
Continuous Flow with Reagent Injection on an Inlaid Microfluidic Platform Applied to Nitrite Determination
 ,
,
Abstract
:1. Introduction
Theory
2. Materials and Methods
2.1. Chemical and Reagent Preparation
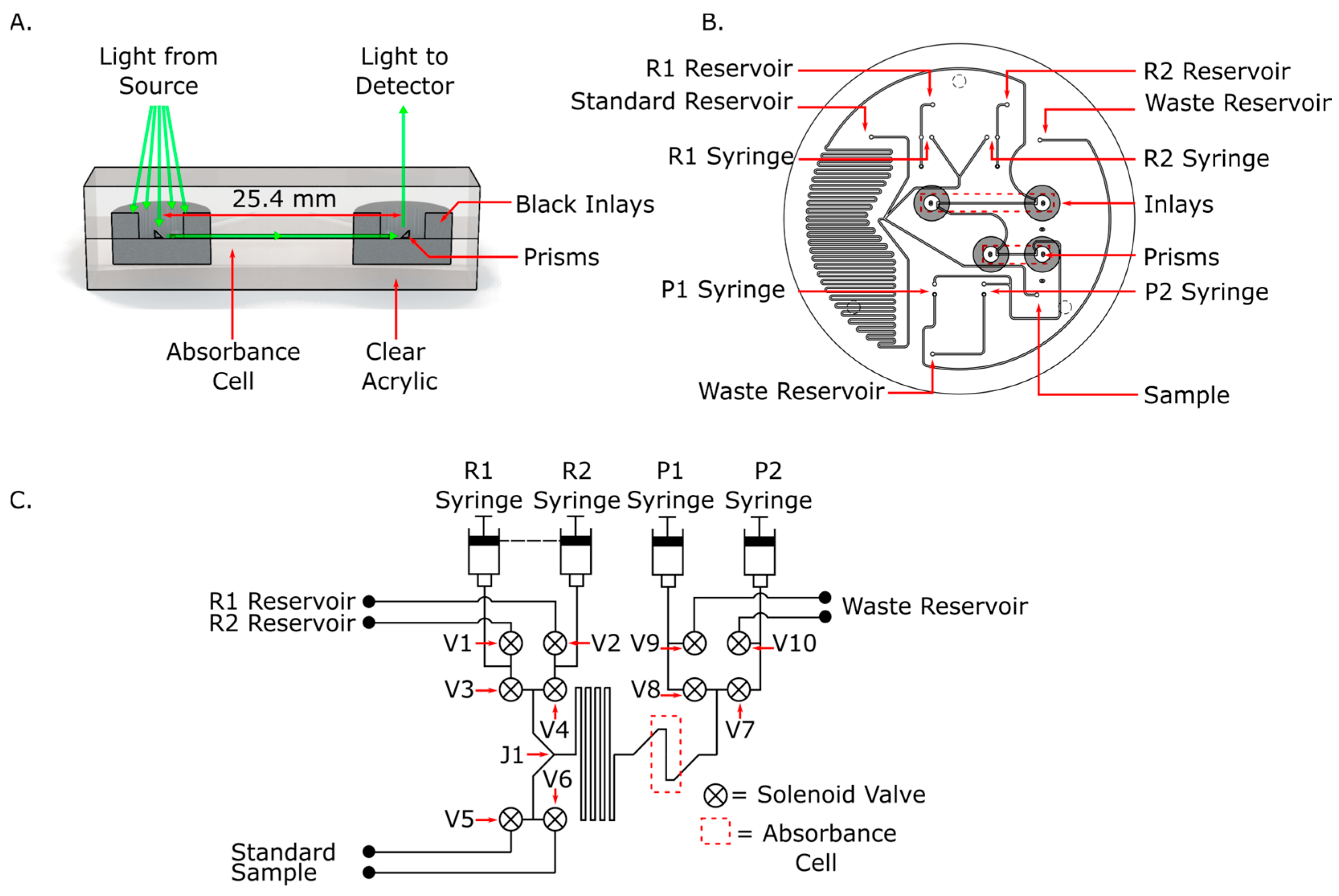
2.2. Microfluidic Chip with Novel Inlaid Optical Cell Fabrication
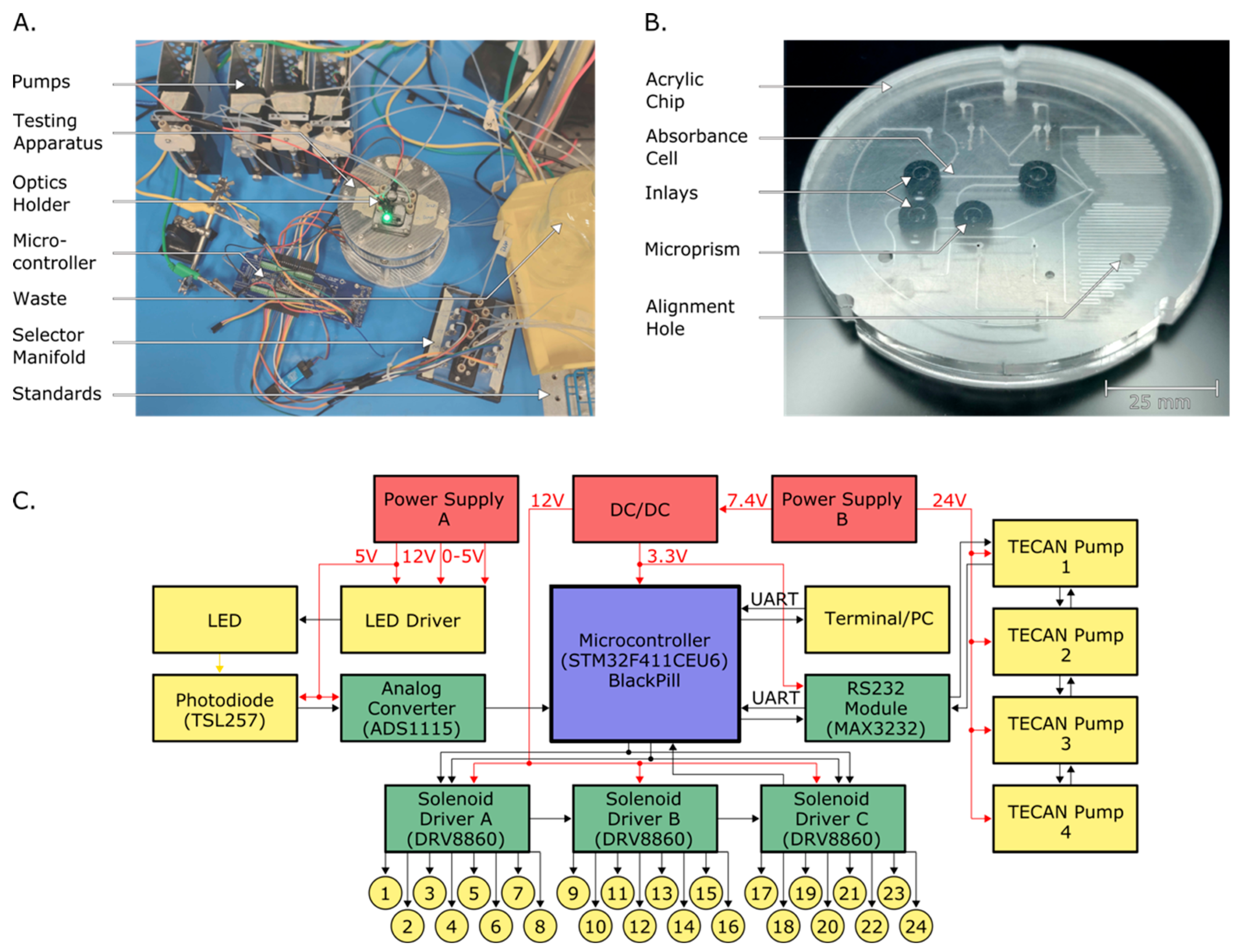
2.3. System Design and Testing Apparatus
2.3.1. Chip Design
2.3.2. System Design
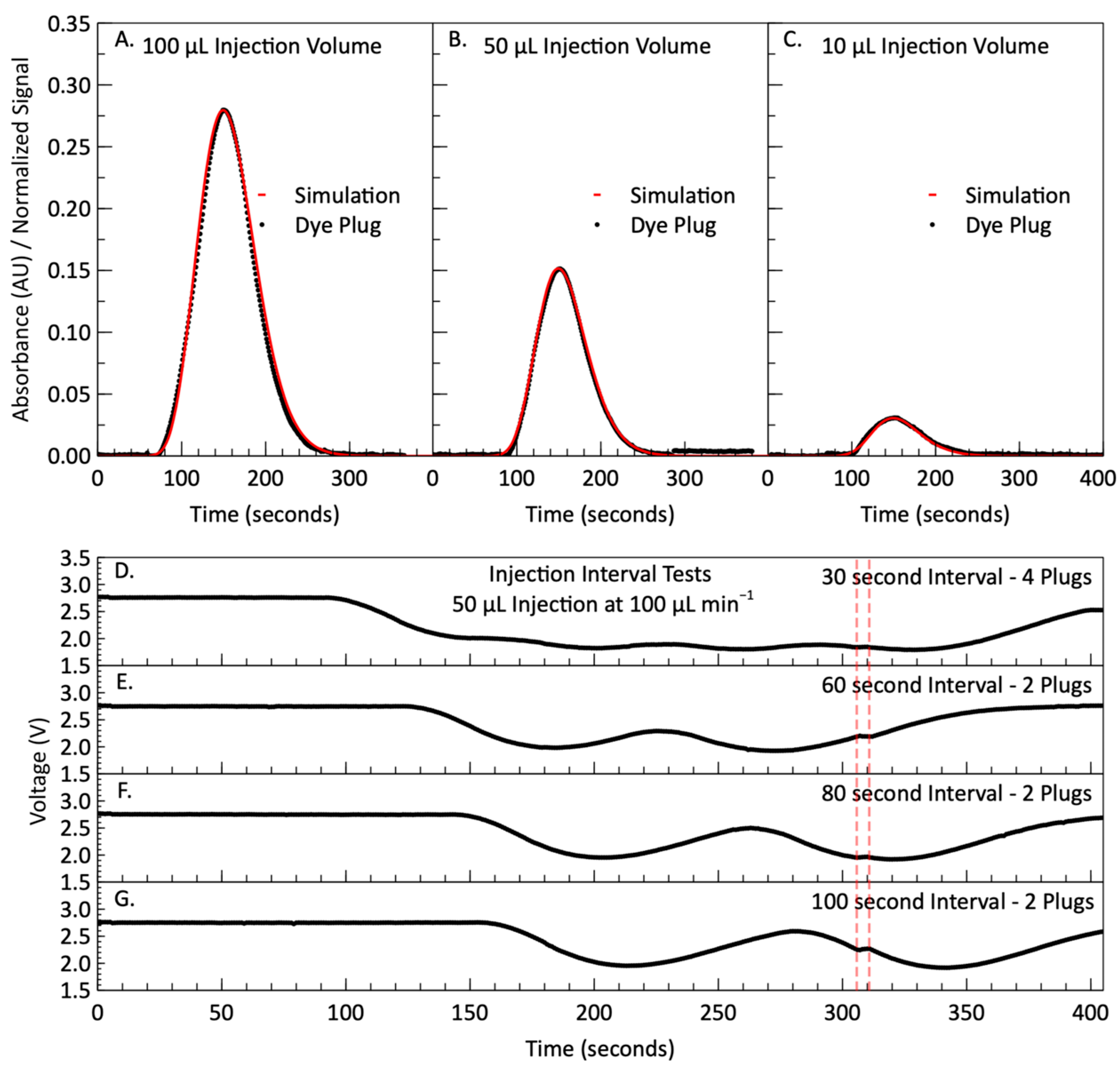
2.4. Simulation and Dye Testing Methods
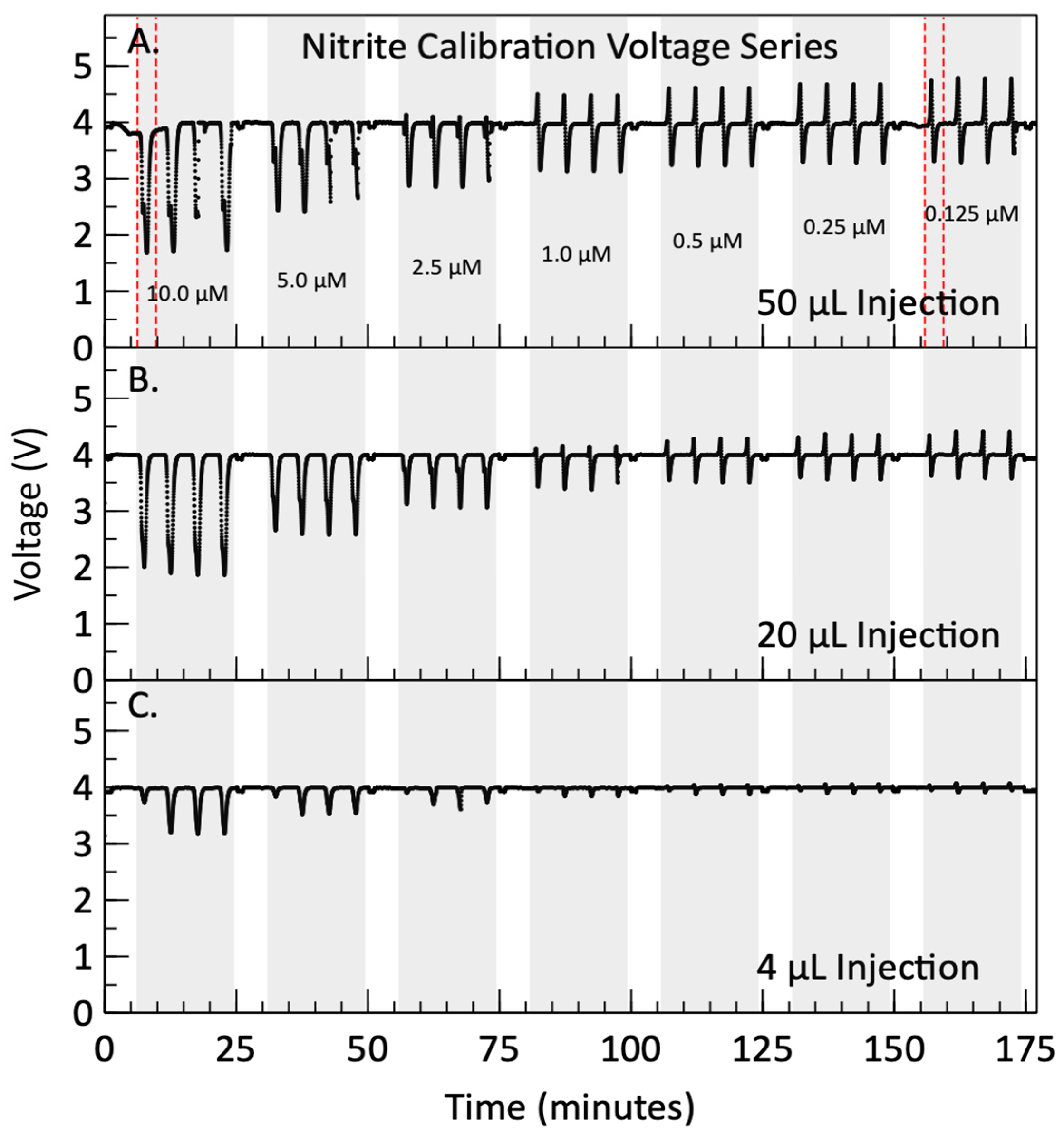
2.5. Nitrite Calibrations Methods
3. Results
3.1. Simulation and Dye Testing Results
3.2. Nitrite Calibration Results
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Sieben, V.J.; Floquet, C.F.A.; Ogilvie, I.R.G.; Mowlem, M.C.; Morgan, H. Microfluidic colourimetric chemical analysis system: Application to nitrite detection. Anal. Methods 2010, 2, 484–491. [Google Scholar] [CrossRef]
- Li, Z.; Liu, H.; Wang, D.; Zhang, M.; Yang, Y.; Ren, T. Recent advances in microfluidic sensors for nutrients detection in water. TrAC Trends Anal. Chem. 2023, 158, 116790. [Google Scholar] [CrossRef]
- Luy, E.; Smith, J.; Grundke, I.; Sonnichsen, C.; Furlong, A.; Sieben, V. Two chemistries on a single lab-on-chip: Nitrate and orthophosphate sensing underwater with inlaid microfluidics. Front. Sens. 2022, 3, 1080020. [Google Scholar] [CrossRef]
- Quan, T.M.; Falkowski, P.G. Redox control of N:P ratios in aquatic ecosystems. Geobiology 2009, 7, 124–139. [Google Scholar] [CrossRef]
- Miller, J. Diurnal and Seasonal Variation in Groundwater Nitrate-n Concentration in a Saturated Buffer Zone. Master’s Thesis, Illinois State University, Normal, IL, USA, 2017. [Google Scholar] [CrossRef]
- Burt, T.P.; Howden, N.J.K.; Worrall, F.; Whelan, M.J. Long-term monitoring of river water nitrate: How much data do we need? J. Environ. Monit. 2010, 12, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Mowlem, M.; Hartman, S.; Harrison, S.; Larkin, K.E. Intercomparison of Biogeochemical Sensors at Ocean Observatories; National Oceanography Centre: Southampton, UK, 2008. [Google Scholar]
- Beaton, A.D.; Schaap, A.M.; Pascal, R.; Hanz, R.; Martincic, U.; Cardwell, C.L.; Morris, A.; Clinton-Bailey, G.; Saw, K.; Hartman, S.E.; et al. Lab-on-Chip for In Situ Analysis of Nutrients in the Deep Sea. ACS Sens. 2022, 7, 89–98. [Google Scholar] [CrossRef]
- Johnson, K.S.; Sakamoto-Arnold, C.M.; Beehler, C.L. Continuous determination of nitrate concentrations in situ. Deep Sea Res. Part A Oceanogr. Res. Pap. 1989, 36, 1407–1413. [Google Scholar] [CrossRef]
- Vuillemin, R.; Le Roux, D.; Dorval, P.; Bucas, K.; Sudreau, J.P.; Hamon, M.; Le Gall, C.; Sarradin, P.M. CHEMINI: A new in situ CHEmical MINIaturized analyzer. Deep Sea Res. Part I Oceanogr. Res. Pap. 2009, 56, 1391–1399. [Google Scholar] [CrossRef]
- Daniel, A.; Birot, D.; Blain, S.; Tréguer, P.; Leïldé, B.; Menut, E. A submersible flow-injection analyser for the in-situ determination of nitrite and nitrate in coastal waters. Mar. Chem. 1995, 51, 67–77. [Google Scholar] [CrossRef]
- Le Bris, N.; Sarradin, P.-M.; Birot, D.; Alayse-Danet, A.-M. A new chemical analyzer for in situ measurement of nitrate and total sulfide over hydrothermal vent biological communities. Mar. Chem. 2000, 72, 1–15. [Google Scholar] [CrossRef]
- Thouron, D.; Vuillemin, R.; Philippon, X.; Lourenço, A.; Provost, C.; Cruzado, A.; Garçon, V. An Autonomous Nutrient Analyzer for Oceanic Long-Term in Situ Biogeochemical Monitoring. Anal. Chem. 2003, 75, 2601–2609. [Google Scholar] [CrossRef]
- Gould, J.; Roemmich, D.; Wijffels, S.; Freeland, H.; Ignaszewsky, M.; Jianping, X.; Pouliquen, S.; Desaubies, Y.; Send, U.; Radhakrishnan, K.; et al. Argo profiling floats bring new era of in situ ocean observations. Eos Trans. Am. Geophys. Union 2004, 85, 185–191. [Google Scholar] [CrossRef]
- Johnson, K.S.; Needoba, J.A. Mapping the spatial variability of plankton metabolism using nitrate and oxygen sensors on an autonomous underwater vehicle. Limnol. Oceanogr. 2008, 53, 2237–2250. [Google Scholar] [CrossRef]
- Webb, D.C.; Simonetti, P.J.; Jones, C.P. SLOCUM: An underwater glider propelled by environmental energy. IEEE J. Ocean. Eng. 2001, 26, 447–452. [Google Scholar] [CrossRef]
- Beaton, A.D.; Cardwell, C.L.; Thomas, R.S.; Sieben, V.J.; Legiret, F.-E.; Waugh, E.M.; Statham, P.J.; Mowlem, M.C.; Morgan, H. Lab-on-Chip Measurement of Nitrate and Nitrite for In Situ Analysis of Natural Waters. Environ. Sci. Technol. 2012, 46, 9548–9556. [Google Scholar] [CrossRef]
- Schierenbeck, T.M.; Smith, M.C. Path to Impact for Autonomous Field Deployable Chemical Sensors: A Case Study of in Situ Nitrite Sensors. Environ. Sci. Technol. 2017, 51, 4755–4771. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, D.; Li, D. Electrochemical and Other Methods for Detection and Determination of Dissolved Nitrite: A Review. Int. J. Electrochem. Sci. 2015, 10, 1144–1168. [Google Scholar] [CrossRef]
- Haldorai, Y.; Kim, J.Y.; Vilian, A.T.E.; Heo, N.S.; Huh, Y.S.; Han, Y.-K. An enzyme-free electrochemical sensor based on reduced graphene oxide/Co3O4 nanospindle composite for sensitive detection of nitrite. Sens. Actuators B Chem. 2016, 227, 92–99. [Google Scholar] [CrossRef]
- Shi, H.; Fu, L.; Chen, F.; Zhao, S.; Lai, G. Preparation of highly sensitive electrochemical sensor for detection of nitrite in drinking water samples. Environ. Res. 2022, 209, 112747. [Google Scholar] [CrossRef]
- Almeida, M.G.; Serra, A.; Silveira, C.M.; Moura, J.J.G. Nitrite Biosensing via Selective Enzymes—A Long but Promising Route. Sensors 2010, 10, 11530–11555. [Google Scholar] [CrossRef]
- Wang, J.; Zhan, G.; Yang, X.; Zheng, D.; Li, X.; Zhang, L.; Huang, T.; Wang, X. Rapid detection of nitrite based on nitrite-oxidizing bacteria biosensor and its application in surface water monitoring. Biosens. Bioelectron. 2022, 215, 114573. [Google Scholar] [CrossRef] [PubMed]
- Griess, P. Bemerkungen zu der Abhandlung der HH. Weselsky und Benedikt “Ueber einige Azoverbindungen”. Ber. Dtsch. Chem. Ges. 1879, 12, 426–428. [Google Scholar] [CrossRef]
- Mikuška, P.; Večeřa, Z. Simultaneous determination of nitrite and nitrate in water by chemiluminescent flow-injection analysis. Anal. Chim. Acta 2003, 495, 225–232. [Google Scholar] [CrossRef]
- Masserini, R.T.; Fanning, K.A. A sensor package for the simultaneous determination of nanomolar concentrations of nitrite, nitrate, and ammonia in seawater by fluorescence detection. Mar. Chem. 2000, 68, 323–333. [Google Scholar] [CrossRef]
- Sandford, R.C.; Exenberger, A.; Worsfold, P.J. Nitrogen Cycling in Natural Waters using In Situ, Reagentless UV Spectrophotometry with Simultaneous Determination of Nitrate and Nitrite. Environ. Sci. Technol. 2007, 41, 8420–8425. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.S.; Coletti, L.J. In situ ultraviolet spectrophotometry for high resolution and long-term monitoring of nitrate, bromide and bisulfide in the ocean. Deep Sea Res. Part I Oceanogr. Res. Pap. 2002, 49, 1291–1305. [Google Scholar] [CrossRef]
- Bittig, H.C.; Maurer, T.L.; Plant, J.N.; Schmechtig, C.; Wong, A.P.S.; Claustre, H.; Trull, T.W.; Udaya Bhaskar, T.V.S.; Boss, E.; Dall’Olmo, G.; et al. A BGC-Argo Guide: Planning, Deployment, Data Handling and Usage. Front. Mar. Sci. 2019, 6, 502. [Google Scholar] [CrossRef]
- TriOS OPUS Datasheet. Available online: https://www.trios.de/en/files/D02-049en202009-Brochure-OPUS.pdf (accessed on 19 March 2024).
- Johnson, K.S.; Plant, J.N.; Coletti, L.J.; Jannasch, H.W.; Sakamoto, C.M.; Riser, S.C.; Swift, D.D.; Williams, N.L.; Boss, E.; Haëntjens, N.; et al. Biogeochemical sensor performance in the SOCCOM profiling float array. J. Geophys. Res. Ocean. 2017, 122, 6416–6436. [Google Scholar] [CrossRef]
- Plant, J.N.; Johnson, K.S.; Sakamoto, C.M.; Jannasch, H.W.; Coletti, L.J.; Riser, S.C.; Swift, D.D. Net community production at Ocean Station Papa observed with nitrate and oxygen sensors on profiling floats. Glob. Biogeochem. Cycles 2016, 30, 859–879. [Google Scholar] [CrossRef]
- Johnson, K.S.; Riser, S.C.; Karl, D.M. Nitrate supply from deep to near-surface waters of the North Pacific subtropical gyre. Nature 2010, 465, 1062–1065. [Google Scholar] [CrossRef]
- Pidcock, R.; Srokosz, M.; Allen, J.; Hartman, M.; Painter, S.; Mowlem, M.; Hydes, D.; Martin, A. A Novel Integration of an Ultraviolet Nitrate Sensor On Board a Towed Vehicle for Mapping Open-Ocean Submesoscale Nitrate Variability. J. Atmos. Ocean. Technol. 2010, 27, 1410–1416. [Google Scholar] [CrossRef]
- SUNA User Manual. Available online: https://www.seabird.com/asset-get.download.jsa?id=54627862534 (accessed on 19 March 2024).
- Sakamoto, C.M.; Johnson, K.S.; Coletti, L.J. Improved algorithm for the computation of nitrate concentrations in seawater using an in situ ultraviolet spectrophotometer. Limnol. Oceanogr. Methods 2009, 7, 132–143. [Google Scholar] [CrossRef]
- Christensen, J.P.; Melling, H. Correcting Nitrate Profiles Measured by the In Situ Ultraviolet Spectrophotometer in Arctic Ocean Waters. Open Oceanogr. J. 2009, 3, 59–66. [Google Scholar] [CrossRef]
- Johnson, K.S.; Beehler, C.L.; Sakamoto-Arnold, C.M. A submersible flow analysis system. Anal. Chim. Acta 1986, 179, 245–257. [Google Scholar] [CrossRef]
- Jannasch, H.W.; Johnson, K.S.; Sakamoto, C.M. Submersible, Osmotically Pumped Analyzer for Continuous Determination of Nitrate in situ. Anal. Chem. 1994, 66, 3352–3361. [Google Scholar] [CrossRef]
- Yang, Z.; Li, C.; Chen, F.; Liu, C.; Cai, Z.; Cao, W.; Li, Z. An in situ analyzer for long-term monitoring of nitrite in seawater with versatile liquid waveguide capillary cells: Development, optimization and application. Mar. Chem. 2022, 245, 104149. [Google Scholar] [CrossRef]
- Ogilvie, I.R.G.; Sieben, V.J.; Mowlem, M.C.; Morgan, H. Temporal Optimization of Microfluidic Colorimetric Sensors by Use of Multiplexed Stop-Flow Architecture. Anal. Chem. 2011, 83, 4814–4821. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, A.M.; Hassan, S.; Warren, B.M.; Makris, K.; Evans, G.W.H.; Papadopoulou, E.; Coleman, S.; Niu, X. A Droplet Microfluidic-Based Sensor for Simultaneous in Situ Monitoring of Nitrate and Nitrite in Natural Waters. Environ. Sci. Technol. 2019, 53, 9677–9685. [Google Scholar] [CrossRef] [PubMed]
- Clinton-Bailey, G.S.; Grand, M.M.; Beaton, A.D.; Nightingale, A.M.; Owsianka, D.R.; Slavik, G.J.; Connelly, D.P.; Cardwell, C.L.; Mowlem, M.C. A Lab-on-Chip Analyzer for in Situ Measurement of Soluble Reactive Phosphate: Improved Phosphate Blue Assay and Application to Fluvial Monitoring. Environ. Sci. Technol. 2017, 51, 9989–9995. [Google Scholar] [CrossRef]
- Birchill, A.J.; Beaton, A.D.; Hull, T.; Kaiser, J.; Mowlem, M.; Pascal, R.; Schaap, A.; Voynova, Y.G.; Williams, C.; Palmer, M. Exploring Ocean Biogeochemistry Using a Lab-on-Chip Phosphate Analyser on an Underwater Glider. Front. Mar. Sci. 2021, 8, 698102. [Google Scholar] [CrossRef]
- Fukuba, T.; Fujii, T. Lab-on-a-chip technology for in situ combined observations in oceanography. Lab Chip 2021, 21, 55–74. [Google Scholar] [CrossRef] [PubMed]
- Greenway, G.M.; Haswell, S.J.; Petsul, P.H. Characterisation of a micro-total analytical system for the determination of nitrite with spectrophotometric detection. Anal. Chim. Acta 1999, 387, 1–10. [Google Scholar] [CrossRef]
- Beaton, A.D.; Sieben, V.J.; Floquet, C.F.A.; Waugh, E.M.; Abi Kaed Bey, S.; Ogilvie, I.R.G.; Mowlem, M.C.; Morgan, H. An automated microfluidic colourimetric sensor applied in situ to determine nitrite concentration. Sens. Actuators B Chem. 2011, 156, 1009–1014. [Google Scholar] [CrossRef]
- Manbohi, A.; Ahmadi, S.H. Portable smartphone-based colorimetric system for simultaneous on-site microfluidic paper-based determination and mapping of phosphate, nitrite and silicate in coastal waters. Environ. Monit. Assess. 2022, 194, 190. [Google Scholar] [CrossRef] [PubMed]
- Bowden, M.; Diamond, D. The determination of phosphorus in a microfluidic manifold demonstrating long-term reagent lifetime and chemical stability utilising a colorimetric method. Sens. Actuators B Chem. 2003, 90, 170–174. [Google Scholar] [CrossRef]
- Jayawardane, B.M.; Wei, S.; McKelvie, I.D.; Kolev, S.D. Microfluidic Paper-Based Analytical Device for the Determination of Nitrite and Nitrate. Anal. Chem. 2014, 86, 7274–7279. [Google Scholar] [CrossRef] [PubMed]
- Duffy, G.; Maguire, I.; Heery, B.; Nwankire, C.; Ducrée, J.; Regan, F. PhosphaSense: A fully integrated, portable lab-on-a-disc device for phosphate determination in water. Sens. Actuators B Chem. 2017, 246, 1085–1091. [Google Scholar] [CrossRef]
- Jang, A.; Zou, Z.; Lee, K.K.; Ahn, C.H.; Bishop, P.L. State-of-the-art lab chip sensors for environmental water monitoring. Meas. Sci. Technol. 2011, 22, 032001. [Google Scholar] [CrossRef]
- Yücel, M.; Beaton, A.D.; Dengler, M.; Mowlem, M.C.; Sohl, F.; Sommer, S. Nitrate and Nitrite Variability at the Seafloor of an Oxygen Minimum Zone Revealed by a Novel Microfluidic In-Situ Chemical Sensor. PLoS ONE 2015, 10, e0132785. [Google Scholar] [CrossRef]
- Beaton, A.D.; Wadham, J.L.; Hawkings, J.; Bagshaw, E.A.; Lamarche-Gagnon, G.; Mowlem, M.C.; Tranter, M. High-Resolution in Situ Measurement of Nitrate in Runoff from the Greenland Ice Sheet. Environ. Sci. Technol. 2017, 51, 12518–12527. [Google Scholar] [CrossRef]
- Vincent, A.G.; Pascal, R.W.; Beaton, A.D.; Walk, J.; Hopkins, J.E.; Woodward, E.M.S.; Mowlem, M.; Lohan, M.C. Nitrate drawdown during a shelf sea spring bloom revealed using a novel microfluidic in situ chemical sensor deployed within an autonomous underwater glider. Mar. Chem. 2018, 205, 29–36. [Google Scholar] [CrossRef]
- James, D.; Oag, B.; Rushworth, C.M.; Lee, J.W.; Davies, J.; Cabral, J.T.; Vallance, C. High-sensitivity online detection for microfluidics via cavity ringdown spectroscopy. RSC Adv. 2012, 2, 5376–5384. [Google Scholar] [CrossRef]
- Dore, J.E.; Karl, D.M. Nitrite distributions and dynamics at Station ALOHA. Deep Sea Res. Part II Top. Stud. Oceanogr. 1996, 43, 385–402. [Google Scholar] [CrossRef]
- Luy, E.A.; Morgan, S.C.; Creelman, J.J.; Murphy, B.J.; Sieben, V.J. Inlaid microfluidic optics: Absorbance cells in clear devices applied to nitrite and phosphate detection. J. Micromech. Microeng. 2020, 30, 095001. [Google Scholar] [CrossRef]
- Morgan, S.; Luy, E.; Furlong, A.; Sieben, V. A submersible phosphate analyzer for marine environments based on inlaid microfluidics. Anal. Methods 2021, 14, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Bowden, M.; Sequiera, M.; Peter Krog, J.; Gravesen, P.; Diamond, D. Analysis of river water samples utilising a prototype industrial sensing system for phosphorus based on micro-system technology. J. Environ. Monit. 2002, 4, 767–771. [Google Scholar] [CrossRef] [PubMed]
- Slater, C.; Cleary, J.; Lau, K.-T.; Snakenborg, D.; Corcoran, B.; Kutter, J.P.; Diamond, D. Validation of a fully autonomous phosphate analyser based on a microfluidic lab-on-a-chip. Water Sci. Technol. 2010, 61, 1811–1818. [Google Scholar] [CrossRef] [PubMed]
- McGraw, C.M.; Stitzel, S.E.; Cleary, J.; Slater, C.; Diamond, D. Autonomous microfluidic system for phosphate detection. Talanta 2007, 71, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cisneros, C.; da Rocha, Z.; Seabra, A.; Valdés, F.; Alonso-Chamarro, J. Highly integrated autonomous lab-on-a-chip device for on-line and in situ determination of environmental chemical parameters. Lab Chip 2018, 18, 1884–1890. [Google Scholar] [CrossRef]
- Lin, K.; Xu, J.; Guo, H.; Huo, Y.; Zhang, Y. Flow injection analysis method for determination of total dissolved nitrogen in natural waters using on-line ultraviolet digestion and vanadium chloride reduction. Microchem. J. 2021, 164, 105993. [Google Scholar] [CrossRef]
- Fornells, E.; Murray, E.; Waheed, S.; Morrin, A.; Diamond, D.; Paull, B.; Breadmore, M. Integrated 3D printed heaters for microfluidic applications: Ammonium analysis within environmental water. Anal. Chim. Acta 2020, 1098, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Miró, M.; Estela, J.M.; Cerdà, V. Application of flowing stream techniques to water analysis. Part I. Ionic species: Dissolved inorganic carbon, nutrients and related compounds. Talanta 2003, 60, 867–886. [Google Scholar] [CrossRef]
- Worsfold, P.J.; Clough, R.; Lohan, M.C.; Monbet, P.; Ellis, P.S.; Quétel, C.R.; Floor, G.H.; McKelvie, I.D. Flow injection analysis as a tool for enhancing oceanographic nutrient measurements—A review. Anal. Chim. Acta 2013, 803, 15–40. [Google Scholar] [CrossRef]
- Socolofsky, S.A.; Jirka, G.H. Environmental Fluid Mechanics. Part I: Mass Transfer and Diffusion. Engineering-Lectures [online]. Available online: https://publikationen.bibliothek.kit.edu/1542004 (accessed on 19 March 2024).
- Hendricks, A.; Mackie, C.M.; Luy, E.; Sonnichsen, C.; Smith, J.; Grundke, I.; Tavasoli, M.; Furlong, A.; Beiko, R.G.; LaRoche, J.; et al. Compact and automated eDNA sampler for in situ monitoring of marine environments. Sci. Rep. 2023, 13, 5210. [Google Scholar] [CrossRef]
- Kreft, J.-U.; Picioreanu, C.; Wimpenny, J.W.T.; van Loosdrecht, M.C.M. Individual-based modelling of biofilms. Microbiology 2001, 147, 2897–2912. [Google Scholar] [CrossRef] [PubMed]
- McKelvie, I.D.; Peat, D.M.W.; Matthews, G.P.; Worsfold, P.J. Elimination of the Schlieren effect in the determination of reactive phosphorus in estuarine waters by flow-injection analysis. Anal. Chim. Acta 1997, 351, 265–271. [Google Scholar] [CrossRef]
- Daniel, A.; Birot, D.; Lehaitre, M.; Poncin, J. Characterization and reduction of interferences in flow-injection analysis for the in situ determination of nitrate and nitrite in sea water. Anal. Chim. Acta 1995, 308, 413–424. [Google Scholar] [CrossRef]
- David, V.; Martínez Calatayud, J.; García Mateo, J.V. Signal Processing Algorithm for Schlieren Effect Correction in Flow Analysis. Anal. Lett. 2001, 34, 1553–1567. [Google Scholar] [CrossRef]
- Griess’ Reagent for Nitrite. Available online: https://www.chemicalbook.com/ChemicalProductProperty_EN_CB6734385.htm (accessed on 19 March 2024).
- Dias, A.C.B.; Borges, E.P.; Zagatto, E.A.G.; Worsfold, P.J. A critical examination of the components of the Schlieren effect in flow analysis. Talanta 2006, 68, 1076–1082. [Google Scholar] [CrossRef]
- Gomes, W. Spectrophotometric determination of nitrite and nitrate in doped potassium chloride crystals. Z. Anal. Chem. 1966, 216, 387–391. [Google Scholar] [CrossRef]
- Ridnour, L.A.; Sim, J.E.; Hayward, M.A.; Wink, D.A.; Martin, S.M.; Buettner, G.R.; Spitz, D.R. A Spectrophotometric Method for the Direct Detection and Quantitation of Nitric Oxide, Nitrite, and Nitrate in Cell Culture Media. Anal. Biochem. 2000, 281, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Loock, H.-P.; Wentzell, P.D. Detection limits of chemical sensors: Applications and misapplications. Sens. Actuators B Chem. 2012, 173, 157–163. [Google Scholar] [CrossRef]
- Shrivastava, A.; Gupta, V.B. Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Chron. Young Sci. 2011, 2, 21–25. [Google Scholar] [CrossRef]
- Liu, B.; Su, H.; Wang, S.; Zhang, Z.; Liang, Y.; Yuan, D.; Ma, J. Automated determination of nitrite in aqueous samples with an improved integrated flow loop analyzer. Sens. Actuators B Chem. 2016, 237, 710–714. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
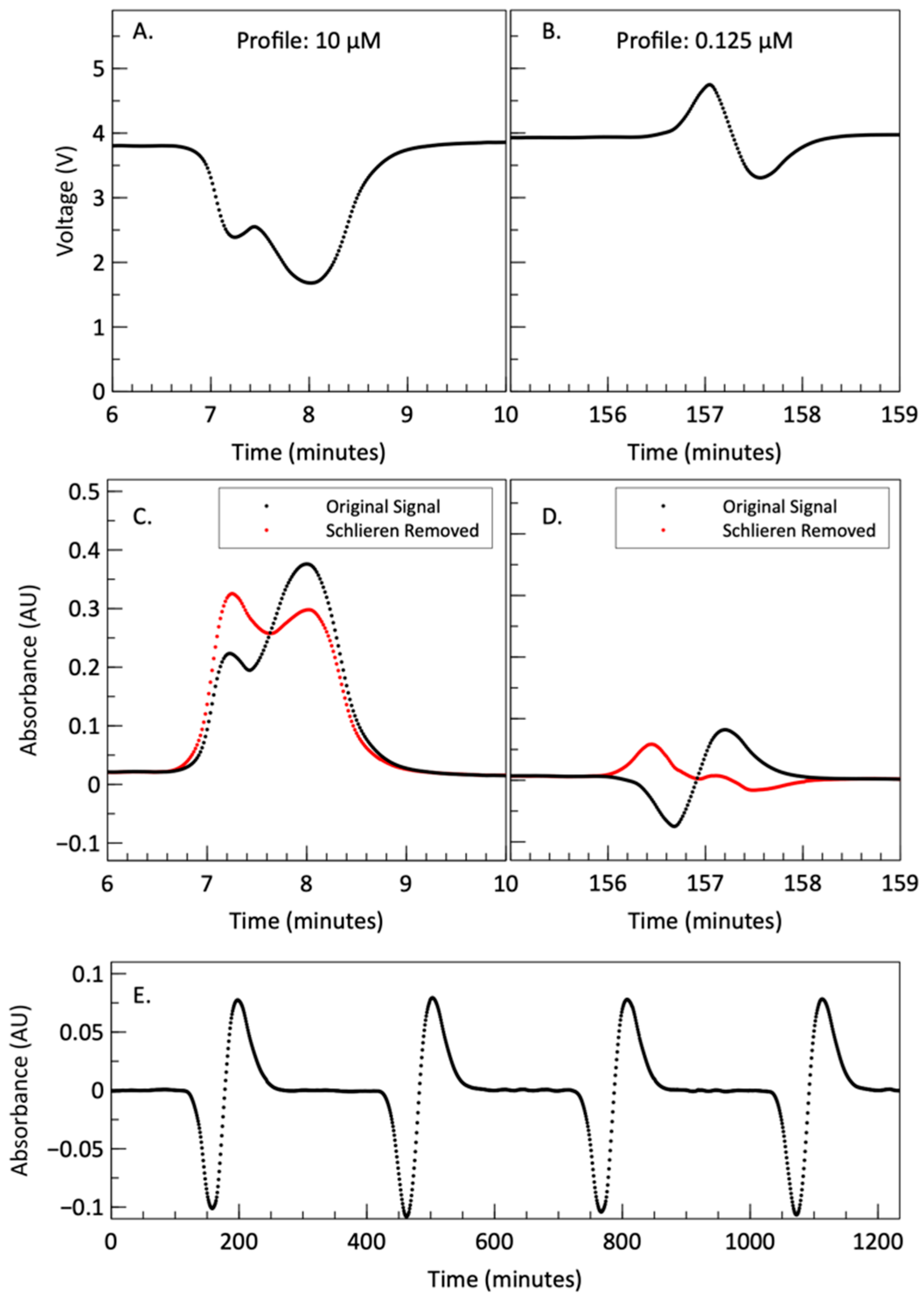{kind=link}
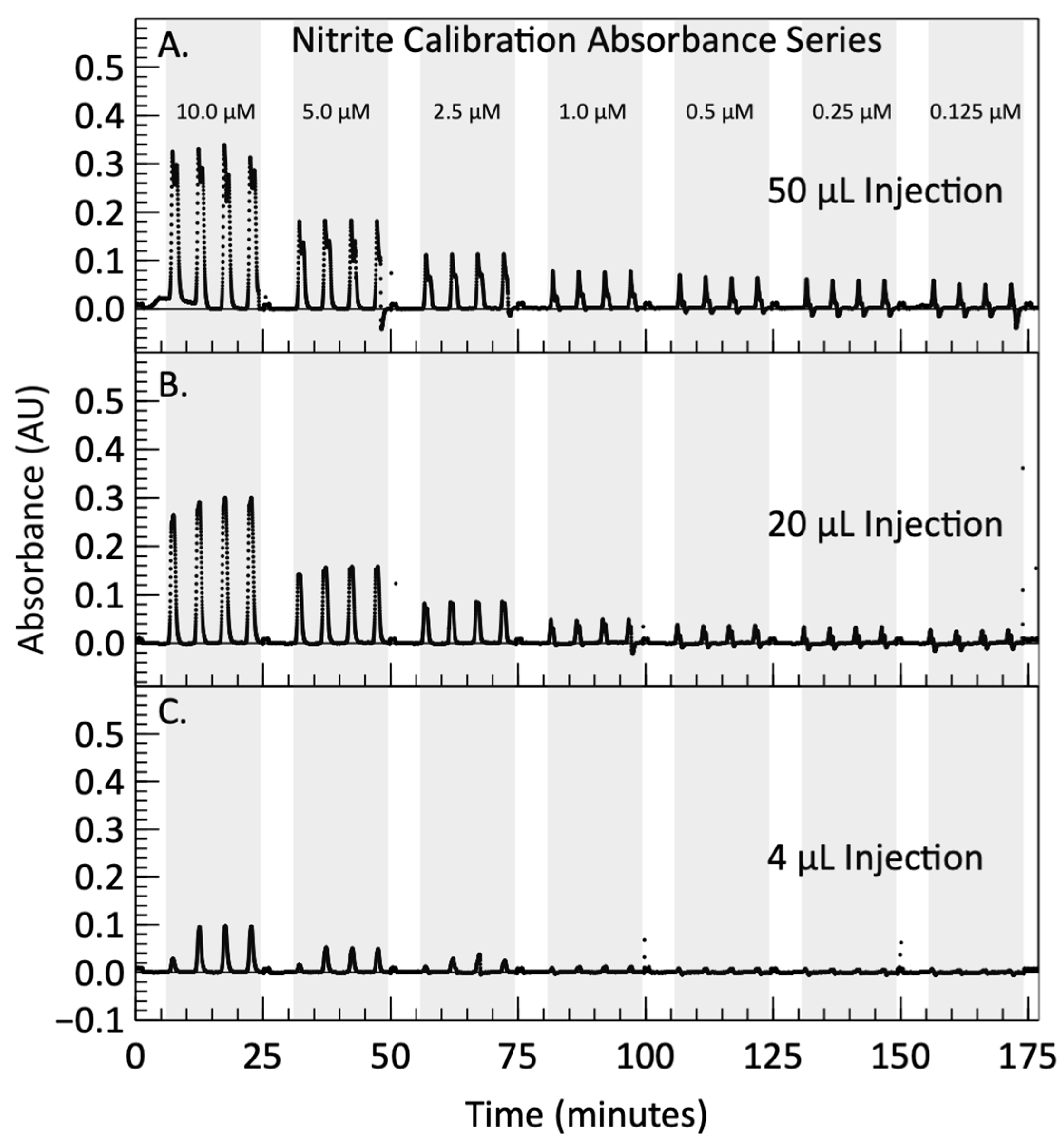{kind=link}
{kind=link}
| Commands | Open | Close | Fill | Push | Pull |
|---|---|---|---|---|---|
| 1. Open valve to draw from environment | V5 | -- | -- | -- | -- |
| 2. Set up to fill reagent syringes | V1, V2 | V3, V4 | -- | -- | -- |
| 3. Fill reagent syringes | -- | -- | R1, R2 | -- | -- |
| 4. Set up to begin flow injection | V3, V4 | V1, V2 | -- | -- | -- |
| 5. Set up push/pull configuration one | V8, V10 | V9, V7 | -- | -- | -- |
| 6. Push/pull configuration one | -- | -- | -- | P2 | P1 |
| 6.1 Delay during step 6 | -- | -- | -- | P2 | P1 |
| 6.2 Reagent injection during step 6 | -- | -- | -- | R1, R2, P2 | P1 |
| 7. Set up push/pull configuration two | V9, V7 | V8, V10 | -- | -- | -- |
| 8. Push/pull configuration two | -- | -- | -- | P1 | P2 |
| 8.1 Delay during step 8 | -- | -- | -- | P1 | P2 |
| 8.2 Reagent injection during step 8 | -- | -- | -- | R1, R2, P1 | P2 |
| 9. Repeat from step 5 until syringes are depleted | |||||
| 10. Repeat from step 2 until sampling is complete | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Motahari, S.; Morgan, S.; Hendricks, A.; Sonnichsen, C.; Sieben, V. Continuous Flow with Reagent Injection on an Inlaid Microfluidic Platform Applied to Nitrite Determination. Micromachines 2024, 15, 519. https://0-doi-org.brum.beds.ac.uk/10.3390/mi15040519
Motahari S, Morgan S, Hendricks A, Sonnichsen C, Sieben V. Continuous Flow with Reagent Injection on an Inlaid Microfluidic Platform Applied to Nitrite Determination. Micromachines. 2024; 15(4):519. https://0-doi-org.brum.beds.ac.uk/10.3390/mi15040519
Chicago/Turabian StyleMotahari, Shahrooz, Sean Morgan, Andre Hendricks, Colin Sonnichsen, and Vincent Sieben. 2024. "Continuous Flow with Reagent Injection on an Inlaid Microfluidic Platform Applied to Nitrite Determination" Micromachines 15, no. 4: 519. https://0-doi-org.brum.beds.ac.uk/10.3390/mi15040519