CRISPR-Cas9 Knockdown and Induced Expression of CD133 Reveal Essential Roles in Melanoma Invasion and Metastasis

 , ,
, ,
Abstract
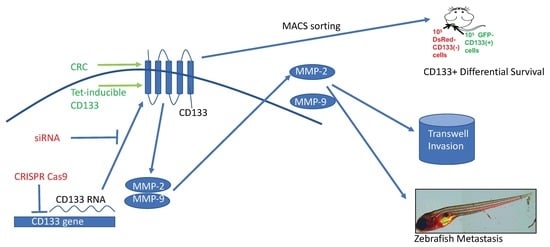
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Magnetically-Sorted (MACS) CD133(+) Melanoma Cells Survive in Xenografts and Give Rise to Tumors in Mixed Populations, But Require Reprogramming to Maintain CD133 Expression in Culture
2.2. MACS-Sorted BAK-P CD133(+) Melanoma Cells and CRC BAK-R Cells Both Exhibit Markers of Cancer Stem Cells, Form Melanospheres, and Are Resistant to Kinase Inhibitors
2.3. CRC BAK-R Melanoma Cells Exhibit Increased Invasiveness and Metastasis Compared to Parental Cells, Which Is Reversed by CD133 siRNA Knockdown
2.4. CRISPR-Cas9 Knockdown of CD133 Significantly Reduces Invasion and Metastasis in BAK-R Cells
2.5. CRISPR-Cas9 Knockdown of CD133 Expression in BAK-P Melanoma Cells Attenuates Cell Invasion and Decreases Levels of MMP9 and MMP2
2.6. Doxycycline-Inducible Expression of CD133 Levels in BAK-P Melanoma Cells Increases Cell Invasion and Levels of Secreted MMP9 and MMP2
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Magnetic Sorting and Pre- and Post-Staining for CD133-Positivity
4.3. Formation of Melanospheres
4.4. Conditional Reprogramming of CD133(+) Cells
4.5. siRNA Knockdown
| Cat # | Sequence | |
| Flexitube siRNA PROM1 1 | SI00083741 | CACGTTATAGTCCATGGTCCA |
| Flexitube siRNA PROM1 2 | SI00083748 | CAGGTAAGAACCCGGATCAAA |
| Flexitube siRNA PROM1 3 | SI00083755 | ACCTTTGAGTTTGGTCCCTAA |
| Flexitube siRNA PROM1 4 | SI03098263 | CTGGCTAAGTACTATCGTCGA |
4.6. Transwell Migration/Invasion Assay
4.7. Quantitative Reverse-Transcription PCR (qRT-PCR)
- CD133 forward-5ʹ-CCC GGG GCT GCT GTT TAT A
- CD133 reverse-5ʹ-ATC ACC AAC AGG GAG ATT G
- GAPDH forward 5ʹ-GAA GGT GAA GGT CGG AGT C
- GAPDH reverse 5ʹ-C GAA GAT GGT GAT GGG ATT TC
4.8. Mouse Xenografting
4.9. Zebrafish Injection and Imaging
4.10. CRISPR-Cas9 Knockdown of CD133
- sgRNA1: CAACAGGGAGCCGAGTACGA (complement of Target 1 underlined below)
- sgRNA2: TTCATCCACAGATGCTCCTA (complement of Target 2 underlined below)
- sgRNA3: TTACCTTCTGGGAAATCACGC (complement of Target 3 underlined below)
- ATG GCC CTC GTA CTC GGC TCC CTG TTG CTG CTG GGG CTG TGC GGG AACT1
- TCC TTT TCA GGA GGG CAG CCT TCA TCC ACA GAT GCT CCT AAG GCT TGGT2
- AAT TAT GAA TTG CCT GCA ACA AAT TAT GAG ACC CAA GAC TCC CAT AAA GCT GGA CCC ATT GGC ATT CTC TTT GAA CTA GTG CAT ATC TTT CTT ATG TGG TAC AGC CGC GTG ATT TCC CAG AAG GTA AT3
- Target 1-forward (F1a) TTCCCCAAGGCTTCCAGAAG
- Target 1-reverse (R1a) GCCCTCCTGAAAAGGAGTTC
- Target 2-forward (F2a) GAACTCCTTTTCAGGAGGGC
- Target 2-reverse (R2a) GAGAATGCCAATGGGTCCAG
- Target 3-forward (F3a) CTGGACCCATTGGCATTCTC
- Target 3-reverse (R3a) CATTCT1TCCCTGCCATCAGC
4.11. Generation of Dox-Inducible Cells
4.12. Immunoblot Analysis
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.H.; Miele, M.E.; Hicks, D.J.; Phillips, K.K.; Trent, J.M.; Weissman, B.E.; Welch, D.R. KiSS-1, a Novel Human Malignant Melanoma Metastasis-Suppressor Gene. J. Natl. Cancer Inst. 1996, 88, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Santos, G.C.; Zielenska, M.; Prasad, M.; Squire, J.A. Chromosome 6p amplification and cancer progression. J. Clin. Pathol. 2007, 60, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, R.; Bhutkar, A.; McNamara, M.C.; Lees, J.A. BMI1 induces an invasive signature in melanoma that promotes metastasis and chemoresistance. Genes Dev. 2016, 30, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.X.; Massagué, J. Genetic determinants of cancer metastasis. Nat. Rev. Genet. 2007, 8, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural Stem Cells and the Origin of Gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Yin, A.H.; Miraglia, S.; Zanjani, E.D.; Almeida-Porada, G.; Ogawa, M.; Leary, A.G.; Olweus, J.; Kearney, J.; Buck, D.W. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 1997, 90, 5002–5012. [Google Scholar] [PubMed]
- Yu, Y.; Flint, A.; Dvorin, E.L.; Bischoff, J. AC133-2, a Novel Isoform of Human AC133 Stem Cell Antigen. J. Biol. Chem. 2002, 277, 20711–20716. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Gibson, P.; Currle, D.S.; Tong, Y.; Richardson, R.J.; Bayazitov, I.T.; Poppleton, H.; Zakharenko, S.; Ellison, D.W.; Gilbertson, R.J. Prominin 1 marks intestinal stem cells that are susceptible to neoplastic transformation. Nature 2009, 457, 603–607. [Google Scholar] [CrossRef]
- Mizrak, D.; Brittan, M.; Alison, M. CD133: Molecule of the moment. J. Pathol. 2008, 214, 3–9. [Google Scholar] [CrossRef]
- Shmelkov, S.V.; St.Clair, R.; Lyden, D.; Rafii, S. AC133/CD133/Prominin-1. Int. J. Biochem. Cell Biol. 2005, 37, 715–719. [Google Scholar] [CrossRef]
- Dowland, S.N.; Madawala, R.J.; Poon, C.E.; Lindsay, L.A.; Murphy, C.R. Prominin-1 glycosylation changes throughout early pregnancy in uterine epithelial cells under the influence of maternal ovarian hormones. Reprod. Fertil. Dev. 2017, 29, 1194–1208. [Google Scholar] [CrossRef] [PubMed]
- Maw, M.A. A frameshift mutation in prominin (mouse)-like 1 causes human retinal degeneration. Hum. Mol. Genet. 2000, 9, 27–34. [Google Scholar] [CrossRef]
- Zhang, Q.; Zulfiqar, F.; Xiao, X.; Riazuddin, S.A.; Ahmad, Z.; Caruso, R.; Macdonald, I.; Sieving, P.; Riazuddin, S.; Hejtmancik, J.F. Severe retinitis pigmentosa mapped to 4p15 and associated with a novel mutation in the PROM1 gene. Qual. Life Res. 2007, 122, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Chen, Y.; Lillo, C.; Chien, J.; Yu, Z.; Michaelides, M.; Klein, M.; Howes, K.A.; Li, Y.; Kaminoh, Y.; et al. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J. Clin. Investig. 2008, 118, 2908–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; E Bonn, V.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Ferrandina, G.; Bonanno, G.; Pierelli, L.; Perillo, A.; Procoli, A.; Mariotti, A.; Corallo, M.; Martinelli, E.; Rutella, S.; Paglia, A.; et al. Expression of CD133-1 and CD133-2 in ovarian cancer. Int. J. Gynecol. Cancer 2008, 18, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Suetsugu, A.; Nagaki, M.; Aoki, H.; Motohashi, T.; Kunisada, T.; Moriwaki, H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem. Biophys. Res. Commun. 2006, 351, 820–824. [Google Scholar] [CrossRef]
- Collins, A.T. Prospective Identification of Tumorigenic Prostate Cancer Stem Cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [Green Version]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; de Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Kaidina, A.M.; Chiang, J.-H.; Yarygin, K.N.; Lupatov, A.Y. Cancer stem cell molecular markers verified in vivo. Biochem. Mosc. Suppl. Ser. B Biomed. Chem. 2017, 11, 43–54. [Google Scholar] [CrossRef]
- Lai, G.M.; Yao, C.J.; Yeh, C.T.; Yeh, C.F.; Chang, C.K.; Yan, J.L.; Li, C.H.; Chuang, S.E. Active fraction of Taiwanofungus camphoratus (HS7) exerted anticancer effects through multiple molecule targeting and elimination of cancer stem-like cells in lung cancer and hepatoma cells. Cancer Res. 2009, 69, 615. [Google Scholar]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galli, R. Isolation and Characterization of Tumorigenic, Stem-like Neural Precursors from Human Glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [Green Version]
- Quintana, E.; Shackleton, M.; Sabel, M.S.; Fullen, D.R.; Johnson, T.M.; Morrison, S.J. Efficient tumour formation by single human melanoma cells. Nature 2008, 456, 593–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrix, M.J.C.; Seftor, E.A.; Seftor, R.E.B.; Kasemeier-Kulesa, J.; Kulesa, P.M.; Postovit, L.-M. Reprogramming metastatic tumour cells with embryonic microenvironments. Nat. Rev. Cancer 2007, 7, 246–255. [Google Scholar] [CrossRef]
- Seftor, R.E.; Hess, A.R.; Seftor, E.A.; Kirschmann, D.A.; Hardy, K.M.; Margaryan, N.V.; Hendrix, M.J. Tumor cell vasculogenic mimicry: From controversy to therapeutic promise. Am. J. Pathol. 2012, 181, 1115–1125. [Google Scholar] [CrossRef]
- Kirschmann, D.A.; Seftor, E.A.; Hardy, K.M.; Seftor, R.E.; Hendrix, M.J. Molecular pathways: Vasculogenic mimicry in tumor cells: Diagnostic and therapeutic implications. Clin. Cancer Res. 2012, 18, 2726–2732. [Google Scholar] [CrossRef] [PubMed]
- Mihic-Probst, D.; Ikenberg, K.; Tinguely, M.; Schraml, P.; Behnke, S.; Seifert, B.; Civenni, G.; Sommer, L.; Moch, H.; Dummer, R. Tumor Cell Plasticity and Angiogenesis in Human Melanomas. PLoS ONE 2012, 7, e33571. [Google Scholar] [CrossRef] [PubMed]
- Simbulan-Rosenthal, C.M.; Gaur, A.; Zhou, H.; Abdussamad, M.; Qin, Q.; Dougherty, R.; Aljehane, L.; Kuo, L.W.; Vakili, S.; Karna, K.; et al. CD133 Is Associated with Increased Melanoma Cell Survival after Multikinase Inhibition. J. Oncol. 2019, 2019, 6486173. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.K.; Manglik, V.; O’Connell, M.; Weeraratna, A.; McCarron, E.C.; Broussard, J.N.; Divito, K.A.; Simbulan-Rosenthal, C.M.; Rosenthal, D.S.; Zapas, J.L. Clonal dominance of CD133+ subset population as risk factor in tumor progression and disease recurrence of human cutaneous melanoma. Int. J. Oncol. 2012, 41, 1570–1576. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.C.; Yang, Y.J.; Zhang, J.Q.; Guo, M.; Xiang, L.; Yu, S.F.; Ping, H.; Zhuo, L. microRNA—448 inhibits stemness maintenance and self-renewal of hepatocellular carcinoma stem cells through the MAGEA6-mediated AMPK signaling pathway. J. Cell. Physiol. 2019, 234, 23461–23474. [Google Scholar] [CrossRef]
- Miyabayashi, T.; Kagamu, H.; Koshio, J.; Ichikawa, K.; Baba, J.; Watanabe, S.; Tanaka, H.; Tanaka, J.; Yoshizawa, H.; Nakata, K.; et al. Vaccination with CD133(+) melanoma induces specific Th17 and Th1 cell-mediated antitumor reactivity against parental tumor. Cancer Immunol. Immunother. 2011, 60, 1597–1608. [Google Scholar] [CrossRef]
- Hofmann, U.B.; Westphal, J.R.; Van Muijen, G.N.; Ruiter, D.J. Matrix Metalloproteinases in Human Melanoma. J. Investig. Dermatol. 2000, 115, 337–344. [Google Scholar] [CrossRef] [Green Version]
- Saladi, S.V.; Keenen, B.; Marathe, H.G.; Qi, H.; Chin, K.-V.; De La Serna, I.L. Modulation of extracellular matrix/adhesion molecule expression by BRG1 is associated with increased melanoma invasiveness. Mol. Cancer 2010, 9, 280. [Google Scholar] [CrossRef]
- Sandri, S.; Faião-Flores, F.; Tiago, M.; Pennacchi, P.C.; Massaro, R.R.; Alves-Fernandes, D.K.; Berardinelli, G.N.; Evangelista, A.F.; Vazquez, V.D.L.; Reis, R.M.; et al. Vemurafenib resistance increases melanoma invasiveness and modulates the tumor microenvironment by MMP-2 upregulation. Pharmacol. Res. 2016, 111, 523–533. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular Matrix Degradation and Remodeling in Development and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Tang, Z.Y.; Liu, Y.; Liu, L.X.; Ding, X.Y.; Zhang, H.; Fang, L.Q. RNAi-mediated MMP-9 silencing inhibits mouse melanoma cell invasion and migration in vitro and in vivo. Cell Biol. Int. 2013, 37, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Liu, L.; Liu, L.; Geng, J.; Zhou, Y.; Chen, L. β-Elemene inhibits the metastasis of B16F10 melanoma cells by downregulation of the expression of uPA, uPAR, MMP-2, and MMP-9. Melanoma Res. 2014, 24, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Guarneri, C.; Bevelacqua, V.; Polesel, J.; Falzone, L.; Cannavò, P.S.; Spandidos, D.A.; Malaponte, G.; Libra, M. NFκB inhibition is associated with OPN/MMP9 downregulation in cutaneous melanoma. Oncol. Rep. 2017, 37, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G.M.; Ledee, D.R.; Feuer, W.J.; Fini, M.E. Cytokines and signaling pathways regulating matrix metalloproteinase-9 (MMP-9) expression in corneal epithelial cells. J. Cell. Physiol. 2009, 221, 402–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.-J.; Neoh, C.-A.; Tsao, C.-Y.; Su, J.-H.; Li, H.-H. Sinulariolide Suppresses Human Hepatocellular Carcinoma Cell Migration and Invasion by Inhibiting Matrix Metalloproteinase-2/-9 through MAPKs and PI3K/Akt Signaling Pathways. Int. J. Mol. Sci. 2015, 16, 16469–16482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falzone, L.; Salemi, R.; Travali, S.; Scalisi, A.; McCubrey, J.A.; Candido, S.; Libra, M. MMP-9 overexpression is associated with intragenic hypermethylation of MMP9 gene in melanoma. Aging 2016, 8, 933–940. [Google Scholar] [CrossRef]
- Dange, M.C.; Agarwal, A.K.; Kalraiya, R.D. Extracellular galectin-3 induces MMP9 expression by activating p38 MAPK pathway via lysosome-associated membrane protein-1 (LAMP1). Mol. Cell. Biochem. 2015, 404, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Wu, Y.; Wang, Y.; Zhou, M.; Yan, S.; Chen, Z.; Gu, D.; Cai, Y. Liquiritigenin Potentiates the Inhibitory Effects of Cisplatin on Invasion and Metastasis Via Downregulation MMP-2/9 and PI3 K/AKT Signaling Pathway in B16F10 Melanoma Cells and Mice Model. Nutr. Cancer 2015, 67, 761–770. [Google Scholar] [CrossRef]
- Falzone, L.; Candido, S.; Salemi, R.; Basile, M.S.; Scalisi, A.; McCubrey, J.A.; Torino, F.; Signorelli, S.S.; Montella, M.; Libra, M. Computational identification of microRNAs associated to both epithelial to mesenchymal transition and NGAL/MMP-9 pathways in bladder cancer. Oncotarget 2016, 7, 72758–72766. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Song, X.; Zhu, J.; Ji, Y.; Wu, F.; Chen, Y.; Cui, X.; Hu, J.; Wang, L.; Wei, Y.; et al. Tumor suppressor microRNA-34a inhibits cell migration and invasion by targeting MMP-2/MMP-9/FNDC3B in esophageal squamous cell carcinoma. Int. J. Oncol. 2017, 51, 378–388. [Google Scholar] [CrossRef] [Green Version]
- Spagnolo, F.; Picasso, V.; Lambertini, M.; Ottaviano, V.; Dozin, B.; Queirolo, P. Survival of patients with metastatic melanoma and brain metastases in the era of MAP-kinase inhibitors and immunologic checkpoint blockade antibodies: A systematic review. Cancer Treat. Rev. 2016, 45, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Ugurel, S.; Röhmel, J.; Ascierto, P.A.; Flaherty, K.T.; Grob, J.J.; Hauschild, A.; Larkin, J.; Long, G.V.; Lorigan, P.; McArthur, G.A.; et al. Survival of patients with advanced metastatic melanoma: The impact of novel therapies–update 2017. Eur. J. Cancer 2017, 83, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Kalal, B.S.; Upadhya, D.; Pai, V.R. Chemotherapy Resistance Mechanisms in Advanced Skin Cancer. Oncol. Rev. 2017, 11, 326. [Google Scholar] [CrossRef] [PubMed]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.-P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Hodis, E.; Heffernan, T.P.; Deribe, Y.L.; Lawrence, M.S.; Protopopov, A.; Ivanova, E.; Watson, I.R.; Nickerson, E.; Ghosh, P.; et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012, 485, 502. [Google Scholar] [CrossRef]
- Pleasance, E.D.; Cheetham, R.K.; Stephens, P.J.; McBride, D.J.; Humphray, S.J.; Greenman, C.D.; Varela, I.; Lin, M.L.; Ordóñez, G.R.; Bignell, G.R.; et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2009, 463, 191. [Google Scholar] [CrossRef] [PubMed]
- McCourt, C.; Dolan, O.; Gormley, G. Malignant Melanoma: A Pictorial Review. Ulst. Med J. 2014, 83, 103–110. [Google Scholar]
- Tejera-Vaquerizo, A.; Descalzo-Gallego, M.; Otero-Rivas, M.; Posada-García, C.; Rodríguez-Pazos, L.; Pastushenko, I.; Marcos-Gragera, R.; García-Doval, I. Skin Cancer Incidence and Mortality in Spain: A Systematic Review and Meta-Analysis. Actas Dermo-Sifiliográficas (Engl. Ed.) 2016, 107, 318–328. [Google Scholar] [CrossRef]
- Brunssen, A.; Waldmann, A.; Eisemann, N.; Katalinic, A. Impact of skin cancer screening and secondary prevention campaigns on skin cancer incidence and mortality: A systematic review. J. Am. Acad. Dermatol. 2017, 76, 129–139.e10. [Google Scholar] [CrossRef]
- Balint, K.; Xiao, M.; Pinnix, C.C.; Soma, A.; Veres, I.; Juhász, I.; Brown, E.J.; Capobianco, A.J.; Herlyn, M.; Liu, Z.J. Activation of Notch1 signaling is required for β-catenin–mediated human primary melanoma progression. J. Clin. Investig. 2005, 115, 3166–3176. [Google Scholar] [CrossRef]
- Wang, E.; Voiculescu, S.; Le Poole, I.C.; El-Gamil, M.; Li, X.; Sabatino, M.; Robbins, P.F.; Nickoloff, B.J.; Marincola, F.M. Clonal Persistence and Evolution During a Decade of Recurrent Melanoma. J. Investig. Dermatol. 2006, 126, 1372–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I.; et al. Rosen, BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006, 439, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; Van Herpen, C.M.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef]
- Bhatia, S.; Tykodi, S.S.; Thompson, J.A. Treatment of metastatic melanoma: An overview. Oncology (Williston Park. N.Y.) 2009, 23, 488–496. [Google Scholar]
- Liu, X.; Ory, V.; Chapman, S.; Yuan, H.; Albanese, C.; Kallakury, B.; Timofeeva, O.A.; Nealon, C.; Dakic, A.; Simic, V.; et al. ROCK Inhibitor and Feeder Cells Induce the Conditional Reprogramming of Epithelial Cells. Am. J. Pathol. 2012, 180, 599–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suprynowicz, F.A.; Upadhyay, G.; Krawczyk, E.; Kramer, S.C.; Hebert, J.D.; Liu, X.; Yuan, H.; Cheluvaraju, C.; Clapp, P.W.; Boucher, R.C.; et al. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, 20035–20040. [Google Scholar] [CrossRef] [Green Version]
- Echeverri, C.J.; Beachy, P.A.; Baum, B.; Boutros, M.; Buchholz, F.; Chanda, S.K.; Downward, J.; Ellenberg, J.; Fraser, A.G.; Hacohen, N.; et al. Minimizing the risk of reporting false positives in large-scale RNAi screens. Nat. Methods 2006, 3, 777–779. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Creanga, A.; Lum, L.; Beachy, P.A. Prevalence of off-target effects in Drosophila RNA interference screens. Nature 2006, 443, 359–363. [Google Scholar] [CrossRef]
- Ohnishi, S.; Maehara, O.; Nakagawa, K.; Kameya, A.; Otaki, K.; Fujita, H.; Higashi, R.; Takagi, K.; Asaka, M.; Sakamoto, N.; et al. Hypoxia-Inducible Factors Activate CD133 Promoter through ETS Family Transcription Factors. PLoS ONE 2013, 8, e66255. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, Y.; Feng, H.; Bian, X.; Zhao, W.; Yang, Z.; Gu, B.; Li, Z.; Liu, Y. CD133 Affects the Invasive Ability of HCT116 Cells by Regulating TIMP-2. Am. J. Pathol. 2013, 182, 565–576. [Google Scholar] [CrossRef]
- Wang, S.S.; Gao, X.L.; Liu, X.; Gao, S.Y.; Fan, Y.L.; Jiang, Y.P.; Ma, X.R.; Jiang, J.; Feng, H.; Chen, Q.M.; et al. CD133+ cancer stem-like cells promote migration and invasion of salivary adenoid cystic carcinoma by inducing vasculogenic mimicry formation. Oncotarget 2016, 7, 29051–29062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiVito, K.A.; Simbulan-Rosenthal, C.M.; Chen, Y.S.; Trabosh, V.A.; Rosenthal, D.S. Id2, Id3 and Id4 overcome a Smad7-mediated block in tumorigenesis, generating TGF-beta-independent melanoma. Carcinogenesis 2014, 35, 951–958. [Google Scholar] [CrossRef] [PubMed]
- DiVito, K.A.; Trabosh, V.A.; Chen, Y.-S.; Chen, Y.; Albanese, C.; Javelaud, D.; Mauviel, A.; Simbulan-Rosenthal, C.M.; Rosenthal, D.S. Smad7 restricts melanoma invasion by restoring N-cadherin expression and establishing heterotypic cell-cell interactions in vivo. Pigment. Cell Melanoma Res. 2010, 23, 795–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogh, J.; Trempe, G. New Human Tumor Cell Lines. In Human Tumor Cells in Vitro; Fogh, J., Ed.; Plenum Publishing Corp: New York, NY, USA, 1975. [Google Scholar]
- Simon, H.G.; Risse, B.; Jost, M.; Oppenheimer, S.; Kari, C.; Rodeck, U. Identification of differentially expressed messenger RNAs in human melanocytes and melanoma cells. Cancer Res. 1996, 56, 3112–3117. [Google Scholar] [PubMed]
- Euhus, D.M.; Hudd, C.; LaRegina, M.C.; Johnson, F.E. Tumor measurement in the nude mouse. J. Surg. Oncol. 1986, 31, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simbulan-Rosenthal, C.M.; Dougherty, R.; Vakili, S.; Ferraro, A.M.; Kuo, L.-W.; Alobaidi, R.; Aljehane, L.; Gaur, A.; Sykora, P.; Glasgow, E.; et al. CRISPR-Cas9 Knockdown and Induced Expression of CD133 Reveal Essential Roles in Melanoma Invasion and Metastasis. Cancers 2019, 11, 1490. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101490
Simbulan-Rosenthal CM, Dougherty R, Vakili S, Ferraro AM, Kuo L-W, Alobaidi R, Aljehane L, Gaur A, Sykora P, Glasgow E, et al. CRISPR-Cas9 Knockdown and Induced Expression of CD133 Reveal Essential Roles in Melanoma Invasion and Metastasis. Cancers. 2019; 11(10):1490. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101490
Chicago/Turabian StyleSimbulan-Rosenthal, Cynthia M., Ryan Dougherty, Sahar Vakili, Alexandra M. Ferraro, Li-Wei Kuo, Ryyan Alobaidi, Leala Aljehane, Anirudh Gaur, Peter Sykora, Eric Glasgow, and et al. 2019. "CRISPR-Cas9 Knockdown and Induced Expression of CD133 Reveal Essential Roles in Melanoma Invasion and Metastasis" Cancers 11, no. 10: 1490. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers11101490