventional Preclinical Methods
3.2.2. Comparison with Organoid Models
3.2.3. Comparison with 3D Bioprinting
4. Potential of OoC as a Drug Efficacy Evaluation in Lung and Kidney Cancer Models
4.1. OoC as Advanced Microfluidic Technologies of the Lung and Kidney Model
4.2. OoC as a Disease Model for Drug Efficacy Evaluation
4.3. Elements of the Drug Efficacy Evaluation in Lung and Kidney Cancer
4.3.1. Assays for Drug Efficacy Evaluation in OoC
4.3.2. Epidermal Growth Factor Receptor (EGFR) and Vascular Endothelial Growth Factor (Vegf) Related Molecules as Key Biomarkers of Lung and Kidney Cancer
4.4. Comparison with Conventional Efficacy Testing Methods and Institutional Devices
4.4.1. Comparison with Animal Testing
4.4.2. Comparison with Clinical Trials
4.4.3. Institutional Devices as Alternatives to Animal Testing
4.5. Potential of Drug Efficacy Testing in Tumor-on-a-Chip and Metastasis-on-a-Chip
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Alépée, N.; Bahinski, A.; Daneshian, M.; Wever, B.D.; Fritsche, E.; Goldberg, A.; Hansmann, J.; Hartung, T.; Haycock, J.; Hogberg, H.T.; et al. State-of-the-art of 3D cultures (organs-on-a-chip) in safety testing and pathophysiology. ALTEX 2014, 31, 441–477. [Google Scholar] [CrossRef]
- Abaci, H.E.; Shuler, M.L. Human-on-a-chip design strategies and principles for physiologically based pharmocokinetics/pharmacodynamics modeling. Integr. Biol. 2015, 7, 383–391. [Google Scholar] [CrossRef]
- Low, L.A.; Tagle, D.A. Tissue Chips—innovative tools for drug development and disease modeling. Lab Chip 2017, 17, 3026–3036. [Google Scholar] [CrossRef]
- Al-Samadia, A.; Poor, B.; Tuomainen, K.; Liu, V.; Hyytiäinen, A.; Suleymanova, I.; Mesimakid, K.; Wilkmand, T.; Mäkitiee, A.; Saavalainen, P.; et al. In vitro humanized 3D microfluidic chip for testing personalized immunotherapeutics for head and neck cancer patients. Exp. Cell Res. 2019, 383, 111508–111514. [Google Scholar] [CrossRef] [PubMed]
- Malaney, P.; Nicosia, S.V.; Davé, V. One mouse, one patient paradigm: New avatars of personalized cancer therapy. Cancer Lett. 2014, 344, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheon, D.J.; Orsulic, S. Mouse models of cancer. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 95–119. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; McCain, M.L.; Yang, L.; He, A.; Pasqualini, F.S.; Agarwal, A.; Yuan, H.; Jiang, D.; Zhang, D.; Zang, L.; et al. Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies. Nat. Med. 2014, 20, 616–623. [Google Scholar] [CrossRef]
- Warren, C.R.; O’Sullivan, JF.; Friesen, M.; Becker, CE.; Zhang, X.; Liu, P.; Wakabayashi, Y.; Morningstar, JE.; Shi, X.; Choi, J.; et al. Induced Pluripotent Stem Cell Differentiation Enables Functional Validation of GWAS Variants in Metabolic Disease. Cell Stem Cell. 2017, 20, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Downing, T.L.; Soto, J.; Morez, C.; Houssin, T.; Fritz, A.; Yuan, F.; Chu, J.; Patel, S.; Schaffer, DV.; Li, S. Biophysical regulation of epigenetic state and cell reprogramming. Nat. Mater. 2013, 12, 1154–1162. [Google Scholar] [CrossRef]
- Lei, Y.; Schaffer, D.V. A fully defined and scalable 3D culture system for human pluripotent stem cell expansion and differentiation. Proc. Natl. Acad. Sci. USA 2013, 110, E5039–E5048. [Google Scholar] [CrossRef] [Green Version]
- Caiazzo, M.; Okawa, Y.; Ranga, A.; Piersigilli, A.; Tabata, Y.; Lutolf, M.P. Defined three-dimensional microenvironments boost induction of pluripotency. Nat. Mater. 2016, 15, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Abilez, O.J.; Wu, J.C. Stem cell reprogramming: A 3D boost. Nat. Mater. 2016, 15, 259–261. [Google Scholar] [CrossRef]
- Gagliano, O.; Elvassore, N.; Luni, C. Microfluidic technology enhances the potential of human pluripotent stem cells. Biochem. Biophys. Res. Commun. 2016, 473, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Zhao, R.; Doi, A.; Ng, K.; Unternaehrer, J.; Cahan, P.; Hongguang, H.; Loh, Y.-H.; Aryee, M.J.; Lensch, M.W.; et al. Donor cell type can influence the epigenome and differentiation potential of human induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 1117–1119. [Google Scholar] [CrossRef] [Green Version]
- Bar-Nur, O.; Russ, H.A.; Efrat, S.; Benvenisty, N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell 2011, 9, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullard, A. Stem-cell discovery platforms yield first clinical candidates. Nat. Rev. Drug Discov. 2015, 14, 589–591. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P. Patient-derived induced pluripotent stem cells in cancer research and precision oncology. Nat. Med. 2016, 22, 1392–1401. [Google Scholar] [CrossRef]
- Low, L.A.; Mummery, C.; Berridge, B.R.; Austin, C.P.; Tagle, D.A. Organs-on-chips: Into the next decade. Nat. Revs. Drug Discov. 2020, 1–17. [Google Scholar]
- Chang, S.-Y.; Weber, E.J.; Sidorenko, V.S.; Chapron, A.; Yeung, C.K.; Gao, C.; Mao, Q.; Shen, D.; Wang, J.; Rosenquist, T.A.; et al. Human liver- kidney model elucidates the mechanisms of aristolochic acid nephrotoxicity. JCI Insight 2017, 2, e95978. [Google Scholar] [CrossRef] [PubMed]
- Phan, D.T.T.; Wang, X.; Cravera, B.M.; Sobrinoa, A.; Zhaoc, D.; Chena, J.C.; Lee, L.Y.N.; Georged, S.C.; Lee, A.P.; Hughes, C.C.W. A vascularized and perfused organ- on-a- chip platform for large- scale drug screening applications. Lab Chip. 2017, 17, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhao, Z.; Rahim, N.A.A.; Noort, D.; Yu, H. Towards a human- on-chip: Culturing multiple cell types on a chip with compartmentalized microenvironments. Lab Chip 2009, 9, 3185–3192. [Google Scholar] [CrossRef] [PubMed]
- Materne, E.M.; Maschmeyer, I.; Lorenz, A.K.; Horland, R.; Schimek, K.M.S.; Busek, M.; Sonntag, F.; Lauster, R.; Marx, U. The multi-organ chip—A microfluidic platform for long- term multi-tissue coculture. J. Vis. Exp. 2015, 98, e52526. [Google Scholar] [CrossRef] [Green Version]
- Maschmeyer, I.; Lorenz, A.K.; Schimek, K.; Hasenberg, T.; Ramme, A.P.; Hübner, J.; Lindner, M.; Drewell, C.; Bauer, S.; Thomas, A.; et al. A four- organ-chip for interconnected long- term co- culture of human intestine, liver, skin and kidney equivalents. Lab Chip 2015, 15, 2688–2699. [Google Scholar] [CrossRef] [Green Version]
- Tsamandouras, N.; Thomas, W.L.K.; Edington, C.D.; Stokes, C.D.; Griffith, L.G.; Cirit, M. Integrated gut and liver microphysiological systems for quantitative in vitro pharmacokinetic studies. AAPS J. 2017, 19, 1499–1512. [Google Scholar] [CrossRef] [Green Version]
- Oleaga, C.; Bernabin, C.; Smith, A.S.T.; Srinivasan, B.; Jackson, M.; McLam, W.; Platt, V.; Bridges, R.; Cai, Y.; Santhanam, N.; et al. Multi-organ toxicity demonstration in a functional human in vitro system composed of four organs. Sci. Rep. 2016, 6, 20030. [Google Scholar] [CrossRef] [PubMed]
- Oleaga, C.; Lavado, A.; Riu, A.; Rothemund, S.; Carmona-Moran, C.A.; Persaud, K.; Yurko, A.; Lear, J.; Narasimban, N.S.; Long, C.J.; et al. Human-on-a-chip systems: Long-term electrical and mechanical function monitoring of a human-on-a-chip system. Adv. Funct. Mater. 2019, 29, 1970049. [Google Scholar] [CrossRef] [Green Version]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caliari, S.R.; Burdick, J.A. A practical guide to hydrogels for cell culture. Nat. Methods 2016, 13, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crapo, P.M.; Tottey, S.; Slivka, P.F.; Badylak, S.F. Effects of biologic scaffolds on human stem cells and implications for CNS tissue engineering. Tissue Eng. A. 2014, 20, 313–323. [Google Scholar] [CrossRef]
- Safaee, H.; Bakooshli, M.A.; Davoudi, S.; Cheng, R.Y.; Martowirogo, A.J.; Li, E.W.; Simmons, C.A.; Gilbert, P.M. Tethered jagged-1 synergizes with culture substrate stiffness to modulate notch- induced myogenic progenitor differentiation. Cell. Mol. Bioeng. 2017, 10, 501–513. [Google Scholar] [CrossRef]
- Trappmann, B.; Baker, B.M.; Polacheck, W.J.; Choi, C.K.; Burdick, J.A.; Chen, C.S. Matrix degradability controls multicellularity of 3D cell migration. Nat. Commun. 2017, 8, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Tokuda, E.Y.; Leight, J.L.; Miksch, C.E.; Brown, T.E.; Anseth, K.S. Synthesis of microgel sensors for spatial and temporal monitoring of protease activity. ACS Biomater. Sci. Eng. 2018, 4, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Wikswo, J.P.; Curtis, E.; Eagleton, Z.E.; Evans, B.C.; Kole, A.; Hofmeister, L.H.; Matloff, W.J. Scaling and systems biology for integrating multiple organs-on-a-chip. J. Lab Chip 2013, 13, 3496–3511. [Google Scholar] [CrossRef]
- Stokes, C.L.; Cirit, M.; Lauffenburger, D.A. Physiome-on-a-chip: The challenge of “scaling” in design, operation, and translation of microphysiological systems. CPT Pharmacometr. Syst. Pharmacol. 2015, 4, 559–562. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.B.; Singh, A. Cellular self-assembly and biomaterials-based organoid models of development and diseases. Acta Biomater. 2017, 53, 29–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shehab, N.; Lovegrove, M.C.; Geller, A.I.; Rose, K.O.; Weidle, N.J.; Budnitz, D.S. Emergency visits for oral anticoagulant bleeding. JAMA 2016, 316, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Ha, G.; Tseng, Y.-Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Belmonte, J.C.I. Organoids—preclinical models of human disease. New Engl. J. Med. 2019, 380, 569–579. [Google Scholar] [CrossRef]
- Huch, M.; Gehart, H.; Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.A.; Ellis, E.; Wenum, M.; Fuchs, S.A.; Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Duarte, A.A.; Gogola, E.; Sachs, N.; Barazas, M.; Annunziato, S.; Ruiter, J.R.; Velds, A.; Blatter, S.; Houthuijzen, J.M.; Ven, M.; et al. BRCA-deficient mouse mammary tumor organoids to study cancer-drug resistance. Nat. Methods 2018, 15, 134–140. [Google Scholar] [CrossRef]
- Behjati, S.; Huch, M.; Boxte, R.; Karthaus, W.; Wedge, D.C.; Tamuri, A.U.; Martincorena, I.; Petljak, M.; Alexandrov, L.B.; et al. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 2014, 513, 422–425. [Google Scholar] [CrossRef]
- Li, Z.; Araoka, T.; Wu, J.; Liao, H.-K.; Li, M.; Lazo, M.; Zhou, B.; Sui, Y.; Wu, M.-Z.; Tamura, I.; et al. 3D culture supports long-term expansion of mouse and human nephrogenic progenitors. Cell Stem Cell 2016, 19, 516–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetering, M.; Francies, H.E.; Francies, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; Houdt, W.; Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [PubMed] [Green Version]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Wetering, M.; Baker, N.; Stange, D.E.; Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymalniche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Es, J.H.; Kuipers, J.; Kujala, P.; Born, M.; Cozijnsen, M.; Haegebarth, A.; Korving, J.; Begthel, H.; Peters, P.J.; et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 2007, 449, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.; Baker, N.; Low, T.Y.; Koo, B.-K.; Li, V.S.W.; Teunissen, H.; Kujala, P.; Haegebarth, A.; Peters, P.J.; Wetering, M.; et al. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature 2011, 476, 293–297. [Google Scholar] [CrossRef]
- Hofmann, C.; Obermeier, F.; Artinger, M.; Hausmann, M.; Falk, W.; Schoelmerich, J.; Rogler, G.; Grossmann, J. Cell-cell contacts prevent anoikis in primary human colonic epithelial cells. Gastroenterology 2007, 132, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E.; Georgescu, A.; Huh, D. Organoids-on-a-chip. Science 2019, 364, 960–965. [Google Scholar] [CrossRef]
- Knowlton, S.; Onal, S.; Yu, C.H.; Zhao, J.J.; Tasoglu, S. Bioprinting for cancer research. Trends Biotechnol. 2015, 33, 504–513. [Google Scholar] [CrossRef]
- Albritton, J.L.; Miller, J.S. 3D bioprinting: Improving in vitro models of metastasis with heterogeneous tumor microenvironments. Dis. Models Mech. 2017, 10, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.-G.; Lee, H.; Cho, D.-W. 3D printing of organs-on-chips. Bioengineering 2017, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Guo, Y.; Yu, Y.; Xu, C.; Xu, H.; Qin, J. Assessment of metabolism-dependent drug efficacy and toxicity on a multilayer organs-on-a-chip. Integr. Biol. 2016, 8, 1022–1029. [Google Scholar] [CrossRef]
- Caballero, D.; Blackburn, S.M.; Pablo, M.; Samitier, J.; Albertazzi, L. Tumour-vessel-on-a-chip models for drug delivery. Lab Chip. 2017, 17, 3760–3771. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Li, K.; Zhang, X.; Liu, C.; Bingkun Guo, B.; Wen, W.; Gao, X. Nanofiber membrane supported lung-on-a-chip microdevice for anti-cancer drug testing. Lab Chip 2018, 18, 486–495. [Google Scholar] [CrossRef]
- Bovard, D.; Antonin Sandoz, A.; Luettich, K.; Frentzel, S.; Iskandar, A.; Marescotti, D.; Trivedi, K.; Guedj, E.; Dutertre, Q.; Peitsch, M.C.; et al. A lung/liver-on-a-chip platform for acute and chronic toxicity studies. Lab Chip 2018, 18, 3814–3829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; LesherPerez, S.C.; Kim, B.; Yamanishi, C.; Labuz, J.M.; Leung, B.; Takayama, S. Pharmacokinetic profile that reduces nephrotoxicity of gentamicin in a perfused Kidney-on-a-chip. Biofabrication. 2016, 8, 015021. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S. Kidney-on-a-chip: A new technology for predicting drug efficacy, interactions, and drug-induced nephrotoxicity. Curr. Drug Metab. 2018, 19, 577–583. [Google Scholar] [CrossRef]
- Arrowsmith, J. Trial watch: Phase III and submission failures: 2007–2010. Nat. Rev. Drug Discov. 2011, 10, 87–87. [Google Scholar] [CrossRef]
- Kimmelman, J.; Federico, C. Consider drug efficacy before first-in-human trials. Nature 2017, 542, 25–27. [Google Scholar] [CrossRef]
- Cook, D.; Brown, D.; Alexander, R.; March, R.; Morgan, P.; Satterthwaite, G.; Pangalos, M.N. Lessons learned from the fate of AstraZeneca’s drug pipeline: A five-dimensional framework. Nat. Rev. Drug Discov. 2014, 13, 419–431. [Google Scholar] [CrossRef]
- Giaever, I.; Keese, C.R. Use of electric fields to monitor the dynamical aspect of cell behavior in tissue culture. IEEE Trans. Biomed. Eng. 1986, 2, 242–247. [Google Scholar] [CrossRef]
- Keese, C.R.; Bhawe, K.; Wegener, J.; Giaever, I. Real-time impedance assay to follow the invasive activities of metastatic cells in culture. Biotechniques 2002, 33, 842–851. [Google Scholar] [CrossRef] [Green Version]
- Asphahani, F.; Zhang, M. Cellular impedance biosensors for drug screening and toxin detection. Analyst 2007, 132, 835–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arias, L.R.; Perry, C.A.; Yang, L. Real-time electrical impedance detection of cellular activities of oral cancer cells. Biosens. Bioelectron. 2010, 25, 2225–2231. [Google Scholar] [CrossRef]
- Wu, Q.; Wei, X.; Pan, Y.; Zou, Y.; Hu, N.; Wang, P. Bionic 3D spheroids biosensor chips for high-throughput and dynamic drug screening. Biomed. Microdevices 2018, 20, 82–90. [Google Scholar] [CrossRef]
- Bersini, S.; Jeon, J.S.; Dubini, G.; Arrigoni, C.; Chung, S.; Charest, J.L.; Moretti, M.; Kamm, R.D. A microfluidic 3D in vitro model for specificity of breast cancer metastasis to bone. Biomaterials 2014, 35, 2454–2461. [Google Scholar] [CrossRef]
- Choucha-Snouber, L.; Aninat, C.; Grsicom, L.; Madalinski, G.; Brochot, C.; Poleni, P.E.; Razan, F.; Guillouzo, C.G.; Legallais, C.; Corlu, A. Investigation of ifosfamide nephrotoxicity induced in a liver–kidney co-culture biochip. Biotechnol Bioeng. 2013, 110, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Ingber, D.E. Gut-on-a-chip microenvironment induces human intestinal cells to undergo villus differentiation. Integr. Biol. (Camb). 2013, 5, 1130–1140. [Google Scholar] [CrossRef] [Green Version]
- Esch, E.W.; Bahinski, A.; Huh, D. Organs-on-chips at the frontiers of drug discovery. Nat. Rev. Drug Discov. 2015, 14, 248–260. [Google Scholar] [CrossRef] [Green Version]
- Yano, S.; Kondo, K.; Yamaguchi, M.; Richmond, G.; Hutchison, M.; Wakeling, A.; Averbuch, S.; Wadsworth, P. Distribution and function of EGFR in human tissue and the effect of EGFR tyrosine kinase inhibition. Anticancer Res. 2003, 23, 3639–3650. [Google Scholar] [PubMed]
- Juliane, H.; Raschke, M.; Rütschle, I.; Gräßle, S.; Hasenberg, T.; Schirrmann, K.; Lorenz, A.; Schnurre, S.; Lauste, R.; Maschmeyer, I.; et al. Simultaneous evaluation of anti-EGFR-induced tumour and adverse skin effects in a microfluidic human 3D co-culture model. Sci Rep. 2018, 8, 15010–15021. [Google Scholar]
- Liu, T.-C.; Jin, X.; Wang, Y.; Wang, K. Role of epidermal growth factor receptor in lung cancer and targeted therapies. Am. J. Cancer Res. 2017, 7, 187–202. [Google Scholar]
- Silva, A.P.S.; Coelho, P.V.; Anazetti, M.; Simioni, P.U. Targeted therapies for the treatment of non-small-cell lung cancer: Monoclonal antibodies and biological inhibitors. Hum. Vaccin. Immunother. 2017, 13, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Gomes, J.R.; Cruz, M.R.S. Combination of afatinib with cetuximab in patients with EGFR-mutant non-small-cell lung cancer resistant to EGFR inhibitors. OncoTargets Ther. 2015, 8, 1137–1142. [Google Scholar] [CrossRef] [Green Version]
- Lacouture, M.E. Mechanisms of cutaneous toxicities to EGFR inhibitors. Nat. Rev. Cancer. 2006, 6, 803–812. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Motzer, R.J. Systemic therapy for metastatic renal-cell carcinoma. New Engl. J. Med. 2017, 376, 354–366. [Google Scholar] [CrossRef]
- Garcia, J.A.; Rini, B.I. Recent progress in the management of advanced renal cell carcinoma. CA Cancer J. Clin. 2007, 57, 112–125. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Clark, P.E. The role of VHL in clear-cell renal cell carcinoma and its relation to targeted therapy. Kidney Int. 2009, 76, 939–945. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.V.; Tran, C.; Mellinghoff, I.K.; Welsbie, D.S.; Chan, E.; Fueger, B.; Czernin, J.; Sawyers, C.L. Hypoxia-inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat. Med. 2006, 12, 122–127. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef]
- Greek, R.; Menache, A. Systematic reviews of animal models: Methodology versus epistemology. Int. J. Med. Sci. 2013, 10, 206–221. [Google Scholar] [CrossRef] [Green Version]
- Wnorowski, A.; Yang, H.; Wu, J.C. Progress, obstacles, and limitations in the use of stem cells in organ-on-a-chip models. Adv. Drug Deliv. Rev. 2018, 13311, 1–9. [Google Scholar] [CrossRef]
- Blumenrath, S.H.; Lee, B.Y.; Low, L.; Prithviraj, R.; Tagle, D. Tackling rare diseases: Clinical trials on chips. Exp. Biol. Med. 2020, 245, 1155–1162. [Google Scholar] [CrossRef]
- Liebsch, M.; Traue, D.; Barrabas, C.; Spielmann, H.; Uphill, P.; Wilkins, S.; McPherson, J.P.; Wiemann, C.; Kaufmann, T.; Remmele, M.; et al. The ECVAM prevalidation study on the use of EpiDerm for skin corrosivity testing. Altern. Lab. Anim. 2000, 28, 371–401. [Google Scholar] [CrossRef]
- Barroso, J.; Ahn, I.Y.; Caldeira, C.; Carmichael, P.L.; Casey, W.; Coecke, S.; Curren, R.; Desprez, B.; Eskes, C.; Griesinger, C.; et al. International harmonization and cooperation in the validation of alternative methods. Adv. Exp. Biol. 2016, 856, 343–386. [Google Scholar]
- Kojima, H.; Yamaguchi, H.; Sozu, T.; Kleinstreuer, N.; Lim, J.-H.; Chen, W.; Watanabe, M.; Fukuda, T.; Yamashita, K.; Takezawa, T. Multi-laboratory validation study of the Vitrigel-Eye irritancy test method as an alternative to in vivo eye irritation testing. Altern. Lab. Anim. 2019, 47, 140–157. [Google Scholar] [CrossRef]
- Livingston, C.A.; Fabre, K.M.; Tagle, D.A. Facilitating the commercialization and use of organ platforms generated by the microphysiological systems (Tissue Chip) program through public-private partnerships. Comput. Struct. Biotechnol. J. 2016, 14, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Low, L.A.; Tagle, D.A. Tissue chips to aid drug development and modeling for rare diseases. Expert Opin. Orphan Drugs 2016, 4, 1113–1121. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-J.; Lee, H.-A. Trends in the development of human stem cell-based non-animal drug testing models. Korean J. Physiol. Pharmacol. 2020, 24, 441–452. [Google Scholar] [CrossRef]
- Shirure, V.S.; Bi, Y.; Curtis, M.B.; Lezia, A.; Goedegebuure, M.M.; Goedegebuure, S.P.; Aft, R.; Fields, R.C.; George, S.C. Tumor-on-a-chip platform to investigate progression and drug sensitivity in cell lines and patient-derived organoids. Lab Chip 2018, 18, 3687–3702. [Google Scholar] [CrossRef]
- Chramiec, A.; Teles, D.; Yeager, K.; Marturano-Kruik, A.; Pak, J.; Chen, T.; Hao, L.; Wang, M.; Lock, R.; Tavakol, D.N.; et al. Integrated human organ-on-a-chip model for predictive studies of anti-tumor drug efficacy and cardiac safety. Lab Chip 2020, 20, 4357–4372. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, D.; Wu, G.; Wu, J.; Li, S.; Lo, J.; He, Y.; Zhao, C.; Zhao, X.; Zhang, H.; et al. Metastasis-on-a-chip mimicking the progression of kidney cancer in the liver for predicting treatment efficacy. Theranostics 2020, 10, 300–311. [Google Scholar] [CrossRef]
- Xu, M.; Wang, Y.; Duan, W.; Xia, S.; Wei, S.; Liu, W.; Wang, Q. Proteomic Reveals Reasons for Acquired Drug esistance in Lung Cancer Derived Brain Metastasis Based on a Newly Established Multi-Organ Microfluidic Chip Model. Front. Bioeng. Biotechnol. 2020, 8, 612091–612100. [Google Scholar] [CrossRef]
- Oliver, C.R.; Westerhof, T.M.; Castro, M.G.; Merajver, S.D. Quantifying the Brain Metastatic Tumor Micro-Environment using an Organ-On-A Chip 3D Model, Machine Learning, and Confocal Tomography. J. Vis. Exp. 2020, 16, 61654. [Google Scholar]
- Lee, S.; Kim, S.; Koo, D.-J.; Yu, J.; Cho, H.; Lee, H.; Song, J.M.; Kim, S.-Y.; Min, D.-H.; Jeon, N.L. 3D Microfluidic Platform and Tumor Vascular Mapping for Evaluating Anti-Angiogenic RNAi-Based Nanomedicine. ACS Nano 2021, 15, 338–350. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, S.-H.; Lee, S.; Park, J.Y.; Jeon, J.S.; Cho, Y.-J.; Kim, S. Potential of Drug Efficacy Evaluation in Lung and Kidney Cancer Models Using Organ-on-a-Chip Technology. Micromachines 2021, 12, 215. https://0-doi-org.brum.beds.ac.uk/10.3390/mi12020215
Hwang S-H, Lee S, Park JY, Jeon JS, Cho Y-J, Kim S. Potential of Drug Efficacy Evaluation in Lung and Kidney Cancer Models Using Organ-on-a-Chip Technology. Micromachines. 2021; 12(2):215. https://0-doi-org.brum.beds.ac.uk/10.3390/mi12020215
Chicago/Turabian StyleHwang, Seong-Hye, Sangchul Lee, Jee Yoon Park, Jessie Sungyun Jeon, Young-Jae Cho, and Sejoong Kim. 2021. "Potential of Drug Efficacy Evaluation in Lung and Kidney Cancer Models Using Organ-on-a-Chip Technology" Micromachines 12, no. 2: 215. https://0-doi-org.brum.beds.ac.uk/10.3390/mi12020215