Tune Up In Situ Autovaccination against Solid Tumors with Oncolytic Viruses


1
Department of Neuro-Oncology, The University of Texas MD Anderson Cancer Center, 6767 Bertner St., Houston, TX 77030, USA
2
Neurosurgery Research, Houston Methodist Research Institute, Houston, TX 77030, USA
3
Pediatric Department, Clínica Universidad de Navarra, 31008 Pamplona, Spain
*
Author to whom correspondence should be addressed.
Cancers 2018, 10(6), 171; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10060171
Submission received: 26 April 2018
/
Revised: 17 May 2018
/
Accepted: 29 May 2018
/
Published: 31 May 2018
(This article belongs to the Special Issue Oncolytic Virotherapy)
Abstract
:With the progress of immunotherapy in cancer, oncolytic viruses (OVs) have attracted more and more attention during the past decade. Due to their cancer-selective and immunogenic properties, OVs are considered ideal candidates to be combined with immunotherapy to increase both specificity and efficacy in cancer treatment. OVs preferentially replicate in and lyse cancer cells, resulting in in situ autovaccination leading to adaptive anti-virus and anti-tumor immunity. The main challenge in OV approaches is how to redirect the host immunity from anti-virus to anti-tumor and optimize the clinical outcome of cancer patients. Here, we summarize the conceptual updates on oncolytic virotherapy and immunotherapy in cancer, and the development of strategies to enhance the virus-mediated anti-tumor immune response, including: (1) arm OVs with cytokines to modulate innate and adaptive immunity; (2) combining OVs with immune checkpoint inhibitors to release T cell inhibition; (3) combining OVs with immune co-stimulators to enhance T cell activation. Future studies need to be enforced on developing strategies to augment the systemic effect on metastasized tumors.
1. Introduction
The history of cancer therapy is a witness of toxicity and failure of efficacy despite numerous efforts to identify druggable cancer targets for personalized and targeted treatments. Emerging evidence indicates that the main challenges in developing efficacious and safe cancer therapeutics are heterogeneity, even within a single cancer, and the evolution of cancer cells during therapy [1,2]. Thus, it is imperative to develop novel strategies to overcome these obstacles.
The immune system is capable of initiating effective responses specifically toward certain molecular targets, such as antigens from pathogens. This type of reaction would be promising for cancer therapy if it could be redirected efficiently against all cancer cell populations. To modulate the immunity against cancers, some pro-inflammatory cytokines, including interleukin-2 (IL-2), interleukin-12 (IL-12), tumor necrosis factor (TNF), and interferon (IFN), have been used to treat malignancies [3,4]. However, the systemic administration of these agents often leads to dose-dependent side-effects (e.g., hypotension, flu-like symptoms, nausea, capillary leak), preventing the escalation to doses that are therapeutically active [4]. During the past two decades, immune checkpoint blockade profoundly changed cancer immunotherapy [5]. Clinical studies have demonstrated the efficacy of these types of therapies in a variety of malignancies, although it is more effective in cancers with an immunogenic tumor microenvironment compared to those with a non-immunogenic microenvironment [5,6]. Though to a less extent, like cytokine therapy, immune checkpoint blockade can cause immune-related adverse events (irAEs) in many patients due to the overstimulation of immune reactivity that may result in autoimmunity [7]. Moreover, to increase the efficacy in patients who are refractory to single antibody blockade, different immune checkpoint blocking antibodies have been combined to treat these patients [8,9]. This may unavoidably increase the risk for irAEs.
To pursue the specificity and safety of immunotherapy, efforts have been made to define cancer-associated antigens and develop therapeutic cancer vaccines. Currently, therapeutic cancer vaccination is only effective as monotherapy for the treatment of premalignant or minimal residual disease, but not in established cancers [10]. Vaccine strategies can increase the frequency and activity of tumor-specific T cells. However, they have failed to ensure that these T cells could infiltrate into tumors and/or exert their function within the tumor due to the immunosuppression in the tumor [10]. Moreover, since cancer vaccines only target a limited number of antigens in the cancer antigen repertoire, after immune editing during the therapy, cancer cells without expression of these antigens can escape and give rise to new tumor cell populations that are resistant to the same vaccine therapy [10,11].
Oncolytic viruses (OVs) are genetically modified or naturally occurring viruses that selectively replicate in and disrupt cancer cells [12,13,14]. Theoretically, these viruses can cause a cascading oncolytic effect in the entire tumor [12,13,14], resulting in the eradication of the infected tumors. However, the viruses have hardly reached their full therapeutic potential due to the antiviral immune responses of the patients and the dynamic immune suppression within the tumor environment [12,13,14]. Nevertheless, OVs have been clinically demonstrated to initiate systemic antitumor immunity due to the in situ cancer vaccination effect of the therapy [12,13,14]. That is, during virotherapy, the in situ viral infection, replication, and subsequent tumor necrosis cooperate to disrupt immunosuppression within the tumor microenvironment, resulting in T cell reactivity against cancer neo-antigens [15,16,17]. Taken together, it seems there could be an opportunity to take advantage of the above strategies to disrupt the immunosuppression within the tumor, upregulate the activity of tumor-specific T cell, and thus further develop more efficacious and safe therapies for cancer patients.
2. In Situ Autovaccination against Cancers Induced by Oncolytic Viruses
Immunity is a double-edged sword in cancer therapy. Thus, it is as important to steer the immune response specifically to cancer cells as to disrupt them efficiently. Unlike cytokine therapy or immune checkpoint blockade that modulate the whole population of certain types of immune cells, cancer vaccines induce immunity specifically against cancer cells. However, in addition to its inefficiency in established cancers [10], this strategy is also challenged by the identification of universal tumor-associated antigens (TAAs) and the difficulty in isolating and preparing individualized vaccines ex vivo [18,19]. During the process of initiation and progression, cancers acquire tens to thousands of nonsynonymous mutations [20,21]. These mutations (either driver or passenger) result in changes of the amino acid sequence or protein structure to produce neo-antigens [22,23]. Since these antigens are not expressed by normal cells, they are predicted to be recognizable by the immune system and be specific targets of immunotherapy. Thus, to improve the effectiveness of cancer vaccine therapy, immunomodulatory agents have been delivered directly into tumors to cause an in situ autovaccination effect. It enhances the immunogenicity of the treated tumor, generates tumor infiltrating lymphocytes (TILs), and triggers a systemic anti-tumor immune response [19].
Accumulating evidence demonstrates that the efficacy of many OVs is at least partly related to the induction of potent antitumor immunity as a result of the in situ vaccination effect of the treatment [12]. When OVs are delivered intratumorally, the infection gives rise to pathogen-associated molecular patterns (PAMPs) [24]. In addition, the replication of the viruses and the consequential lysis of the infected cells, a type of immunogenic cell death, release damage- (or danger-) associated molecular patterns (DAMPs) [17,25,26]. These molecules can be recognized by cells of the innate immune system to initiate an inflammatory immune response [24,27,28]. Thus, the immune suppressive tumor microenvironment is changed to an immune active one, increasing the infiltration of immune cells to the tumor site [16,17]. The tumor-associated antigens (TAAs) from virally lysed cancer cells are released to the tumor milieu and are then cross-presented to T cells by endogenous antigen-presenting cells (APCs), including dendritic cells (DCs) and macrophages. Moreover, tumor cells with or without viral infection can also function as APCs to present TAAs to T cells. In our studies, oncolytic adenoviruses induce autophagy leading to immunogenic cell death, and upregulate proteasome activity and MHC expression in the infected cells, resulting in increased presentation of viral antigens and TAAs to T cells to stimulate their activation [15,17]. The consequential adaptive immunity not only inhibits the treated tumors but also has effect on the distant disseminated tumors and results in immune memory against the same cancer cells [16,17].
Results from clinical trials show that complete responses to OVs as a single agent have rarely been observed [12]. In order to achieve effective virotherapy, induction of potent and sustaining antitumor immunity is critical. During T cell development for adaptive immunity, the T Cell Receptor (TCR) signaling (Signal 1) initiates the reaction, then co-stimulation and/or co-inhibition (Signal 2) shape the outcome of T cells, and cytokines (Signal 3) determine whether Signals 1 and 2 cause tolerance or a productive response leading to potent effector functions, survival, and formation of immune memory [29,30]. Intratumoral delivery of the OVs generates an inflammatory environment in the tumor and initiates the TCR signaling through cross-presenting TAAs to T cells while the reaction is to some extent also modulated by co-signaling and cytokines. Thus, it is logical to combine the viruses with immunotherapeutic strategies to modulate the three signals to enhance the in situ and abscopal antitumor effect (Figure 1).
3. Strategies to Boost Oncolytic Virus-Induced Anti-Cancer Immunity
3.1. Oncolytic Viruses and Cytokines: Modulate the Innate and Adaptive Immune Response
The complex interaction between tumor cells and the tumor microenvironment components, including fibroblasts, extracellular matrix, blood vessels, inflammatory cells, and stimulatory molecules such as chemokines and cytokines, plays an important role in tumor development. Although the interaction mostly occurs via direct cell-cell contact, the secretion of the molecular messenger cytokines stimulates immune cell recruitment to the tumor site. Pro-inflammatory cytokines, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-12, IL-2 and interferons (IFNs), are highly considered for anticancer therapeutic applications [31,32,33,34]. For cancer vaccination strategies, the administration or expression of cytokines at the site of tumors has been shown to increase the efficacy of cytokine therapy and decrease the toxic side effects [35]. Thus, intratumoral delivery of the cytokines with OVs were the first to be developed to modulate anti-tumor immunity (Table 1). Some cytokine-expressing OVs have already been tested in clinical trials.
GM-CSF is secreted by many cell types including T cells, macrophages, endothelial cells, and fibroblasts in response to immune stimuli [50]. It mediates antitumor immune responses by recruiting dendritic cells and macrophages [31]. Injections of GM-CSF-secreting tumor cells increased the infiltration of professional APCs, resulting in the recognition of circulating TAAs by CD4+ and CD8+ T cells [51]. GM-CSF-expressing oncolytic virus Ad5-D24-GMCSF have been shown to mediate tumor-specific immunity in an immunocompetent syngeneic hamster model [52]. In patients with advanced solid tumors refractory to standard therapies, the virus induced immune response in injected and non-injected tumors, resulting in both tumor-specific and virus-specific immunity [52]. Another version of this GM-CSF-expressing virus Ad5/3-D24-GMCSF (ONCOS-102) containing a genetically modified fiber with a serotype 3 knob was tested in a Phase I clinical trial for patients with solid tumors refractory to available treatments [36]. The trial showed that this virus was safe and the patients were able to tolerate the tested dose. Furthermore, this virus was associated with an infiltration of CD8+ T cells and upregulation of PD-L1 within the tumor, and caused an antitumor immune response related with the clinical efficacy. [36]. In addition, a first in human Phase I clinical study of CG0070, an oncolytic adenovirus with selective E1A and GM-CSF expression in Rb pathway-defective cells [53], showed that intravesical delivery of the virus was associated with a tolerable safety profile and anti-bladder cancer activity [37]. Furthermore, a Phase II/III trial of the virus for bcg-refractory non-muscle-invasive bladder cancer demonstrated that intravesical CG0070 caused a durable response in a subset of high-risk patients and has an attractive toxicity profile [38].
Herpes simplex virus type 1 (HSV-1)-derived OVs have also been engineered to express GM-CSF and showed significant tumor growth inhibition in vitro using human tumor cell lines and in vivo using mouse cancer models [54,55]. Talimogene laherparepvec (T-VEC, also known as OncoVEXGM-CSF, ImlygicTM) is the first oncolytic virus to be approved for melanoma treatment by FDA in the USA in 2015 and was subsequently approved in Europe and in Australia in 2016 for clinical trials [56]. In patients with stage IIIC and IV melanoma, direct injection of T-VEC induced local and systemic antigen-specific T cell responses and decreased regulatory T cells (Tregs), suppressor T cells, and myeloid-derived suppressor cells (MDSC) in patients displaying therapeutic responses [39]. The virus was also compared with GM-CSF monotherapy in patients with unresected stage IIIB to IV melanoma in a randomized open-label Phase III trial [40]. Intratumorally administered T-VEC was well tolerated and induced significant benefits in tumor regression, a higher durable response rate, and longer median overall survival than GM-CSF monotherapy [40]. These results suggest that improved systemic immunity might potentially lead to an antitumor and antiviral T cell response and prolong the overall survival. This also underlines the necessity of OV for therapeutic efficacy [40]. Based on these observations, several clinical trials of T-VEC in combination with systemic administration of immune checkpoint inhibitors (ICIs) are ongoing.
Additionally, due to its immunogenic nature, vaccinia virus produces a strong cytotoxic T lymphocyte (CTL) response and has been used to express human GM-CSF (JX-594, also known as Pexa-Vec) [57]. Increased antitumor immune response through infiltration of CD4+ and CD8+ T cells and cancer cell-selective replication were shown in an immunocompetent liver tumor model treated with JX-594 [58]. Phase I, II and III clinical trials were carried out with JX-594 in various cancers in adult and pediatric patients [43,44,45,46,47]. The therapy was well-tolerated and resulted in efficient viral replication in metastatic tumors, which also successfully recruited adaptive immune cells at the site of infection [43,44,45]. A paramyxoviruses family member, Newcastle disease virus (NDV), is one of the naturally occurring viruses with inherent oncolytic ability, and was investigated as novel cancer therapy [59,60]. Recombinant NDVs with an inserted GM-CSF gene increased the stimulation of human peripheral blood mononuclear cells (PBMC) by the infected tumor cells and led to a much higher IFN-α production in these cells compared to the control virus with no transgene, suggesting that GM-CSF-armed NDV could be a potential tumor immunotherapy to enhance immune cell infiltration [61].
IL-12 is produced by phagocytic cells and antigen-presenting cells in response to antigenic stimulation [62]. IL-12 targets NK cells, T cells, DCs, and macrophages, and stimulates the production of IFN-γ [62]. It also mediates T helper type 1 (Th1) differentiation and enhances the cytolytic effect of NK cells and CTLs [62]. IL-12 represents a potential candidate for tumor immunotherapy in murine models of melanoma, colon carcinoma, mammary carcinoma, and sarcoma [63,64,65,66,67,68]. To avoid the clinical toxicity and side effects, OVs offer a promising platform for the delivery of IL-12, restricting its expression within the tumor microenvironment. Several adenoviral vectors expressing IL-12 have been studied to investigate the regression of malignant tumors. The studies in a mouse model of mammary adenocarcinoma demonstrated that tumor regression was mediated through the induction of specific antitumor antigen CTLs which secreted IFN-γ [69]. IL-12 co-expressing B7-1 (YKL-IL12/B7), IL-18 (RdB/IL-12/IL-18), and 4-1BBL oncolytic adenoviruses have been generated to evaluate the antitumor effect of the virus in a murine melanoma B16-F10 tumor model [70,71,72]. The antitumor immunity was shown to be associated with increase of Th1/Th2 cytokine ratio and upregulation of IL-12, IL-18, IFN-γ, and GM-CSF within the tumor tissues [70,71,72]. These results suggest that an antitumor immune response is potentially mediated by the increased antitumor CTLs and IFN-γ-releasing immune cells. The intratumoral administration of oncolytic adenovirus co-expressing IL-23 and p35, the subunit of IL-12, stimulated an antitumor effect in a murine B16-F10 syngeneic tumor model, by inducing the up-regulation of IL-12, IL-23, IFN-γ, and TNF-α within the tumor tissues and reducing the Tregs frequency [73]. Oncolytic adenovirus Ad5-yCD/mutTKSR39rep-mIL12, which expresses two suicide genes and IL-12, induced high levels of IL-12 and IFN-γ in serum and tumor, increased natural killer (NK) and CTL lytic activities, and the developed tumor-specific antitumor immunity in prostate adenocarcinoma model, resulting in a significant increase in survival [74]. In addition, based on favorable results in murine tumor models [75,76,77], a Phase I clinical trial has been designed to evaluate the therapeutic effect of oncolytic HSV M032 expressing human IL-12 in in patients with recurrent/progressive glioblastoma multiforme, anaplastic astrocytoma, or gliosarcoma [48]. Consistently, IL-12 expression also enhanced the anti-tumor activity of oncolytic vesicular stomatitis virus [78,79].
IL-2 is secreted by activated T cells [80]. It is crucial for T cell activation, proliferation and differentiation [80]. NDV expressing human IL-2 was generated and demonstrated to express IL-2 upon infection of various human cancer cell line [81]. IL-2 expression further augmented the immunostimulatory properties of NDV to activate human T cells [82]. Intratumoral injections of recombinant NDV expressing IL-2 elicited immune reaction and induced dramatic reduction in tumor growth, resulting in complete and long-lasting remission in the mice bearing colon carcinoma or hepatoma [83,84]. NDV expressing both IL-2 and/or IL-12 was more efficient than NDV in stimulating INF-γ expression and inducing tumor regression in the murine hepatoma carcinoma model, resulting in immune memory against the same tumor rechallenge [85]. It was reported that IL-12 upregulated IL-2R alpha-chain (CD25) expression and stimulated the proliferation of Th1 clones, but not Th2 clones [86]. This explains the collaborating therapeutic effect of these two cytokines. In a small pilot clinical study, IL-2 expressing oncolytic vaccinia virus demonstrated minimal toxicity and persistent IL-2 expression up to 3 weeks post viral injection [49].
IFN-γ is primarily secreted by T lymphocytes, NK cells, and APCs [87]. It is a major stimulator of macrophages and promotes Th1 differentiation of CD4+ T cells by inhibiting Th2 cytokine production (IL-4 and IL-10) [87]. IFN-γ also enhances the expression of MHC I, MHC II [88], thus promoting antigen presentation and antigen processing and destruction of intracellular pathogens [89,90,91]. An engineered vesicular stomatitis virus (VSV) encoding the IFN-γ has been used in 4T1 mammary adenocarcinoma and other murine tumor models [92]. The virus-treated tumors showed increased activation of DCs and T cell, and slowed tumor growth [92]. This improved efficacy was lost in immunocompromised animals, suggesting the T cell-dependent mechanism of action and the role of IFN-γ in tumor immunosurveillance [92].
In summary, GM-CSF-armed adenovirus, vaccinia virus, and especially HSV, have been examined more intensively in clinic. T-VEC has gained approval in USA and Europe to treat advanced melanomas. IL-12-armed adenovirus and HSV have been tested in Phase I clinical trials. The OVs armed with IL-2 or IFN-γ have been mainly tested pre-clinically.
3.2. Oncolytic Viruses and Immune Checkpoint Blockade: Release the “Brake” on T Cell Activation
The co-signaling (stimulatory or inhibitory) together with TCR signaling direct T cell function and determine T cell fate [29]. The inhibitory immune checkpoint molecules mediate tolerance to self-antigens and prevent auto-immunity [5]. These molecules are expressed on T cells in the tumor microenvironment and inhibit T cell function [5], including well studied cytotoxic T-lymphocyte associated protein 4 (CTLA-4, also known as CD152) [93] and programmed cell death protein 1 (PD-1, also known as CD279) [94].
CTLA-4 decreases T cell activation by outcompeting T cell co-stimulatory receptor CD28 in binding CD80 (also known as B7.1) and CD86 (also known as B7.2), actively delivering inhibitory signals to the T cell [5,95,96,97]. CTLA-4 blockade causes a broad enhancement of immune responses [5]. Recent clinical studies using ipilimumab, a CTLA-4-blocking monoclonal antibody, as monotherapy showed promising results in phase II and III studies among metastatic melanoma patients [98,99]. However, irAEs are quite frequent, which is consistent with the proposed mechanism of action of ipilimumab [100,101].
To improve therapeutic efficacy, immune checkpoint blockade has been combined with OVs (Table 2). VSV in combination with a CTLA-4 antibody was able to amplify a reproducible and statistically significant antitumor T cell immunologic response in murine mammary tumors, eliminating established macroscopic tumor implants [102]. It was also shown that the timing of anti-CTLA-4 mAb treatment was important since the effect depends on amplifying T cell responses [102]. NDV combined with CTLA-4 antibody was used to treat mouse B16 melanoma [16]. Intratumoral administration of NDV induced infiltration of activated CD4 and CD8 T cells in distant (non-virally injected) tumors in the absence of distant viral spread [16]. The inflammation made the tumor tissues susceptible to systemic CTLA-4 blockade therapy, leading to tumor rejection and resulted in increased survival rate in the mice treated with the combined therapy [16]. One systemic dose of vaccinia virus followed by three intra-peritoneal doses of anti-CTLA-4 antibody was used in a pre-clinical therapy in renal and colon adenocarcinoma mouse models, and the therapy showed a robust antitumoral response [103]. However, the induction of CD8 T and NK cells was only observed when the anti-CTLA-4 antibody was administered a few days after vaccinia virus injection [103]. Therapeutic potential of a systemically delivered VSV encoding tumor antigens c-Myc, HIF-2α, and Sox-10 in combination with anti-PD-1 or anti-CTLA-4 therapy has been shown in a pre-clinical glioma model [104]. The use of anti-PD-1 or anti-CTLA-4 alone prolonged the survival of mice [104]. However, the combined therapy with VSV-TAA showed an induction of the Th1 IFN-γ memory and TH17 response, and a significant increase in the survival [104]. The results demonstrated that VSV-TAA could induce a robust immune response when combined with ICIs [104].
PD-1 and its ligands, PD-L1 and PD-L2, are also emerging as promising targets for cancer therapy. PD-1, a transmembrane protein absents on resting naïve and memory T cells, plays an important role in regulating T cell activation [94]. It is transiently upregulated in activated T cells during antigen presentation [94]. PD-1 regulates T cell suppression through binding to PD-L1 or PD-L2 ligand, resulting in inhibition of T effector cell functions [94,105,106]. However, unlike CTLA-4, PD-1 can be expressed by other activated non-T-lymphocyte immune cell subsets, including B cells and NK cells [107]. The upregulation of PD-L1 has been observed on melanoma, lung and ovarian cancer, and glioma cells [17,108,109]. In vivo studies using PD-1, PD-L1, and PD-L2-knockout mice demonstrated a milder auto-immune phenotype compared to CTLA-4-knockout mice [106,110,111]. PD-1/PD-L1 blockers, such as pembrolizumab and nivolumab (anti-PD-1), and atezolizumab, durvalumab, and avelumab (anti-PD-L1), have shown activity in clinical trials, and are gaining approval for an expanding array of indications, including metastatic melanoma, advanced non-small-cell lung cancer, renal cell carcinoma, and classic Hodgkin’s lymphoma [112].
A double-stranded RNA virus reovirus (Reolysin, Oncolytics Biotech Inc., Calgary, AB, Canada) has been used as monotherapy or in combination with anti-PD-1 in pre-clinical and clinical studies [113,114]. In a melanoma mouse model, the combined therapy showed an induction of NK cell-mediated cytotoxicity and CD8-dependent antitumor T cell response as well as a reduction in Treg activity [113]. Another oncolytic virus used in combination with PD-1 blockade is Maraba rhabdovirus. Monotherapy of the virus has been shown to induce NK-mediated cytotoxicity, activate DC maturation, and enhance the production of pro-inflammatory cytokines and chemokines [115,116]. It is effective in several mouse tumor models [115] and is currently being tested in phase I and II clinical trials. A recent pre-clinical study used Maraba virus injected intratumorally or intravenously followed by intraperitoneal injections of both anti-PD1 and anti-CTLA-4 to treat triple-negative breast cancer [117]. Although virus treatment alone upregulated tumor cell PD-L1 expression and tumor-specific Tregs, the combination of the virus with ICIs demonstrated stronger inhibition of tumor growth than the individual therapies [117].
The combination of anti-CTLA-4 with T-VEC has been investigated in a Phase Ib clinical trial for the treatment of advanced melanoma where the intratumoral doses of T-VEC are followed by intravenous ipilimumab administration [118]. The combination had a tolerable safety profile, and showed greater efficacy than either T-VEC or ipilimumab monotherapy [118]. A phase II trial using the combination of Delta-24-RGD adenovirus and PD-1 has begun for patients with recurrent glioblastomas or gliosarcomas. One challenge that should be taken into consideration is that these clinical studies have been carried out in unresectable and advanced stages of cancer patients with slowed immune system. The potential of early stage treatment with more robust immune response should also be considered in order to prevent relapses.
Oncolytic viruses have also been engineered to encode antibodies against immune checkpoint receptors. The attenuated strains of measles virus (MV) encoding antibodies against CTLA-4 and PD-L1 (MV-aCTLA-4 and MV-aPD-L1) have been generated and tested in an immunocompetent murine model of malignant melanoma to evaluate the therapeutic efficacy of the virus [120]. The study showed that the viruses were equally efficient as parental MV in oncolytic efficacy against human tumors [120]. MV-aCTLA-4 enhanced antitumor immunity at early time points after treatment, while the effect of MV-aPD-L1 appeared at later phases of T cell activation in the periphery. This is consistent with CTLA-4 function, where immune responses within the lymphoid organs appeared in the early phase [120]. Contrastingly, PD-L1 signaling occurred at later phases of T cell activation in the periphery [120]. Systemic administration of anti-CTLA-4 or anti-PD-L1 together with intratumoral MV delayed tumor progression and prolonged survival, which was also observed with local, MV-mediated ICI expression [120]. Additionally, Ad5/3-Δ24aCTLA4, an oncolytic adenovirus expressing complete human mAb specific for CTLA-4, was evaluated in vitro and in vivo [119]. In this study, T cells of cancer patients, but not those of healthy donors, were activated by an anti-CTLA4 mAb produced by the virus [119]. High mAb concentrations were seen in tumors, compared to lower systemic levels of the antibody in mice [119].
Since T-VEC has been approved for melanoma patient treatment, the combination of this virus with ICIs has more advance in clinical testing than other OVs. Moreover, due to better safety profile of anti-PD-1 antibodies, there are more clinical trials for this ICI combined with OVs than for anti-CTLA-4 antibodies. We may expect encouraging results for this strategy in the near future.
3.3. Oncolytic Viruses and Immune Co-Stimulation: Hit the “Gas” for T Cell Activation
Since the discovery of CD28 to prove the two-signal model of T cell activation, more and more molecules have been reported to have co-signaling function. The agonists for some of the co-stimulating receptors of the immunoglobulin (CD28, ICOS) and tumor necrosis factor (4-1BB, CD27, GITR, OX40) superfamily have been evaluated for cancer therapy [29,121,122,123]. Oncolytic viruses have been combined with 4-1BB, OX40 and ICOS co-stimulating pathways to enhance efficacy of virotherapy (Table 3).
Co-stimulation through 4-1BB and OX40 promotes T cell survival through upregulation of anti-apoptotic factors and activation of AKT to promote cell cycling [29]. OX40 is also implicated with the upregulation of TRAF6, leading to activation of non-canonical NF-κB signaling and induction of IL-9 production in CD4+ T cells [128]. In immune-competent syngeneic mouse models of sarcoma and breast cancer, combining oncolytic vaccinia viruses (intratumoral) and anti-4-1BB agonist antibody (intraperitoneal) significantly reduced the growth of established subcutaneous tumors and the development of pulmonary metastatic lesions compared to either treatment alone [124]. The tumor inhibition was accompanied with an increased frequency of myeloid cells positive for CD11b+ and CD11c+ within the tumor draining lymph nodes, a heightened infiltration of CD8+ effector T and natural killer (NK) cells, and a maintained presence of neutrophils at the tumor site. [124]. Oncolytic vaccinia virus was also used to express 4-1BB ligand (4-1BBL) [125]. The recombinant virus rV-4-1BBL induced modest tumor regression in the poorly immunogenic B16 murine melanoma model [125]. Furthermore, in the context of lymphodepletion, rV-4-1BBL injection promoted MHC I expression, reduced antiviral antibody titers, promoted viral persistence, and rescued effector memory CD8+ T cells, leading to significant improvement of the therapeutic effectiveness of the oncolytic vector [125]. In the same murine melanoma model, an oncolytic adenovirus co-expressing IL-12 and 4-1BBL greatly enhanced the antitumor effect [72]. Co-administration of the virus with DCs elicited greater antitumor and anti-metastatic effects than either treatment alone [72]. The effect of the combination arms is associated with enhanced type-1 antitumor immune response and higher migratory abilities of DCs in tumors [72]. In addition, another oncolytic adenovirus LOAd703, which expresses both CD40L and 4-1BBL, was shown to be a potent immune activator that modulates the stroma to support antitumor responses when it was injected peritumorally in human pancreatic xenografts in nude mice [126].
Recently, we developed an oncolytic adenovirus Delta-24-RGDOX that expresses co-stimulator OX40 ligand (OX40L) [17]. Its receptor OX40 is expressed only in activated effector T cells [129], which means targeting this molecule spares the inactivated naïve T cells. The agonist antibody of OX40 has shown therapeutic effects in both mouse cancer models and late-stage human cancer patients [130,131]. In this new virus, the OX40L-expressing cassette replaced E3 region in the human adenovirus type 5 (Ad5) genome [17]. The therapeutic effect of this new construct was tested in syngeneic murine intracranial glioma models using immune competent mice. Compared to its predecessor Delta-24-RGD, Delta-24-RGDOX augmented the activation of lymphocytes by tumor cells and the expansion of the CD8+ T cells recognizing tumor-associated antigens, resulting in a tumor-specific immunity in immunocompetent mouse glioma models [17]. Consequently, Delta-24-RGDOX induced more potent anti-glioma activity [17]. Importantly, the virus synergized with intratumoral injections of anti-programmed death ligand 1 (PD-L1) antibody to reject gliomas in mice [17].
During CTLA-4 blockade therapy in cancer patients, a marked increase was found in the frequency of T cells expressing inducible co-stimulator (ICOS) in both tumor tissues and blood [132,133]. It was later found that ICOS pathway enhanced the efficacy of CTLA-4 blockade [122,123]. In addition to activation of PI3K-AKT pathway, ICOS also engaged the C-MAF pathway to induce secretion of IL-4 and IL-21 [29,134]. Its signaling is crucial for the induction of the transcriptional repressor BCL-6 which cooperates with C-MAF to promote the Tfh phenotype [135]. Lately, a recombinant NDV expressing ICOS ligand (NDV-ICOSL) was tested in the bilateral flank murine tumor models in immune competent mouse [127]. Intratumoral administration of NDV-ICOSL caused an enhanced infiltration of activated T cells in both virus-injected and distant tumors; importantly, treatment with NDV-ICOSL results in effective rejection of both tumors when used in combination with systemic CTLA-4 blockade [127].
Currently, compared to the other two strategies, this strategy is less-developed and is still at pre-clinical stage. We believe that the OVs armed with immune co-stimulator are worthy to be tested clinically due to the promising pre-clinical results and more tumor-specific immune stimulation.
4. Conclusions and Perspectives
For the last decade, the field of oncolytic virotherapy has experienced a rapid progress. The paradigm has changed from oncolysis to virus-mediated anti-tumor immunity. With the approval of T-VEC for intratumoral therapy of non-resectable metastatic melanoma, oncolytic virotherapy finally starts to deliver its promise as an alternative therapy for cancers that are refractory to traditional chemotherapy, radiotherapy, targeted therapy, and immunotherapy. It provides versatile platforms for designing and optimizing targeted molecular anticancer agents to execute cancer-selective killing through multiplex mechanisms, reducing the chance for cancer cells to develop resistance.
Although T-VEC has gained approval for treatment in advanced melanoma, it doesn’t mean it will be a one-fits-all cancer virotherapy. Currently, OVs from various viral species are under clinical evaluation, including herpesvirus, adenovirus, measles virus, vaccinia virus, reovirus, poliovirus, coxsackievirus, VSV, parvovirus, and retrovirus [136]. Due to the complexity of each viral species, the researchers are usually experts on one virus but not on the others. There is a lack of knowledge in comparing the effect of individual viruses. Thus, it is hard to conclude which virus is advantageous over others for cancer therapy. In addition, different mechanisms of cancer selectivity for each OV also determine the diversified feasibility of OVs in various malignancies. However, when several OVs are suitable for a certain type of cancer, it may be more effective to choose a virus with low seroprevalence in the patients. Moreover, to avoid antiviral immunity developed during virotherapy to dampen the efficacy of the treatment, sequential use of immunologically non-cross-reactive multiple viruses may be adopted [137]. Therefore, before it is proven that one oncolytic viral platform is efficacious in all tumor types, the diversity of virotherapy will continue. Meanwhile, as our knowledge in virology and cancer biology expands, novel virotherapeutic strategies may appear as well. In a recent report, limited clinical data indicate that adenovirus and HSV have demonstrated better safety profile than reovirus and NDV with respect to virus-mediated adverse events in cancer patients [138]. With accumulating data from clinical testing, we should expect more statistically meaningful comparison of the safety and efficacy of different OVs in cancer patients.
The in situ autovaccination of OVs has shown systemic effect on metastatic tumors. But there are still questions to be answered before we can further improve the efficacy to benefit more cancer patients. The disseminated metastatic tumors may already have well established immunosuppressive tumor microenvironment and/or include cancer cells differentiated from the ones in the virally treated tumor, and thus can become non-responsive to the anti-tumor immunity mediated by the intratumoral viral injection at one or limited tumor sites. Will systemically delivered OVs be able to induce in situ vaccination at each lesion site, resulting in more efficacious and sustainable antitumor immunity? To achieve this, OVs need to avoid rapid clearance from the circulation before they reach the tumor sites. One strategy is to circumvent preexisting immunity through choosing OVs with low seroprevalence, such as VSV [137], or modifying OVs to make them less immunogenic and recognizable by pre-existing immunity and/or redirected to the tumor sites [139,140,141]. Another strategy is to use cellular carriers to shield OVs from the adverse conditions in the blood stream. CTLs as well as other cell types, such as dendritic cells and mesenchymal stem cells, have been used as OV carriers [142,143]. However, before this “Trojan Horse” strategy can be efficiently utilized in clinic, several issues need to be addressed. The first is the tumor homing efficiency and specificity. The second is the cytotoxicity of OVs toward the carriers. The third is carrier cell engraftment efficiency.
Currently, we are excited to witness OVs moving from the benchtop to the bedside of cancer patients. There is still a lot to expect in the improvement of the efficacy of OVs by themselves or through combination with other therapies to increase anti-tumor immunity. Meanwhile, cautions should always be given to the safety of virotherapy through confining the viral effect only on tumors as much as possible. Therefore, optimizing virus-mediated in situ autovaccination to improve anti-tumor immunity is a promising strategy for this purpose.
Funding
This research was funded by J.P. Harris Brain Tumor Research Fund, NIH Brain Cancer Spore (P50 CA127001), Cancer Prevention and Research Institute of Texas (RP170066).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Lengauer, T. Managing drug resistance in cancer: Lessons from HIV therapy. Nat. Rev. Cancer 2012, 12, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Dhupkar, P.; Gordon, N. Interleukin-2: Old and New Approaches to Enhance Immune-Therapeutic Efficacy. Adv. Exp. Med. Biol. 2017, 995, 33–51. [Google Scholar] [PubMed]
- Neri, D.; Sondel, P.M. Immunocytokines for cancer treatment: Past, present and future. Curr. Opin. Immunol. 2016, 40, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Michot, J.M.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Lichty, B.D.; Breitbach, C.J.; Stojdl, D.F.; Bell, J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Gomez-Manzano, C.; Rivera-Molina, Y.; Lang, F.F.; Conrad, C.A.; Fueyo, J. Oncolytic adenovirus research evolution: From cell-cycle checkpoints to immune checkpoints. Curr. Opin. Virol. 2015, 13, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W.; Bell, J.C. Oncolytic virotherapy. Nat. Biotechnol. 2012, 30, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Clise-Dwyer, K.; Ruisaard, K.E.; Fan, X.; Tian, W.; Gumin, J.; Lamfers, M.L.; Kleijn, A.; Lang, F.F.; Yung, W.K.; et al. Delta-24-RGD oncolytic adenovirus elicits anti-glioma immunity in an immunocompetent mouse model. PLoS ONE 2014, 9, e97407. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Rivera-Molina, Y.; Gomez-Manzano, C.; Clise-Dwyer, K.; Bover, L.; Vence, L.M.; Yuan, Y.; Lang, F.F.; Toniatti, C.; Hossain, M.B.; et al. Oncolytic Adenovirus and Tumor-Targeting Immune Modulatory Therapy Improve Autologous Cancer Vaccination. Cancer Res. 2017, 77, 3894–3907. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L.; Binder, A.; Brody, J.D. In situ vaccination: Cancer immunotherapy both personalized and off-the-shelf. Mol. Oncol. 2015, 9, 1966–1981. [Google Scholar] [CrossRef] [PubMed]
- Pierce, R.H.; Campbell, J.S.; Pai, S.I.; Brody, J.D.; Kohrt, H.E. In-situ tumor vaccination: Bringing the fight to the tumor. Hum. Vaccines Immunother. 2015, 11, 1901–1909. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Segal, N.H.; Parsons, D.W.; Peggs, K.S.; Velculescu, V.; Kinzler, K.W.; Vogelstein, B.; Allison, J.P. Epitope landscape in breast and colorectal cancer. Cancer Res. 2008, 68, 889–892. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, C.; Gale, M., Jr. Recognition of viruses by cytoplasmic sensors. Curr. Opin. Immunol. 2010, 22, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Workenhe, S.T.; Mossman, K.L. Oncolytic virotherapy and immunogenic cancer cell death: Sharpening the sword for improved cancer treatment strategies. Mol. Ther. 2014, 22, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.E.; Mossman, K.L. Danger, diversity and priming in innate antiviral immunity. Cytokine Growth Factor Rev. 2014, 25, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Fueyo, J. Healing after death: Antitumor immunity induced by oncolytic adenoviral therapy. Oncoimmunology 2014, 3, e947872. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Curtsinger, J.M.; Mescher, M.F. Inflammatory cytokines as a third signal for T cell activation. Curr. Opin. Immunol. 2010, 22, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Borrello, I.; Pardoll, D. GM-CSF-based cellular vaccines: A review of the clinical experience. Cytokine Growth Factor Rev. 2002, 13, 185–193. [Google Scholar] [CrossRef]
- Eklund, J.W.; Kuzel, T.M. A review of recent findings involving interleukin-2-based cancer therapy. Curr. Opin. Oncol. 2004, 16, 542–546. [Google Scholar] [CrossRef] [PubMed]
- Portielje, J.E.; Gratama, J.W.; van Ojik, H.H.; Stoter, G.; Kruit, W.H. IL-12: A promising adjuvant for cancer vaccination. Cancer Immunol. Immunother. 2003, 52, 133–144. [Google Scholar] [PubMed]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. Paracrine Cytokine Adjuvants in Cancer-Immunotherapy. Annu. Rev. Immunol. 1995, 13, 399–415. [Google Scholar] [CrossRef] [PubMed]
- Ranki, T.; Pesonen, S.; Hemminki, A.; Partanen, K.; Kairemo, K.; Alanko, T.; Lundin, J.; Linder, N.; Turkki, R.; Ristimaki, A.; et al. Phase I study with ONCOS-102 for the treatment of solid tumors—An evaluation of clinical response and exploratory analyses of immune markers. J. Immunother. Cancer 2016, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.M.; Lamm, D.L.; Meng, M.V.; Nemunaitis, J.J.; Stephenson, J.J.; Arseneau, J.C.; Aimi, J.; Lerner, S.; Yeung, A.W.; Kazarian, T.; et al. A First in Human Phase 1 Study of CG0070, a GM-CSF Expressing Oncolytic Adenovirus, for the Treatment of Nonmuscle Invasive Bladder Cancer. J. Urol. 2012, 188, 2391–2397. [Google Scholar] [CrossRef] [PubMed]
- Packiam, V.T.; Campanile, A.N.; Barocas, D.A.; Chamie, K.; Davis, R.L.; Kader, A.K.; Lamm, D.L.; Yeung, A.W.; Steinberg, G.D. A Phase II/III Trial of Cg0070, an Oncolytic Adenovirus, for Bcg-Refractory Non-Muscle-Invasive Bladder Cancer (Nmibc). J. Urol. 2016, 195, E142. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2798. [Google Scholar] [CrossRef] [PubMed]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II Clinical Trial of a Granulocyte-Macrophage Colony-Stimulating Factor–Encoding, Second-Generation Oncolytic Herpesvirus in Patients with Unresectable Metastatic Melanoma. J. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef] [PubMed]
- Toda, M.; Rabkin, S.D.; Kojima, H.; Martuza, R.L. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum. Gene Ther. 1999, 10, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Park, B.H.; Hwang, T.; Liu, T.C.; Sze, D.Y.; Kim, J.S.; Kwon, H.C.; Oh, S.Y.; Han, S.Y.; Yoon, J.H.; Hong, S.H.; et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: A phase 1 trial. Lancet Oncol. 2008, 9, 533–542. [Google Scholar] [CrossRef]
- Lattime, E.C.; Mastrangelo, M.J. Intratumoral recombinant GM-CSF encoding vaccinia virus as gene therapy in patients with cutaneous melanoma. Cancer Gene Ther. 2000, 7, S22. [Google Scholar]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat. Med. 2013, 19, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Cripe, T.P.; Ngo, M.C.; Geller, J.I.; Louis, C.U.; Currier, M.A.; Racadio, J.M.; Towbin, A.J.; Rooney, C.M.; Pelusio, A.; Moon, A.; et al. Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol. Ther. 2015, 23, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.J.; Moon, A.; Burke, J.; Hwang, T.H.; Kirn, D.H. A Phase 2, Open-Label, Randomized Study of Pexa-Vec (JX-594) Administered by Intratumoral Injection in Patients with Unresectable Primary Hepatocellular Carcinoma. Methods Mol. Biol. 2015, 1317, 343–357. [Google Scholar] [PubMed]
- Patel, D.M.; Foreman, P.M.; Nabors, L.B.; Riley, K.O.; Gillespie, G.Y.; Markert, J.M. Design of a Phase I Clinical Trial to Evaluate M032, a Genetically Engineered HSV-1 Expressing IL-12, in Patients with Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, or Gliosarcoma. Hum. Gene Ther. Clin. Dev. 2016, 27, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Haenel, T.; Himbeck, R.; Scott, B.; Ramshaw, I.; Lake, R.A.; Harnett, G.; Phillips, P.; Morey, S.; Smith, D.; et al. Replication-restricted vaccinia as a cytokine gene therapy vector in cancer: Persistent transgene expression despite antibody generation. Cancer Gene Ther. 2000, 7, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, C.H.; Roberts, A.I.; Das, J.; Xu, G.; Ren, G.; Zhang, Y.; Zhang, L.; Yuan, Z.R.; Tan, H.S.; et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: What we do and don’t know. Cell Res. 2006, 16, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Jackson, V.; Hamada, H.; Pardoll, D.; Mulligan, R.C. Vaccination with Irradiated Tumor-Cells Engineered to Secrete Murine Granulocyte-Macrophage Colony-Stimulating Factor Stimulates Potent, Specific, and Long-Lasting Antitumor Immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef] [PubMed]
- Cerullo, V.; Pesonen, S.; Diaconu, I.; Escutenaire, S.; Arstila, P.T.; Ugolini, M.; Nokisalmi, P.; Raki, M.; Laasonen, L.; Sarkioja, M.; et al. Oncolytic Adenovirus Coding for Granulocyte Macrophage Colony-Stimulating Factor Induces Antitumoral Immunity in Cancer Patients. Cancer Res. 2010, 70, 4297–4309. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, N.; Ge, Y.; Ennist, D.L.; Zhu, M.Z.; Mina, M.; Ganesh, S.; Reddy, P.S.; Yu, D.C. CG0070, a conditionally replicating granulocyte macrophage colony-stimulating factor-armed oncolytic adenovirus for the treatment of bladder cancer. Clin. Cancer Res. 2006, 12, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.C.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A phase I study of OncoVEX(GM-CSF), a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed]
- BLA125518 Talimogene Lahaparepvec FDA Briefing Document. 2015. Available online: https://www.fda.gov/downloads/BiologicsBloodVaccines/CellularGeneTherapyProducts/ApprovedProducts/UCM469670.pdf (accessed on 17 May 2018).
- Kirn, D.H.; Thorne, S.H. Targeted and armed oncolytic poxviruses: A novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer 2009, 9, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Oh, J.Y.; Park, B.H.; Lee, D.E.; Kim, J.S.; Park, H.E.; Roh, M.S.; Je, J.E.; Yoon, J.H.; Thorne, S.H.; et al. Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF. Mol. Ther. 2006, 14, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Reichard, K.W.; Lorence, R.M.; Cascino, C.J.; Peeples, M.E.; Walter, R.J.; Fernando, M.B.; Reyes, H.M.; Greager, J.A. Newcastle-Disease Virus Selectively Kills Human Tumor-Cells. J. Surg. Res. 1992, 52, 448–453. [Google Scholar] [CrossRef]
- Lech, P.J.; Russell, S.J. Use of attenuated paramyxoviruses for cancer therapy. Expert Rev. Vaccines 2010, 9, 1275–1302. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Wang, W.J.; Xu, Q.; Harper, J.; Carroll, D.; Galinski, M.S.; Suzich, J.; Jin, H. Genetic Modification of Oncolytic Newcastle Disease Virus for Cancer Therapy. J. Virol. 2016, 90, 5343–5352. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.B.; Chamberlain, R.S.; Bronte, V.; Carroll, M.W.; Irvine, K.R.; Moss, B.; Rosenberg, S.A.; Restifo, N.P. IL-12 is an effective adjuvant to recombinant vaccinia virus-based tumor vaccines—Enhancement by simultaneous B7-1 expression. J. Immunol. 1996, 156, 3357–3365. [Google Scholar] [PubMed]
- Strengell, M.; Lehtonen, A.; Matikainen, S.; Julkunen, I. IL-21 enhances SOCS gene expression and inhibits LPS-induced cytokine production in human monocyte-derived dendritic cells. J. Leukoc. Biol. 2006, 79, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Carr, A.L.; Donald, E.J.; Skitzki, J.J.; Okuyama, R.; Stoolman, L.M.; Chang, A.E. Synergistic effects of IL-12 and IL-18 in skewing tumor-reactive T-cell responses towards a type 1 pattern. Cancer Res. 2005, 65, 1063–1070. [Google Scholar] [PubMed]
- Kaufman, H.L.; Flanagan, K.; Lee, C.S.D.; Perretta, D.J.; Horig, H. Insertion of interleukin-2 (IL-2) and interleukin-12 (IL-12) genes into vaccinia virus results in effective anti-tumor responses without toxicity. Vaccine 2002, 20, 1862–1869. [Google Scholar] [CrossRef]
- Yoshimoto, T.; Nagai, N.; Ohkusu, K.; Ueda, H.; Okamura, H.; Nakanishi, K. LPS-stimulated SJL macrophages produce IL-12 and IL-18 that inhibit IgE production in vitro by induction of IFN-gamma production from CD3(int)IL-2R beta(+) T cells. J. Immunol. 1998, 161, 1483–1492. [Google Scholar] [PubMed]
- Yoshimoto, T.; Takeda, K.; Tanaka, T.; Ohkusu, K.; Kashiwamura, S.; Okamura, H.; Akira, S.; Nakanishi, K. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: Synergism with IL-18 for IFN-gamma production. J. Immunol. 1998, 161, 3400–3407. [Google Scholar] [PubMed]
- Addison, C.L.; Bramson, J.L.; Hitt, M.M.; Muller, W.J.; Gauldie, J.; Graham, F.L. Intratumoral coinjection of adenoviral vectors expressing IL-2 and IL-12 results in enhanced frequency of regression of injected and untreated distal tumors. Gene Ther. 1998, 5, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.K.; Lee, J.S.; Zhang, S.N.; Park, J.; Sonn, C.H.; Lee, K.M.; Yun, C.O. Oncolytic adenovirus co-expressing IL-12 and IL-18 improves tumor-specific immunity via differentiation of T cells expressing IL-12R beta(2) or IL-18R alpha. Gene Ther. 2011, 18, 898–942. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kim, J.H.; Choi, K.J.; Choi, I.K.; Kim, H.; Cho, S.; Cho, B.C.; Yun, C.O. Enhanced antitumor effect of oncolytic adenovirus expressing interleukin-12 and B7-1 in an immunocompetent murine model. Clin. Cancer Res. 2006, 12, 5859–5868. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.H.; Zhang, S.N.; Choi, K.J.; Choi, I.K.; Kim, J.H.; Lee, M.G.; Kim, H.; Yun, C.O. Therapeutic and Tumor-specific Immunity Induced by Combination of Dendritic Cells and Oncolytic Adenovirus Expressing IL-12 and 4-1BBL. Mol. Ther. 2010, 18, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.K.; Li, Y.; Oh, E.; Kim, J.; Yun, C.O. Oncolytic Adenovirus Expressing IL-23 and p35 Elicits IFN-gamma- and TNF-alpha-Co-Producing T Cell-Mediated Antitumor Immunity. PLoS ONE 2013, 8, e67512. [Google Scholar]
- Freytag, S.O.; Barton, K.N.; Zhang, Y. Efficacy of oncolytic adenovirus expressing suicide genes and interleukin-12 in preclinical model of prostate cancer. Gene Ther. 2013, 20, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Stafman, L.L.; Garner, E.F.; Mruthyunjayappa, S.; Stewart, J.E.; Friedman, G.K.; Coleman, J.M.; Markert, J.M.; Gillespie, G.Y.; Beierle, E.A. Effect of Repeat Dosing of Engineered Oncolytic Herpes Simplex Virus on Preclinical Models of Rhabdomyosarcoma. Transl. Oncol. 2016, 9, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.D.; Meza-Perez, S.; Bevis, K.S.; Randall, T.D.; Gillespie, G.Y.; Langford, C.; Alvarez, R.D. IL-12 Expressing oncolytic herpes simplex virus promotes anti-tumor activity and immunologic control of metastatic ovarian cancer in mice. J. Ovarian Res. 2016, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Gillory, L.A.; Megison, M.L.; Stewart, J.E.; Mroczek-Musulman, E.; Nabers, H.C.; Waters, A.M.; Kelly, V.; Coleman, J.M.; Markert, J.M.; Gillespie, G.Y.; et al. Preclinical evaluation of engineered oncolytic herpes simplex virus for the treatment of neuroblastoma. PLoS ONE 2013, 8, e77753. [Google Scholar] [CrossRef] [PubMed]
- Jackaman, C.; Nelson, D.J. Cytokine-armed vaccinia virus infects the mesothelioma tumor microenvironment to overcome immune tolerance and mediate tumor resolution. Cancer Gene Ther. 2010, 17, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.J.; Wanna, G.B.; Choi, B.; Aguila, D.; Ebert, O.; Genden, E.M.; Woo, S.L. Interleukin-12 expression enhances vesicular stomatitis virus oncolytic therapy in murine squamous cell carcinoma. Laryngoscope 2007, 117, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Janke, M.; Fournier, P.; Schirrmacher, V. Recombinant Newcastle disease virus expressing human interleukin-2 serves as a potential candidate for tumor therapy. Virus Res. 2008, 136, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Janke, M.; Peeters, B.; Zhao, H.; De Leeuw, O.; Moorman, R.; Arnold, A.; Ziouta, Y.; Fournier, P.; Schirrmacher, V. Activation of human T cells by a tumor vaccine infected with recombinant Newcastle disease virus producing IL-2. Int. J. Oncol. 2008, 33, 823–832. [Google Scholar] [PubMed]
- Vigil, A.; Park, M.S.; Martinez, O.; Chua, M.A.; Xiao, S.; Cros, J.F.; Martinez-Sobrido, L.; Woo, S.L.C.; Garcia-Sastre, A. Use of reverse genetics to enhance the oncolytic properties of newcastle disease virus. Cancer Res. 2007, 67, 8285–8292. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.Z.; He, J.J.; An, Y.; Wang, X.; Liu, Y.Y.; Yan, S.J.; Ye, X.L.; Qi, J.Y.; Zhu, S.L.; Yu, Q.Z.; et al. Recombinant Newcastle disease virus (NDV/Anh-IL-2) expressing human IL-2 as a potential candidate for suppresses growth of hepatoma therapy. J. Pharmacol. Sci. 2016, 132, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.P.; Tian, G.Y.; Liu, Y.Y.; He, J.J.; Gao, X.Y.; Yu, Y.H.; Liu, X.; Zhang, X.; Sun, T.; Liu, S.Q.; et al. Recombinant Newcastle Disease Virus Encoding IL-12 and/or IL-2 as Potential Candidate for Hepatoma Carcinoma Therapy. Technol. Cancer Res. Treat. 2016, 15, Np83–Np94. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, T.; Kato, T.; Igarashi, O.; Inoue, T.; Nariuchi, H., 2nd. Signal Activity of Il-12 on the Proliferation and Il-2r Expression of T-Helper Cell-1 Clone. J. Immunol. 1994, 152, 4919–4928. [Google Scholar] [PubMed]
- Frucht, D.M.; Fukao, T.; Bogdan, C.; Schindler, H.; O’Shea, J.J.; Koyasu, S. IFN-gamma-production by antigen-presenting cells: Mechanisms emerge. Trends Immunol. 2001, 22, 556–560. [Google Scholar] [CrossRef]
- Boehm, U.; Klamp, T.; Groot, M.; Howard, J.C. Cellular responses to interferon-gamma. Annu. Rev. Immunol. 1997, 15, 749–795. [Google Scholar] [CrossRef] [PubMed]
- Nimal, S.; McCormick, A.L.; Thomas, M.S.; Heath, A.W. An interferon gamma-gp120 fusion delivered as a DNA vaccine induces enhanced priming. Vaccine 2005, 23, 3984–3990. [Google Scholar] [CrossRef] [PubMed]
- Heath, A.W.; Devey, M.E.; Brown, I.N.; Richards, C.E.; Playfair, J.H.L. Interferon-Gamma as an Adjuvant in Immunocompromised Mice. Immunology 1989, 67, 520–524. [Google Scholar] [PubMed]
- Kaplan, D.H.; Shankaran, V.; Dighe, A.S.; Stockert, E.; Aguet, M.; Old, L.J.; Schreiber, R.D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7556–7561. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois-Daigneault, M.C.; Roy, D.G.; Falls, T.; Twumasi-Boateng, K.; St-Germain, L.E.; Marguerie, M.; Garcia, V.; Selman, M.; Jennings, V.A.; Pettigrew, J.; et al. Oncolytic vesicular stomatitis virus expressing interferon-gamma has enhanced therapeutic activity. Mol. Ther. Oncolytics 2016, 3, 16001. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpel, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.H. Costimulation of Lymphocytes-T—The Role of Cd28, Ctla-4, and B7/Bb1 in Interleukin-2 Production and Immunotherapy. Cell 1992, 71, 1065–1068. [Google Scholar] [CrossRef]
- Schneider, H.; Mandelbrot, D.A.; Greenwald, R.J.; Ng, F.; Lechler, R.; Sharpe, A.H.; Rudd, C.E. Cutting edge: CTLA-4 (CD152) differentially regulates mitogen-activated protein kinases (extracellular signal-regulated kinase and c-Jun N-terminal kinase) in CD4(+) T cells from receptor/ligand-deficient mice. J. Immunol. 2002, 169, 3475–3479. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Downey, J.; Smith, A.; Zinselmeyer, B.H.; Rush, C.; Brewer, J.M.; Wei, B.; Hogg, N.; Garside, P.; Rudd, C.E. Reversal of the TCR stop signal by CTLA-4. Science 2006, 313, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Hodi, F.S.; Weber, J.S.; Allison, J.P.; Urba, W.J.; Robert, C.; O’Day, S.J.; Hoos, A.; Humphrey, R.; Berman, D.M.; et al. Development of ipilimumab: A novel immunotherapeutic approach for the treatment of advanced melanoma. Ann. N. Y. Acad. Sci. 2013, 1291, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Neyns, B.; Linette, G. Ipilimumab monotherapy in patients with pretreated advanced melanoma: A randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010, 11, 155–164. [Google Scholar] [CrossRef]
- Thompson, J.A.; Berman, D.; Siegal, J.; Minor, D.; Amin, A.; Ron, I.; Ridolfi, R.; Assi, H.; Hamid, O.; Weber, J. Effect of prior treatment status on the efficacy and safety of ipilimumab monotherapy in treatment-naive and previously treated patients with advanced melanoma. J. Clin. Oncol. 2008, 26, 4134–4140. [Google Scholar] [CrossRef]
- Gao, Y.; Whitaker-Dowling, P.; Griffin, J.A.; Barmada, M.A.; Bergman, I. Recombinant vesicular stomatitis virus targeted to Her2/neu combined with anti-CTLA4 antibody eliminates implanted mammary tumors. Cancer Gene Ther. 2009, 16, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Rojas, J.J.; Sampath, P.; Hou, W.Z.; Thorne, S.H. Defining Effective Combinations of Immune Checkpoint Blockade and Oncolytic Virotherapy. Clin. Cancer Res. 2015, 21, 5543–5551. [Google Scholar] [CrossRef] [PubMed]
- Cockle, J.V.; Rajani, K.; Zaidi, S.; Kottke, T.; Thompson, J.; Diaz, R.M.; Shim, K.; Peterson, T.; Parney, I.F.; Short, S.; et al. Combination viroimmunotherapy with checkpoint inhibition to treat glioma, based on location-specific tumor profiling. Neuro-Oncology 2016, 18, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Latchman, Y.; Wood, C.; Chemova, T.; Iwai, Y.; Malenkovich, N.; Long, A.; Bourque, K.; Boussiotis, V.; Nishimura, H.; Honjo, T.; et al. PD-L2, a novel B7 homologue, is a second ligand for PD-1 and inhibits T cell activation. Faseb J. 2001, 15, A345. [Google Scholar]
- Gianchecchi, E.; Delfino, D.V.; Fierabracci, A. Recent insights into the role of the PD-1/PD-L1 pathway in immunological tolerance and autoimmunity. Autoimmun. Rev. 2013, 12, 1091–1100. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.D.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.F.; Tamada, K.; et al. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C.; et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chung, Y.; Bishop, C.; Daugherty, B.; Chute, H.; Hoist, P.; Kurahara, C.; Lott, F.; Sun, N.; Welcher, A.A.; et al. Regulation of T cell activation and tolerance by PDL2. Proc. Natl. Acad. Sci. USA 2006, 103, 11695–11700. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Rajani, K.; Parrish, C.; Kottke, T.; Thompson, J.; Zaidi, S.; Ilett, L.; Shim, K.G.; Diaz, R.M.; Pandha, H.; Harrington, K.; et al. Combination Therapy With Reovirus and Anti-PD-1 Blockade Controls Tumor Growth Through Innate and Adaptive Immune Responses. Mol. Ther. 2016, 24, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.J.; Vile, R.G.; Melcher, A.; Chester, J.; Pandha, H.S. Clinical trials with oncolytic reovirus: Moving beyond phase I into combinations with standard therapeutics. Cytokine Growth Factor Rev. 2010, 21, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Brun, J.; McManus, D.; Lefebvre, C.; Hu, K.; Falls, T.; Atkins, H.; Bell, J.C.; McCart, J.A.; Mahoney, D.; Stojdl, D.F. Identification of Genetically Modified Maraba Virus as an Oncolytic Rhabdovirus. Mol. Ther. 2010, 18, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.G.; Zhang, L.; Bridle, B.W.; Stephenson, K.B.; Resseguier, J.; Hanson, S.; Chen, L.; Kazdhan, N.; Bramson, J.L.; Stojdl, D.F.; et al. Maraba virus as a potent oncolytic vaccine vector. Mol. Ther. 2014, 22, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois-Daigneault, M.C.; Roy, D.G.; Aitken, A.S.; El Sayes, N.; Martin, N.T.; Varette, O.; Falls, T.; St-Germain, L.E.; Pelin, A.; Lichty, B.D.; et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.S.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene Laherparepvec in Combination with Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.D.; Hemminki, O.; Diaconu, I.; Hirvinen, M.; Bonetti, A.; Guse, K.; Escutenaire, S.; Kanerva, A.; Pesonen, S.; Loskog, A.; et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012, 19, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [PubMed]
- Sturgill, E.R.; Redmond, W.L. A Review of Current Biologics Targeting OX40, 4–1BB, CD27, and GITR. Am. J. Hematol. Oncol. 2017, 13, 4–15. [Google Scholar]
- Fan, X.; Quezada, S.A.; Sepulveda, M.A.; Sharma, P.; Allison, J.P. Engagement of the ICOS pathway markedly enhances efficacy of CTLA-4 blockade in cancer immunotherapy. J. Exp. Med. 2014, 211, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Fu, T.; He, Q.; Sharma, P. The ICOS/ICOSL pathway is required for optimal antitumor responses mediated by anti-CTLA-4 therapy. Cancer Res. 2011, 71, 5445–5454. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Howland, L.J.; Flynn, J.K.; West, A.C.; Devaud, C.; Duong, C.P.; Stewart, T.J.; Westwood, J.A.; Guo, Z.S.; Bartlett, D.L.; et al. Oncolytic virus and anti-4-1BB combination therapy elicits strong antitumor immunity against established cancer. Cancer Res. 2012, 72, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim-Schulze, S.; Kim, D.W.; Kaufman, H.L. Host lymphodepletion enhances the therapeutic activity of an oncolytic vaccinia virus expressing 4-1BB ligand. Cancer Res. 2009, 69, 8516–8525. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, E.; Milenova, I.; Wenthe, J.; Stahle, M.; Leja-Jarblad, J.; Ullenhag, G.; Dimberg, A.; Moreno, R.; Alemany, R.; Loskog, A. Shaping the Tumor Stroma and Sparking Immune Activation by CD40 and 4-1BB Signaling Induced by an Armed Oncolytic Virus. Clin. Cancer Res. 2017, 23, 5846–5857. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Ricca, J.; Plitt, T.; Palese, P.; Sharma, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat. Commun. 2017, 8, 14340. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Balasubramanian, S.; Liu, W.; Chu, X.; Wang, H.; Taparowsky, E.J.; Fu, Y.X.; Choi, Y.; Walsh, M.C.; Li, X.C. OX40 signaling favors the induction of T(H)9 cells and airway inflammation. Nat. Immunol. 2012, 13, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, M. Control of immunity by the TNFR-related molecule OX40 (CD134). Annu. Rev. Immunol. 2010, 28, 57–78. [Google Scholar] [CrossRef] [PubMed]
- Kjaergaard, J.; Tanaka, J.; Kim, J.A.; Rothchild, K.; Weinberg, A.; Shu, S. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer Res. 2000, 60, 5514–5521. [Google Scholar] [PubMed]
- Curti, B.D.; Kovacsovics-Bankowski, M.; Morris, N.; Walker, E.; Chisholm, L.; Floyd, K.; Walker, J.; Gonzalez, I.; Meeuwsen, T.; Fox, B.A.; et al. OX40 is a potent immune-stimulating target in late-stage cancer patients. Cancer Res. 2013, 73, 7189–7198. [Google Scholar] [CrossRef] [PubMed]
- Liakou, C.I.; Kamat, A.; Tang, D.N.; Chen, H.; Sun, J.; Troncoso, P.; Logothetis, C.; Sharma, P. CTLA-4 blockade increases IFNgamma-producing CD4+ICOShi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc. Natl. Acad. Sci. USA 2008, 105, 14987–14992. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liakou, C.I.; Kamat, A.; Pettaway, C.; Ward, J.F.; Tang, D.N.; Sun, J.; Jungbluth, A.A.; Troncoso, P.; Logothetis, C.; et al. Anti-CTLA-4 therapy results in higher CD4+ICOShi T cell frequency and IFN-gamma levels in both nonmalignant and malignant prostate tissues. Proc. Natl. Acad. Sci. USA 2009, 106, 2729–2734. [Google Scholar] [CrossRef] [PubMed]
- Simpson, T.R.; Quezada, S.A.; Allison, J.P. Regulation of CD4 T cell activation and effector function by inducible costimulator (ICOS). Curr. Opin. Immunol. 2010, 22, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Kroenke, M.A.; Eto, D.; Locci, M.; Cho, M.; Davidson, T.; Haddad, E.K.; Crotty, S. Bcl6 and Maf cooperate to instruct human follicular helper CD4 T cell differentiation. J. Immunol. 2012, 188, 3734–3744. [Google Scholar] [CrossRef] [PubMed]
- Lawler, S.E.; Speranza, M.C.; Cho, C.F.; Chiocca, E.A. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017, 3, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Peng, K.W. Oncolytic Virotherapy: A Contest between Apples and Oranges. Mol. Ther. 2017, 25, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Karube, H.; Aruga, A. A comparative Safety profile assessment of oncolytic virus therapy based on clinical Trials. Ther. Innov. Regul. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Conner, J.; Braidwood, L.; Brown, S.M. A strategy for systemic delivery of the oncolytic herpes virus HSV1716: Redirected tropism by antibody-binding sites incorporated on the virion surface as a glycoprotein D fusion protein. Gene Ther. 2008, 15, 1579–1592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Krimmel, J.; Zhang, Z.; Hu, Z.; Seth, P. Systemic delivery of a novel liver-detargeted oncolytic adenovirus causes reduced liver toxicity but maintains the antitumor response in a breast cancer bone metastasis model. Hum. Gene. Ther. 2011, 22, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhang, Z.; Yang, Y.; Hu, Z.; Wang, C.H.; Morgan, M.; Wu, Y.; Hutten, R.; Xiao, X.; Stock, S.; et al. Ad5/48 hexon oncolytic virus expressing sTGFbetaRIIFc produces reduced hepatic and systemic toxicities and inhibits prostate cancer bone metastases. Mol. Ther. 2014, 22, 1504–1517. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.S.; Lemoine, N.R.; Wang, Y. Systemic delivery of oncolytic viruses: Hopes and hurdles. Adv. Virol. 2012, 2012, 805629. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hall, R.R.; Lesniak, M.S.; Ahmed, A.U. Stem Cell-Based Cell Carrier for Targeted Oncolytic Virotherapy: Translational Opportunity and Open Questions. Viruses 2015, 7, 6200–6217. [Google Scholar] [CrossRef] [PubMed]
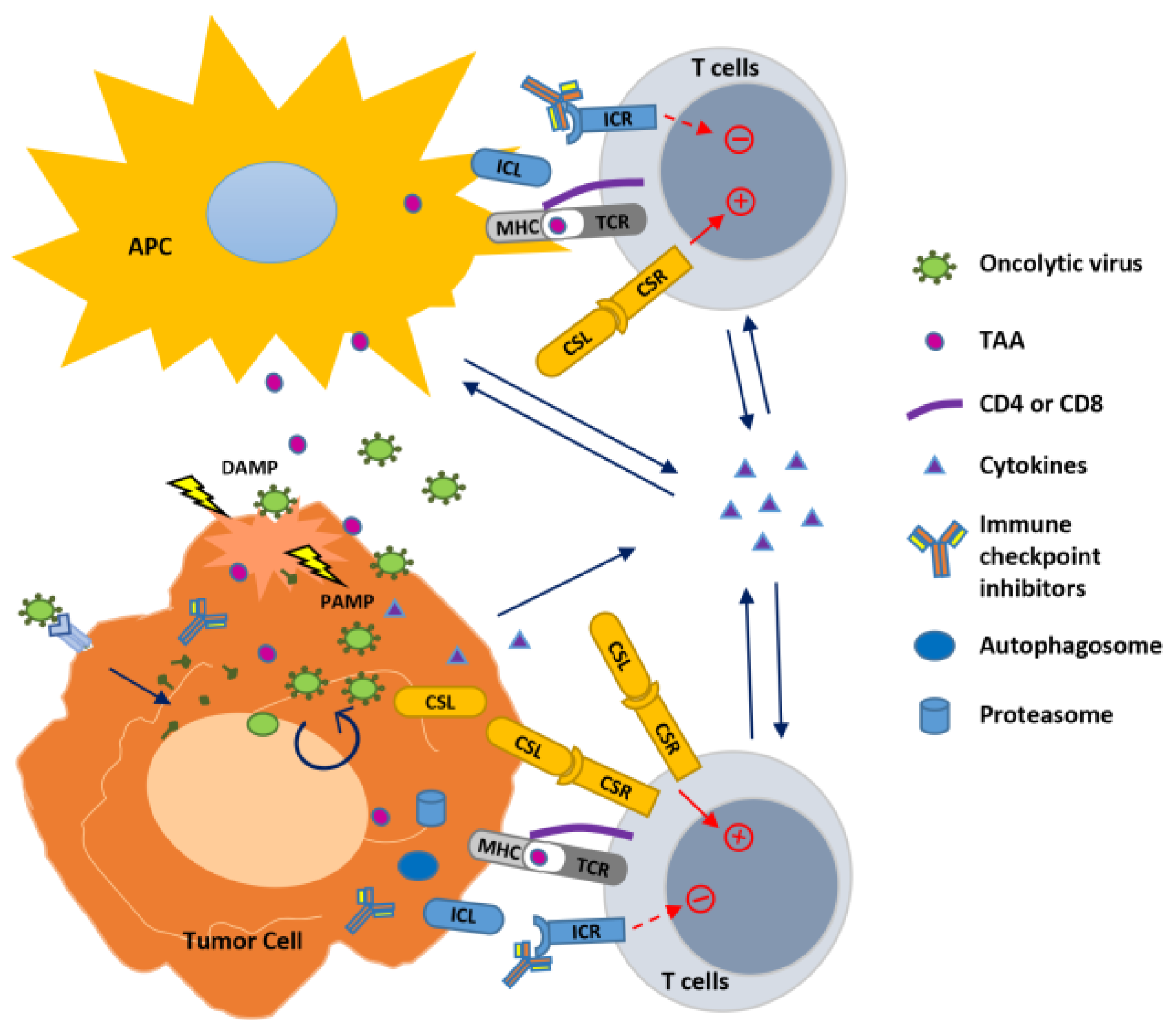
Figure 1.
Strategies to improve oncolytic virus-mediated anti-tumor immunity. Oncolytic viruses (OVs) infect and replicate inside cancer cells, resulting in cell lysis and propagation of virions to infect nearby cancer cells. This process generates pathogen-associated molecular patterns (PAMPs) and damage- (or danger-) associated molecular patterns (DAMPs) that trigger an innate immune response to modulate the tumor microenvironment, resulting in in situ autovaccination leading to adaptive anti-virus and anti-tumor immunity. In the infected tumor cells, OVs also induce autophagy (autophagosome formation) and activity of proteasome to increase their capability to function as APC to present tumor-associated antigens (TAAs) to T cells. OVs have been combined with immune modulators to enhance immunity against the tumor. Cytokines expressed by OVs stimulate innate and adaptive immunity within the tumor. Combination of OVs with immune checkpoint blockade through antibodies to inhibit the interaction between immune checkpoint ligand (ICL) and receptor (ICR), or with agonist antibody or expression of the co-stimulatory ligand (CSL) to bind with the co-stimulatory receptor (CSR) augments T cell receptor (TCR) signaling initiated by the virus through presenting TAAs with major histocompatibility complex (MHC), leading to enhanced T cell activation against the tumor.
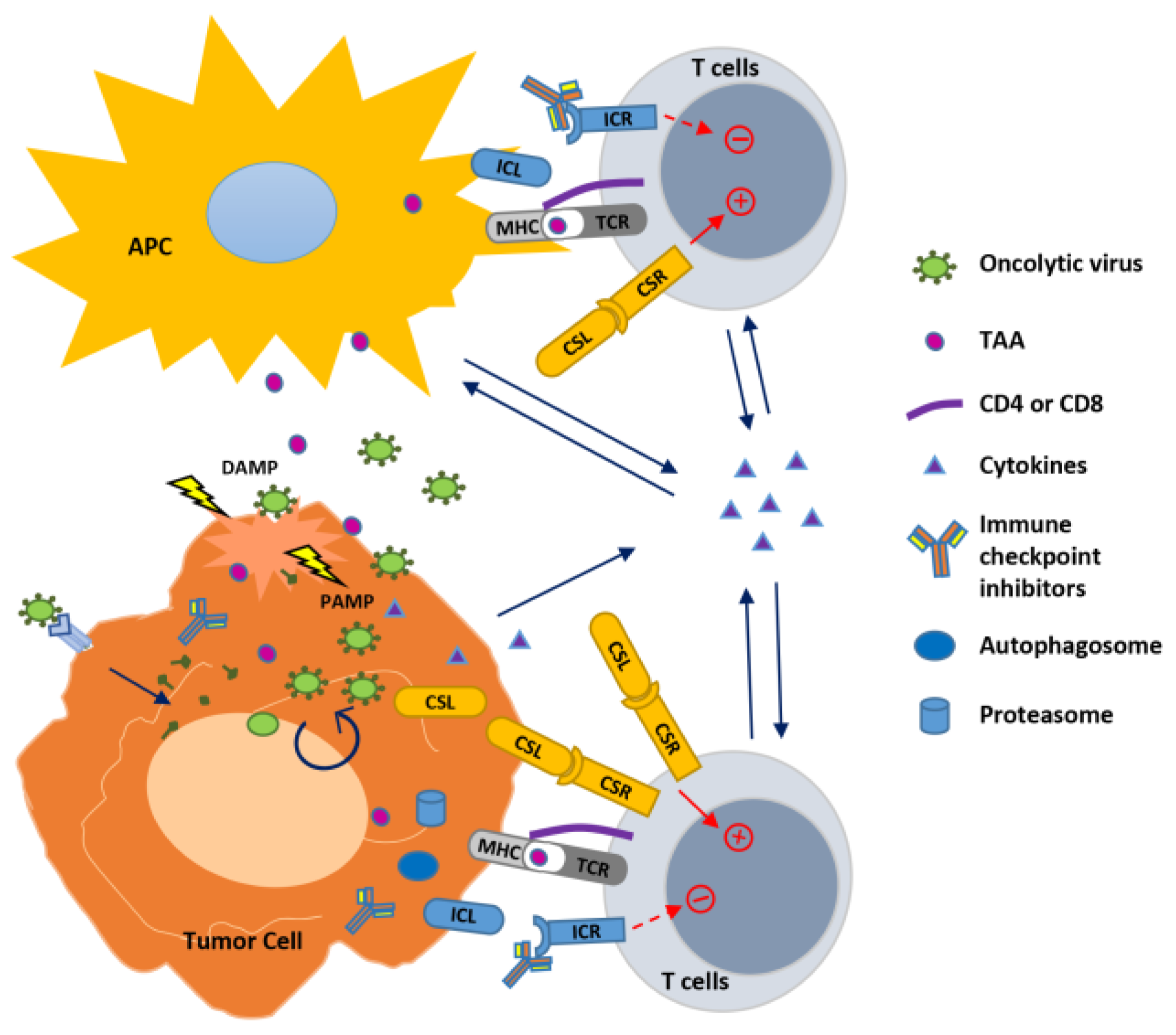
Figure 1.
Strategies to improve oncolytic virus-mediated anti-tumor immunity. Oncolytic viruses (OVs) infect and replicate inside cancer cells, resulting in cell lysis and propagation of virions to infect nearby cancer cells. This process generates pathogen-associated molecular patterns (PAMPs) and damage- (or danger-) associated molecular patterns (DAMPs) that trigger an innate immune response to modulate the tumor microenvironment, resulting in in situ autovaccination leading to adaptive anti-virus and anti-tumor immunity. In the infected tumor cells, OVs also induce autophagy (autophagosome formation) and activity of proteasome to increase their capability to function as APC to present tumor-associated antigens (TAAs) to T cells. OVs have been combined with immune modulators to enhance immunity against the tumor. Cytokines expressed by OVs stimulate innate and adaptive immunity within the tumor. Combination of OVs with immune checkpoint blockade through antibodies to inhibit the interaction between immune checkpoint ligand (ICL) and receptor (ICR), or with agonist antibody or expression of the co-stimulatory ligand (CSL) to bind with the co-stimulatory receptor (CSR) augments T cell receptor (TCR) signaling initiated by the virus through presenting TAAs with major histocompatibility complex (MHC), leading to enhanced T cell activation against the tumor.

{kind=link}
Table 1.
Cytokine-armed OVs under clinical investigation.
| Cytokine | Virus | Modification in Viral Genome | Tested Disease | Route of Administration | Clinical Status |
|---|---|---|---|---|---|
| GM-CSF | Human adenovirus 5 (ONCOS-102) | 24-bp deletion in E1A; modified fiber with a serotype 3 knob | solid tumors refractory to available treatments | Intratumoral and intravenous | Phase I [36] |
| Human adenovirus 5 (CG0070) | E2F-1 promoter /E1A gene, human GM-CSF insertion | Non-muscle invasive bladder cancer after BCG failure | Bladder instillation | Phase II/III [37,38] | |
| HSV-1 (T-VEC) | Deletion of ICP34.5, ICP47, human GM-CSF insertion | Unresected stage IIIB/C to IV melanoma with various metastasis | Intratumoral | Approved in the USA and Europe [39,40,41,42] | |
| Vaccinia virus (JX-594) | Thymidine kinase, human GM-CSF, lacZ insertion | Various cancers in adult and pediatric patients | Intravenous | Phase III trial [43,44,45,46,47] | |
| IL-12 | Human adenovirus 5 (Ad5-yCD/mutTKSR39rep-hIL12) | IL-12, yeast cytosine deaminase (CD), TKSR39 (thymidine kinase mutant) insertions | Non-small cell lung carcinoma (NSCLC) Prostate cancer | Intratumoral | Phase I (NSCLC) Phase II (prostate cancer) |
| HSV-1 (M032) | Deletion of ICP34.5, IL-12 insertion | Recurrent/Progressive Glioblastoma Multiforme, Anaplastic Astrocytoma, Gliosarcoma | Intracerebral | Phase I [48] | |
| IL-2 | Vaccinia virus (VV-IL-2) | Deletion of thymidine kinase, insertion of IL-2 | malignant mesothelioma | Intratumoral | Small pilot study with six patients [49] |
Table 2.
Combinational therapies with ICIs and OVs.
| Antibodies | Virus | Modification in Viral Genome | Tested Disease | Route of Administration | Clinical Status |
|---|---|---|---|---|---|
| Anti-CTLA-4 | VSV | Breast cancer | Intraperitoneally | Pre-clinical [102] | |
| NDV | Colon carcinoma and melanoma | Intratumoral dose of OV followed by intraperitoneal ICIs | Pre-clinical [16] | ||
| HSV-1 (T-VEC) | Deletion of ICP34.5, ICP47, human GM-CSF insertion | Malignant melanoma | Intratumoral dose of OV followed by intravenous ICIs | Phase II [118] | |
| Human adenovirus 5 (Ad5/3-Delta24aCTLA4) | 24-bp deletion in E1A; modified fiber with a serotype 3 knob; anti-CTLA-4 mAb insertion | Advanced solid tumors | Subcutaneous dose of OV followed by intraperitoneal ICIs | Pre-clinical [119] | |
| Anti-CTLA-4 + anti-CD25 | Vaccinia virus | Renal adenocarcinoma | Intravenous dose of OV followed by intraperitoneal ICIs | Pre-clinical [103] | |
| Anti-CTLA-4 + anti-PD-1 | VSV (VSV-HIF-2a, VSV-Sox-10, VSV-c-Myc) | c-Myc, HIF-2α, and Sox-10 insertion | Glioma | Intravenous dose of OV followed by intracranial ICIs | Pre-clinical [104] |
| Maraba virus MG1 | Triple-negative breast cancer | Intratumoral or intravenous dose of OV followed by intraperitoneal ICIs | Pre-clinical [117] | ||
| Anti-CTLA-4 or anti-PD-L1 | Measles virus (MV-aCTLA-4, MV-aPD-L1) | Anti-CTLA-4 p4F10-γ1 or anti-PD-L1 mAb insertion | Melanoma | Intratumoral injection of OV | Pre-clinical [120] |
| Anti-PD-1 | Reovirus | Melanoma | Intratumoral dose of OV followed by systemic ICIs | Pre-clinical [113] | |
| HSV-1 (T-VEC) | Deletion of ICP34.5, ICP47, human GM-CSF insertion | Unresectable Stage IIIB to IVM1c Melanoma | Intratumoral dose of OV followed by intravenous ICIs | Phase Ib/3 | |
| HSV-1 (T-VEC) | Deletion of ICP34.5, ICP47, human GM-CSF insertion | Head and neck squamous cell carcinoma | Intratumoral dose of OV followed by intravenous ICIs | Phase Ib/3 | |
| Human adenovirus 5 (DNX-2401) | 24-bp deletion in E1A, RGD-4C motif insertion in fiber | Recurrent glioblastoma or gliosarcoma | Intratumoral dose of OV followed by intravenous ICIs | Phase II | |
| Human adenovirus 5 (ONCOS-102) | Insertion of human GM-CSF | Advanced or Unresectable Melanoma | Intratumoral dose of OV followed by intravenous ICIs | Phase I | |
| Maraba virus (MG1-MAGEA3) | Insertion of human melanoma antigen A3 (MAGE-A3) | Non-Small Cell Lung Cancer | Intratumoral dose of OV followed by intravenous ICIs | Phase I/II | |
| Human adenovirus 5 (ADV/HSV-tk) | Insertion of herpes simplex virus thymidine kinase (HSV-tk) | Metastatic triple negative breast cancer and metastatic non-small cell lung cancer | Intratumoral dose of OV followed by intravenous ICIs | Phase II |
Table 3.
Combinational therapies including immune co-stimulatory agents and OVs.
| Virus | Modification in Viral Genome | Tested Disease | Route of Administration | Clinical Status |
|---|---|---|---|---|
| Vaccinia virus | Sarcoma and breast cancer | OV: intratumoral; anti-4-1BB: intraperitoneal | Pre-clinical [124] | |
| Vaccinia virus (rV-4-1BBL) | Insertion of 4-1BBL | Melanoma | Intratumoral | Pre-clinical [125] |
| Human adenovirus 5 (Ad-ΔB7/IL-12/4-1BBL) | Insertion of IL-12 and 4-1BB | Melanoma | Intratumoral | Pre-clinical [72] |
| Human adenovirus 5 (LOAd703) | Co-insertion of CD40L and 4-1BBL | Human pancreatic xenografts in nude mice | Peritumoral injection | Pre-clinical [126] |
| Human adenovirus 5 (Delta-24-RGDOX) | 24-bp deletion in E1A, RGD-4C motif insertion in fiber, insertion of OX40L | Glioma | OV: intratumoral; anti-PD-L1: intratumoral | Pre-clinical [124] |
| NDV (NDV-ICOSL) | Insertion of ICOSL | Melanoma | OV: Intratumoral; anti-CTLA-4: Intraperitoneal | Pre-clinical [127] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nguyen, T.; Avci, N.G.; Shin, D.H.; Martinez-Velez, N.; Jiang, H. Tune Up In Situ Autovaccination against Solid Tumors with Oncolytic Viruses. Cancers 2018, 10, 171. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10060171
AMA Style
Nguyen T, Avci NG, Shin DH, Martinez-Velez N, Jiang H. Tune Up In Situ Autovaccination against Solid Tumors with Oncolytic Viruses. Cancers. 2018; 10(6):171. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10060171
Chicago/Turabian StyleNguyen, Teresa, Naze G. Avci, Dong Ho Shin, Naiara Martinez-Velez, and Hong Jiang. 2018. "Tune Up In Situ Autovaccination against Solid Tumors with Oncolytic Viruses" Cancers 10, no. 6: 171. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers10060171
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.